

Abstract
BACKGROUND AND PURPOSE
Interstitial fibrosis plays a causal role in the development of heart failure after chronic myocardial infarction (MI), and anti-fibrotic therapy represents a promising strategy to mitigate this pathological process. The purpose of this study was to investigate the effect of long-term administration of scutellarin (Scu) on cardiac interstitial fibrosis of myocardial infarct rats and the underlying mechanisms.
EXPERIMENTAL APPROACH
Scu was administered to rats that were subjected to coronary artery ligation. Eight weeks later, its effects on cardiac fibrosis were assessed by examining cardiac function and histology. The number and collagen content of cultured cardiac fibroblasts exposed to angiotensin II (Ang II) were determined after the administration of Scu in vitro. Protein expression was detected by Western blot technique, and mRNA levels by quantitative reverse transcription-PCR.
KEY RESULTS
The echocardiographic and haemodynamic measurements showed that Scu improved the impaired cardiac function of infarct rats and decreased interstitial fibrosis. Scu inhibited the expression of FN1 and TGFβ1, but produced no effects on inflammatory cytokines (TNFα, IL-1β and IL-6) in the 8 week infarct hearts. Scu inhibited the proliferation and collagen production of cardiac fibroblasts (CFs) and the up-regulation of FN1 and TGFβ1 induced by Ang II. The enhanced phosphorylation of p38-MAPK and ERK1/2 in both infarct cardiac tissue and cultured CFs challenged by Ang II were suppressed by Scu.
CONCLUSIONS AND IMPLICATIONS
Long-term administration of Scu improved the cardiac function of MI rats by inhibiting interstitial fibrosis, and the mechanisms may involve the suppression of pro-fibrotic cytokine TGFβ1 expression and inhibition of p38 MAPK and ERK1/2 phosphorylation.
Keywords: cardiac infarction, ERK1/2, fibrosis, p38-MAPK, scutellarin, TGFβ1
Introduction
Currently, chronic heart failure (CHF) remains one of the leading causes of morbidity and mortality worldwide, with cardiac remodelling after myocardial infarction (MI) as the most common underlying factor (Pfeffer, 1995; Sutton and Sharpe, 2000; Yousef et al., 2000). Significant fibrosis, occurring in both the infarcted and non-infarcted myocardium, represents a characteristic pathological alteration of post-infarct remodelling and is recognized to be a major determinant of the progressive deterioration of ventricular function after MI (Pfeffer, 1995; Sutton and Sharpe, 2000; Yousef et al., 2000). Generally, fibrosis is modulated by a cascade of biochemical intracellular signalling processes that is triggered by necrotic myocardium in the context of MI in combination with increased preload and afterload (Pfeffer, 1995; Sutton and Sharpe, 2000; Yousef et al., 2000).
Transforming growth factor-β1 is a key signalling molecule that induces cardiac fibrosis by activating the proliferation and collagen production of cardiac fibroblasts (Bujak and Frangogiannis, 2007). Intracellular mitogen-activated protein kinase (MAPK) signalling cascades were also proven to play an important role in the pathogenesis of cardiac fibrosis (Muslin, 2008). dministration of RWJ-67657, a p38α and p38β inhibitor, to long-term MI rats resulted in suppressed interstitial fibrosis and improved cardiac function (See et al., 2004). Cardiac fibroblasts (CFs), which rapidly proliferate in MI, are responsible for the main deposition of extracellular matrix (ECM). The augmentation of extracellular signal-regulated kinase (ERK1/2) activity caused by the up-regulation of miR-21 enhances CF proliferation and thereby the interstitial fibrosis and cardiac dysfunction (Thum et al., 2008). The activation of the ERK1/2 cascade underlies the angiotensin II (Ang II)-induced proliferation of CFs (Schorb et al., 1995; Stockand and Meszaros, 2003). Although major therapeutic advances have been made in the management of MI, post-infarction CHF is still a common cause of morbidity, hospitalization and premature death. Efficient strategies that could regress fibrosis after infarction would open up a new route for preventing the exacerbation of cardiac function and improving the post-infarct prognosis (Dorn, 2009). Agents that can affect the molecular pathways that are elicited during post-infarct remodelling are promising therapeutic candidates.
Scutellarin (Scu) (Figure 1) is a flavonoid extracted from a traditional Chinese herb, Erigeron breviscapus (vant.) Hand-Mazz. E. breviscapus Hand Mazz is the main component of the Chinese herbal drug that has been used to treat cardiovascular diseases in China for a long time. Nowadays, various preparations of Scu are still widely used clinically to treat cardiovascular diseases such as hypertension, angina pectoris, coronary heart disease and stroke. Recent experimental studies have indicated that Scu exhibits a variety of pharmacological properties, including decreasing the infarct size of rats with myocardial ischaemia (Li et al., 2004; Lin et al., 2007; Jia et al., 2008), suppressing cardiac hypertrophy (Pan et al., 2010), relaxing rat isolated aortic rings (Pan et al., 2008) and protecting PC12 cells from cobalt chloride-induced apoptosis (Wang et al., 2007). However, there remains a lack of evidence for the role of Scu in post-infarct remodelling. The present study was performed to explore the effects of Scu on myocardial fibrosis after long-term MI and the underlying mechanisms in rats.
Figure 1.
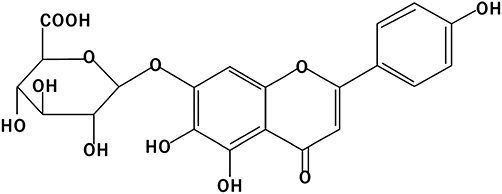
Chemical structure of scutellarin.
Methods
Animals
Healthy male Wistar rats (200–250 g) used in the current study were kept under standard animal room conditions (temperature 21 ± 1°C; humidity 55–60%) with food and water available ad libitum for 1 week before the experiment. All experimental procedures were in accordance with the Institutional Animal Care and Use Committee of Harbin Medical University, China.
MI and Scu administration
Male Wistar rats were subjected to left anterior decending (LAD) coronary artery ligation to induce MI as described previously (Yang et al., 2007). Briefly, rats were anaesthetized with 1% sodium phenobarbital (40–60 mg kg−1, i.p.) and intubated orally with a polyethylene tube for artificial respiration (UGO Basile S.R.L. Biological Research Apparatus, Comerio VA, Lombardy, Italy). A thoracotomy was performed at the fourth intercostal space, and the LAD branch was ligated approximately 2 mm from its origin. Sham-operated animals underwent the same procedure but the coronary ligature was left untied (Sham group, n = 8). The rats with MI that survived were randomly assigned into four groups: MI, MI + Scu (3, 10 and 30 mg kg−1 day−1, i.p.), with nine rats in each group. Drug administration was started 3 days after surgery, and continued for 8 weeks. Sham and MI control rats were injected with the same volume of saline containing DMSO (final concentration = 0.1%). Two additional groups of normal rats (n = 8 each) were injected with Scu (30 mg kg−1) (Scu group) or the same volume of saline containing DMSO (final concentration = 0.1%) (normal group).
Echocardiographic and haemodynamic measurements
Four and eight weeks after drug administration, the changes in left ventricular function were evaluated by transthoracic echocardiography with an ultrasound machine (Vivid 7, GE Medical, USA) equipped with a 10-MHz phased-array transducer. Left ventricular systolic diameter (LVSd) and left ventricular diastolic diameter (LVDd) were measured at the same time, and left ventricular ejection fraction and fractional shortening (FS) were calculated from M-mode recording. Haemodynamic studies were carried out 8 weeks after drug administration by use of a pressure volume control unit (Scisense Inc., London, Ontario, Canada) with the degree of anaesthesia adjusted down to the level where rats just started responding to toe pinch. A pressure-sensing catheter (1.9F, Scisense Inc.) was inserted into the left ventricle of the rats via the right common carotid artery. Parameters including heart rate, left ventricular systolic pressure, left ventricular distolic pressure, +dP/dtmax and −dP/dtmax were collected. After the haemodynamic measurements had been obtained, the rats were killed and the heart was removed rapidly and washed in ice-cold 0.9% saline. Parts of the heart were frozen in liquid nitrogen or fixed in 4% paraformalin for later use.
Histological analysis of collagen deposition and infarct size
Eight weeks after drug administration, rats were anaesthetized with 1% sodium phenobarbital (40–60 mg kg−1, i.p.) and the hearts were collected. The right ventricle and atrium were cut off, and the left ventricle were fixed in 4% paraformalin, subjected to paraffin, then cross-sectionally cut into 5 µm thick sections along the centre of the fibrotic scar. Haematoxylin and eosin staining was used to distinguish the infarct and normal area. Masson's trichrome staining was used to evaluate collagen deposition. Sections were imaged at 200× magnification by bright-field microscopy (IX71, Olympus, Tokyo, Japan). The extent of cardiac fibrosis in the peri-infarct region was assessed by calculating collagen volume fraction. Infarct size was assessed by examining images obtained at low magnification and calculated as the ratio of scar average circumferences to LV average inner circumferences. All quantitative evaluations were carried out by ImagePro Plus software (version 6.0, Media Cybernetics, Bethesda, MD, USA).
Quantitative reverse transcription-PCR
Total RNA was extracted using TRIZOL reagent from cultured neonatal cardiac myocytes and cardiac tissues. After reverse transcription, the cDNA obtained was used in quantitative reverse transcription-PCR analysis to determine the expression of TGFβ1, FN1, IL-1β, IL-6, TNFα. The mRNA levels were quantified by SYBR Green incorporation on ABI 7500 fast Real Time PCR system (Applied Biosystems, Carlsbad, CA, USA), and GAPDH was used as an internal control. The sequences of primers were: TGFβ1, forward, 5′-GCGCCTGCAGAGATTCAAGTCAAC-3′, reverse, 5′-GTATCAGTGGGGGTCAGCAGCC-3′; FN1, forward, 5′- ACTTCTGGTCCTCTCCCGTGTCC -3′, reverse, 5′- CGCCCTCTCCAGGAGGCTAGT -3′; IL-1β, forward, 5′- GCTAGGGAGCCCCCTTGTCGAG-3′, reverse, 5′- AGGCAGGGAGGGAAACACACGTT-3′; IL-6, forward, 5′- TCCGCAAGAGACTTCCAGCCAG-3′, reverse, 5′- TGTGAAGTAGGGAAGGCAGTGGC-3′; TNFα, forward, 5′- GCCTCTTCTCATTCCTGC-3′, reverse, 5′-CTTCTCCTCCTTGTTGGG-3′; GAPDH, forward, 5′- TCTACATGTTCCAGTATGACTC-3′, reverse, 5′-ACTCCACGACATACTCAGCACC-3′.
Isolation and culture of cardiac fibroblasts
The cultured CFs used in these experiments were obtained from neonatal Wistar rats as previously described (Yang et al., 2007), with slight modifications. Briefly, after being treated with 0.25% trypsin solution, fibroblasts were isolated by the removal of myocytes through selective adhesion of non-myocytes at a 1.5 h pre-plating interval. CFs were maintained in DMEM supplemented with penicillin and streptomycin (1%v v−1) and foetal bovine serum (10% v/v). CFs at the 3rd or 4th passage were used in the experiments.
Fibroblast proliferation assay
CF proliferation was measured by counting cell number. CFs were seeded in 24-well tissue culture plates (2 × 104 cells per well) in DMEM supplemented with 10% fetal bovine serum (FBS). After the CFs had been deprived of serum for 24 h by being placed in serum-free DMEM, angiotensin II (Ang II, 1 × 10−7 mol mL−1) alone or in combination with scutellairn (0, 10, 30, 100 µM) was then added to the medium and the CFs were incubated for another 48 h. The CFs were then trypsinized and counted by using a haemocytometer.
Measurement of collagen production
To test the total collagen production, primary cardiac myofibroblasts were seeded in equal numbers in 100-mm cell culture dishes and grown until confluent in DMEM with 10% FBS. The cells were deprived of serum for 48 h before the addition of Ang II and Scu. Then after being incubated for 48 h, the hydroxyproline content in CFs was measured using a hydroxyproline assay kit according to the manufacturer's instruction. The results are expressed as degree of alteration compared with values from control CFs.
Protein isolation and western blot
The total amount of protein was extracted from the cultured CFs and the left ventricular peri-infarct region of rats for immunoblotting analysis, by use of procedures essentially the same as described in detail elsewhere (Yang et al., 2007). Briefly, the protein content was determined by use of a bicinchoninic acid protein assay kit using bovine serum albumin as the standard. Protein samples (100 µg) were subjected to 10% SDS-PAGE and blotted to nitrocellulose. The blots were blocked by 5% non-fat milk and dissolved in phosphate-buffered saline (PBS) for 1 h, then probed with ERK (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-ERK (Santa Cruz Biotechnology), p38-MAPK (Santa Cruz Biotechnology), phospho-p38-MAPK (Cell Signaling Technology, Danvers, MA, USA) antibodies and GAPDH (Kangcheng Inc., Shanghai, China) in PBS and incubated overnight at 4°C. Membranes were washed three times, 15 min each time, with PBS containing 0.5% Tween 20 (PBS-T) and incubated with secondary antibody (Alexa Fluor, Molecular Probes, Eugene, OR, USA) for 1 h. Western blot bands were collected by using Imaging System (LI-COR Biosciences, Lincoln, NE, USA) and quantified with Odyssey v1.2 software (LI-COR Biosciences, Lincoln, NE, USA) by measuring the band intensity (area × OD) in each group and normalizing this to that obtained with GAPDH as an internal control.
Statistical analysis
All data are expressed as mean ± SEM. Statistical analysis was performed using one-way analysis of variance followed by Dunnett's t-test. Differences were considered to be statistically significant when P < 0.05.
Materials
Scu (purity >95%) was purchased from Yunnan Yuxi Wangzilong Pharmaceutical Co., Ltd. (Yuxi City, Yunnan, China). Scu was dissolved in DMSO and diluted with saline before use. The final concentration of DMSO was less than 0.1%. The hydroxyproline assay kit was obtained from Nanjing Jiancheng Bioengineering Research Institute (Nanjing, China). Angiotensin II was obtained from Sigma Chemical Co. (St. Louis, MO, USA).
Results
Mortality, infarct size and heart weight
One rat in the MI group died after the commencement of treatment 3 days post-MI induction; there was no mortality in the Scu-treated or sham groups. Infarct size was similar in all the groups (Figure 2), indicating that the degree of pro-fibrotic stimulation was the same for all the drug-treated groups. Body weight and left ventricle/body weight ratio were not significantly altered among the groups. The increase in heart/body weight ratio was significantly attenuated with Scu treatment (Table 1). Scu produced no effects on these parameters in normal rats.
Figure 2.
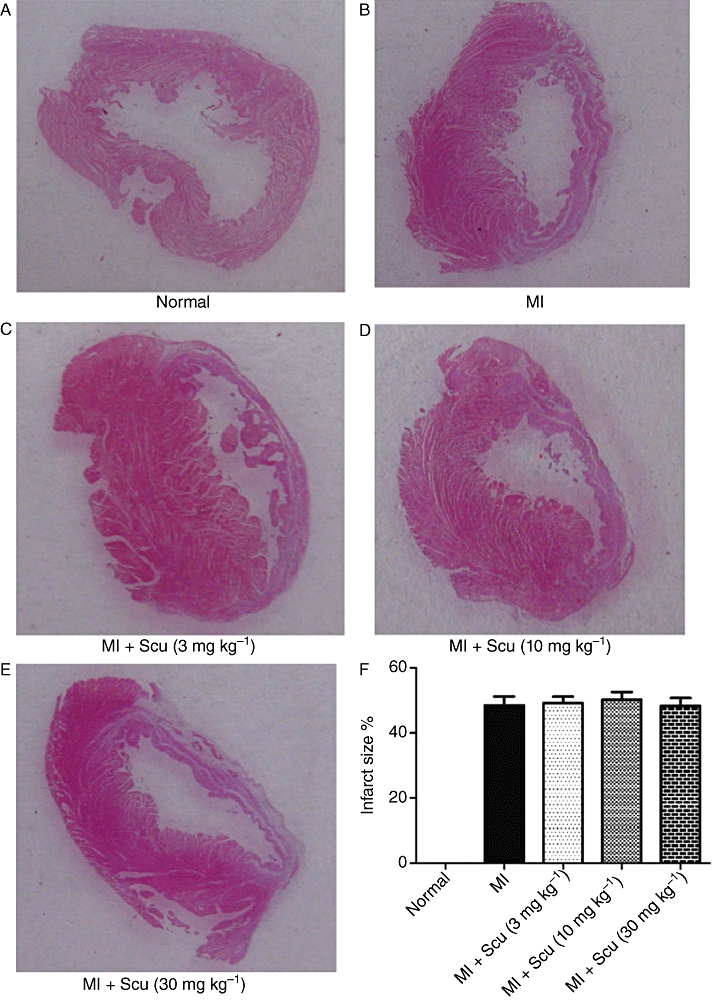
Effects of scutellarin (Scu) on infarct size. (A–E) Representative pictures of infarct heart. (F) Histogram depicting the effects of scutellarin on infarct size. Data are expressed as mean ± SEM, n = 8–9. MI, myocardial infarction.
Table 1.
Tissue weights of rats before and after MI and treatment with scutellarin
| Group | Normal (n = 8) | Scutellarin (30 mg kg−1 day−1n = 8) | Sham (n = 8) | MI (n = 8) | MI + Scu (3 mg kg−1 day−1n = 9) | MI + Scu (10 mg kg−1 day−1n = 9) | MI + Scu (30 mg kg−1 day−1n = 9) |
|---|---|---|---|---|---|---|---|
| Body weight (g) | 366 ± 13 | 361 ± 11 | 367 ± 12 | 348 ± 13 | 362 ± 12 | 358 ± 11 | 368 ± 9 |
| HW/BW ratio (mg g−1) | 3.13 ± 0.14 | 3.15 ± 0.07 | 3.16 ± 0.09 | 3.74 ± 0.11# | 3.61 ± 0.16 | 3.51 ± 0.14 | 3.22 ± 0.08* |
| LVW/BW ratio (mg g−1) | 2.34 ± 0.07 | 2.37 ± 0.1 | 2.36 ± 0.08 | 2.71 ± 0.09 | 2.6 ± 0.09 | 2.47 ± 0.07 | 2.43 ± 0.1 |
P < 0.05 versus sham
P < 0.05 versus MI.
Data are expressed as mean ± SEM.
MI, myocardial infarction; Scu, scutellarin; HW, heart weight; BW, body weight; LVW, left ventricle weight.
Effect of Scu on cardiac function evaluated by echocardiography
Echocardiography was performed at 4 and 8 weeks post-Scu administration (Table 2). At both time points, the MI hearts were significantly dilated as evidenced by an increase in LVDd and LVSd (P < 0.01), while eject fraction (EF) and FS were significantly decreased, indicating impaired cardiac function. Scu treatment significantly attenuated the increase in left ventricle dimension and the deterioration of left ventricular performance as indicated by increased EF and FS (Table 2). Scu (30 mg kg−1) produced no effects on the echocardiographic parameters of normal rats.
Table 2.
Echocardiography of MI rats after treatment with scutellarin for 4 and 8 weeks
| Group | Normal (n = 8) | Scutellarin (30 mg kg−1 day−1n = 8) | Sham (n = 8) | MI (n = 8) | MI + Scu (3 mg kg−1 day−1n = 9) | MI + Scu (10 mg kg−1 day−1n = 9) | MI + Scu (30 mg kg−1 day−1n = 9) |
|---|---|---|---|---|---|---|---|
| 4 weeks | |||||||
| LVDd (mm) | 6.09 ± 0.21 | 6.17 ± 0.13 | 6.15 ± 0.17 | 9.35 ± 0.29## | 8.94 ± 0.66 | 8.57 ± 0.45 | 8.31 ± 0.23 |
| LVSd (mm) | 3.56 ± 0.17 | 3.67 ± 0.18 | 3.73 ± 0.14 | 8.01 ± 0.21## | 7.5 ± 0.59 | 6.87 ± 0.37 | 6.57 ± 0.33 |
| FS (%) | 40.2 ± 2.16 | 38.9 ± 1.87 | 39.5 ± 1.59 | 14.6 ± 1.13## | 16.3 ± 1.14 | 19.8 ± 1.54 | 22 ± 2.04* |
| EF (%) | 75.8 ± 2.21 | 74.3 ± 1.96 | 76.6 ± 1.65 | 27.7 ± 1.5## | 36.9 ± 1.36 | 43.5 ± 2.67** | 50.1 ± 3.66** |
| 8 weeks | |||||||
| LVDd (mm) | 6.49 ± 0.27 | 6.71 ± 0.18 | 6.58 ± 0.2 | 9.84 ± 0.32## | 9.67 ± 0.44 | 9.48 ± 0.45 | 8.9 ± 0.55 |
| LVSd (mm) | 4.04 ± 0.16 | 3.93 ± 0.23 | 3.9 ± 0.21 | 8.36 ± 0.15## | 8.17 ± 0.48 | 7.43 ± 0.27* | 6.92 ± 0.28** |
| FS (%) | 42.4 ± 2.01 | 43.5 ± 1.69 | 41.3 ± 1.07 | 15.1 ± 1.08## | 16.1 ± 1.48 | 20.5 ± 1.37 | 22.9 ± 1.73** |
| EF (%) | 77.5 ± 2.81 | 78.3 ± 1.49 | 79.4 ± 1.38 | 25.1 ± 1.06## | 32.3 ± 1.36 | 42 ± 2.91** | 48.9 ± 2.49** |
P < 0.01 versus sham
P < 0.05 versus MI
P < 0.01 versus MI.
Data are expressed as mean ± SEM.
MI, myocardial infarction; Scu, scutellarin; EF, eject fraction; FS, fractional shortening; LVDd, left ventricle diastolic diameter; LVSd, left ventricle systolic diameter.
Effect of Scu on haemodynamic parameters
Eight weeks after Scu treatment, the haemodynamic parameters of rats were measured before scarification. Compared with control rats, MI rats demonstrated a significant increase in left ventricular end-diastolic pressure (LVEDP) (4.1 ± 0.68 vs. 14.6 ± 1.72 mm Hg for MI, P < 0.01) and decreases in dP/dtmax (7483 ± 135 vs. 5716 ± 199 mm Hg s−1 for MI, P < 0.01) and −dP/dtmax (6456 ± 210 vs. 4816 ± 177 mmHg s−1 for MI, P < 0.01) (Figure 3). These changes were alleviated by Scu in a dose-dependent manner. At the dose of 30 mg kg−1, Scu markedly improved cardiac function by decreasing LVEDP (7.1 ± 0.8 mm Hg) and increasing dP/dtmax (7029 ± 212 mm Hg s−1) and –dP/dtmax (6130 ± 275 mm Hg s−1) (P < 0.01 vs. MI) (Figure 3). Scu (30 mg kg−1) did not affect the haemodynamic parameters of normal rats (Figure 3). The mean arterial pressure among the groups did not change (Figure 3E).
Figure 3.
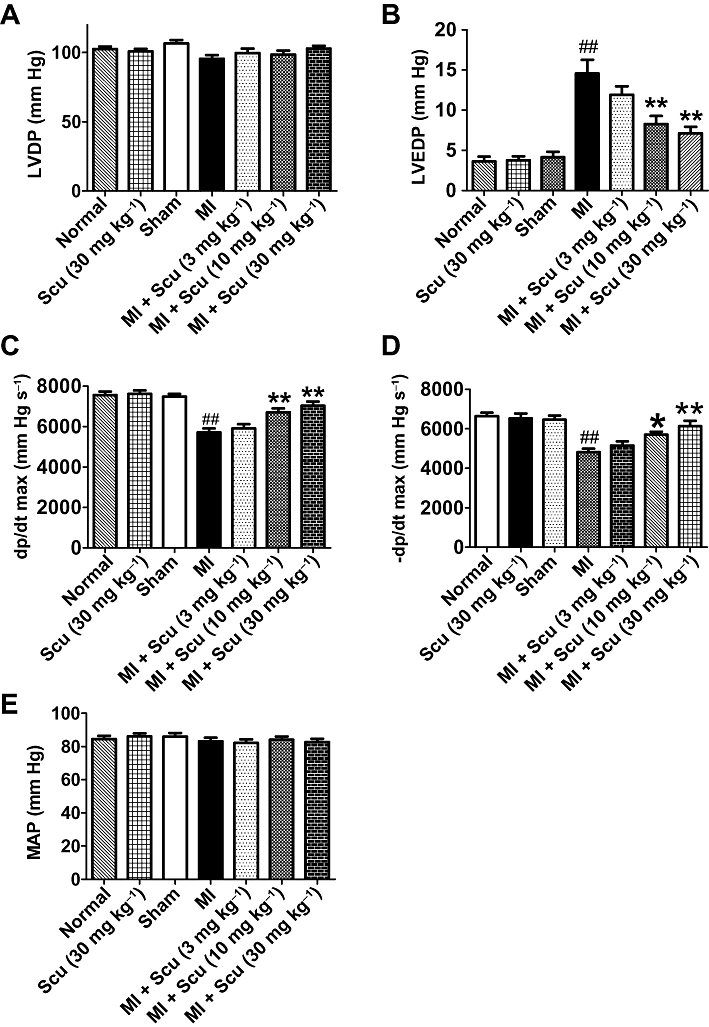
Effects of scutellarin (Scu) treatment on the haemodynamic effects of myocardial infarction (MI) in rats. (A) Left ventricular developed pressure, LVDP; (B) left ventricular end-diastolic pressure, LVEDP; (C) +dP/dt max; (D) –dP/dt max; (E) MAP, mean arterial pressure. Data are expressed as mean ± SEM, n = 8–9. ##P < 0.01 versus sham; *P < 0.05, **P < 0.01 versus MI.
Effect of Scu on cardiac interstitial fibrosis post-myocardial infarction
ECM deposition in the peri-infarct region was assessed using Masson's trichrome staining of the histological sections of the hearts. The interstitial fibrotic area of MI rats increased significantly compared with control animals (0.8 ± 0.1% for control vs. 16 ± 1.5% for MI, P < 0.01) (Figure 4). Treatment with Scu at doses of 3, 10 and 30 mg kg−1 resulted in a dose-dependent reduction in ECM deposition, with a fibrotic area of 2.4 ± 0.3% in the 30 mg kg−1 Scu group (Figure 4). Scu (30 mg kg−1) did not affect the cardiac collagen deposits of normal rats (Figure 4).
Figure 4.
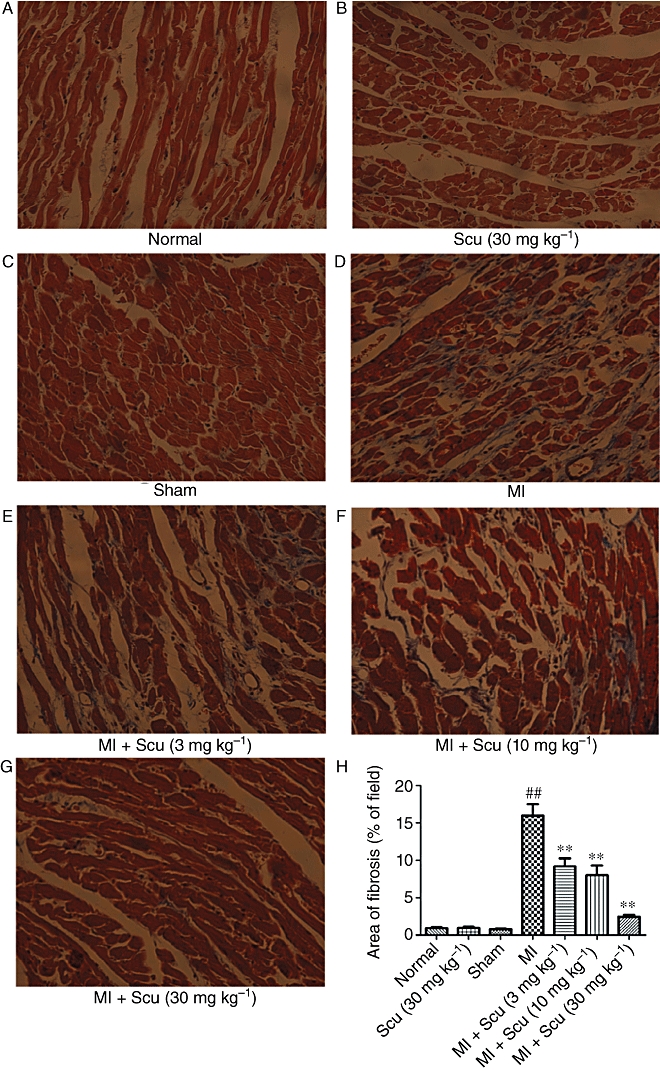
Effects of scutellarin (Scu) on the deposition of collagen in the peri-infarct region during myocardial infarction (MI) in rats. (A–G) Representative sections of heart stained with Masson's trichrome viewed at a magnification of 200×. The fibrotic area is stained blue and the viable area red. (H) Collagen deposition was quantified by automated image analysis and expressed as percentage of tissue area. Data are expressed as mean ± SEM, n = 5. ##P < 0.01 versus sham; **P < 0.01 versus MI.
The mRNA expression of fibronectin 1 (FN1), a glycoprotein component of the ECM, was examined. In the peri-infarct region of 8-week MI rats, FN1 increased by 5.8-fold compared with the sham group (P < 0.01). Treatment with Scu significantly attenuated this increase in FN1. At the doses of 10 and 30 mg kg−1, the FN1 levels decreased by 38 and 71%, respectively, compared with untreated MI rats (Figure 5A).
Figure 5.
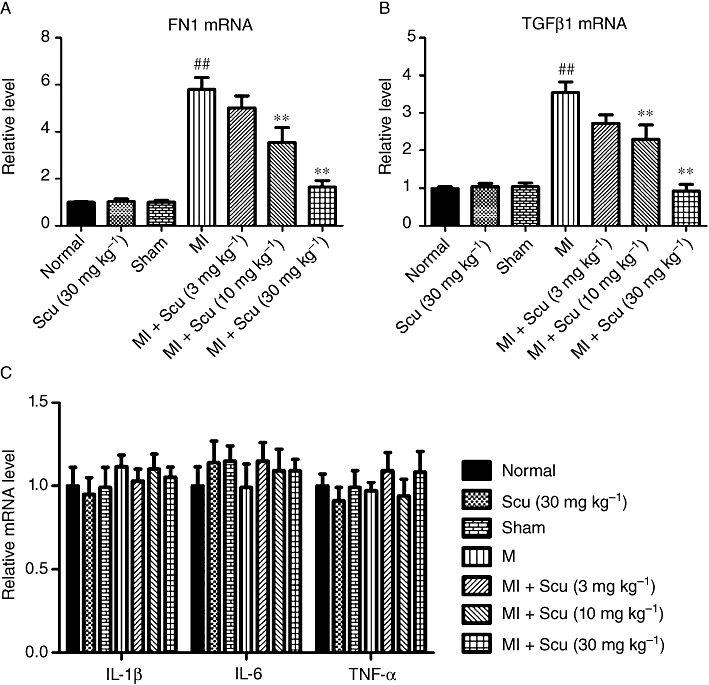
Effects of scutellarin (Scu) on the mRNA levels of fibronectin 1 (FN1) (A), TGFβ1 (B) and pro-inflammatory cytokines (IL-1β, IL-6, TNFα) (C) in peri-infarct cardiac tissues. All values are expressed as mean ± SEM (n = 4). ##P < 0.01 versus sham; **P < 0.01 compared with MI group.
In addition, the mRNA levels of the pro-fibrotic cytokine TGFβ1 and the pro-inflammatory cytokines IL-1β, IL-6, TNFα were determined in the peri-infarct cardiac tissues. In 8-week MI hearts, the expression of TGFβ1 was significantly increased compared with that in sham hearts (P < 0.01), and this increase was attenuated by the administration of Scu (Figure 5B). The expressions of IL-1β, IL-6, TNFα did not differ between the groups (Figure 5C).
Effect of Scu on the proliferation of cultured CFs and the expression of FN1 and TGFβ1
The effect of Scu on the proliferation of CFs was assessed by counting the number of cells. Treatment of CFs with 100 nM Ang II led to a 40% increase in the number of CFs, and the concomitant administration of Scu dose dependently inhibited this proliferation of CFs (Figure 6A). To determine whether the protein level of the main component of ECM (i.e. collagen) is altered by Scu, total collagen content was measured in cultured CFs using a hydroxyproline assay. The exposure of CFs to Ang II (100 nM) caused a significant up-regulation of collagen production (P < 0.01), which was attenuated by the co-application of Scu at doses of 30 and 100 µM respectively (Figure 6B). Scu also significantly suppressed the expression of FN1 (Figure 6C) and TGFβ1 (Figure 6D) at doses of 30 and 100 µM.
Figure 6.
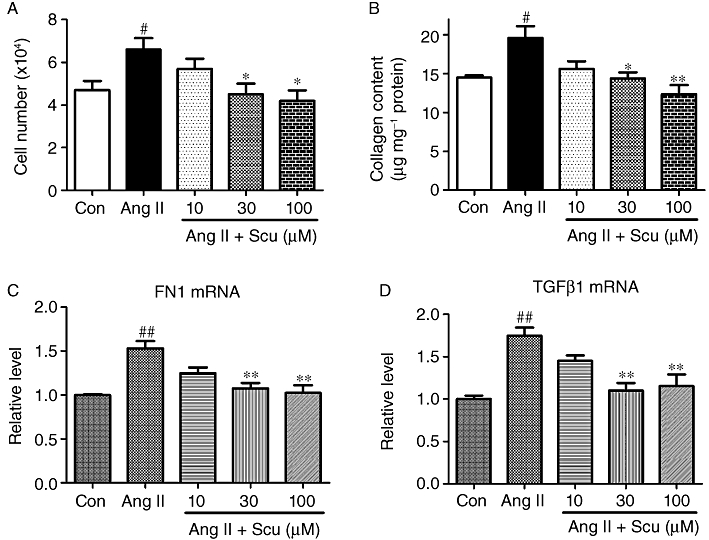
Inhibitory effects of scutellarin on angiotensin II (Ang II, 100 nM)-induced cardiac fibroblast proliferation. (A) Cell number; (B) collagen production; (C) FN1 expression; (D) TGFβ1 expression. Data are expressed as mean ± SEM, n = 4 to 5. #P < 0.05, ##P < 0.01 versus control (Con); *P < 0.05, **P < 0.01 versus Ang II.
Effect of Scu on ERK and p-ERK expression
To further explore the underlying molecular mechanism of Scu's effect on collagen production and deposition, the expression of ERK and p-ERK was examined in the cultured CFs and in the peri-infarct region of cardiac infarct rats. Treatment of CFs with Ang II (100 nM) significantly activated the phosphorylation of ERK, with p-ERK increased by about 68% (P < 0.01) compared with control cells, while the expression of total ERK was not affected (Figure 7A,B). Co-application of Scu significantly inhibited the activation of ERK at doses of 10, 30 and 100 µM. The same qualitative results were obtained from the infarct hearts of rats (Figure 7C,D), with pERK increased in the peri-infarct region and suppressed by the administration of Scu, but with no change in total ERK levels.
Figure 7.
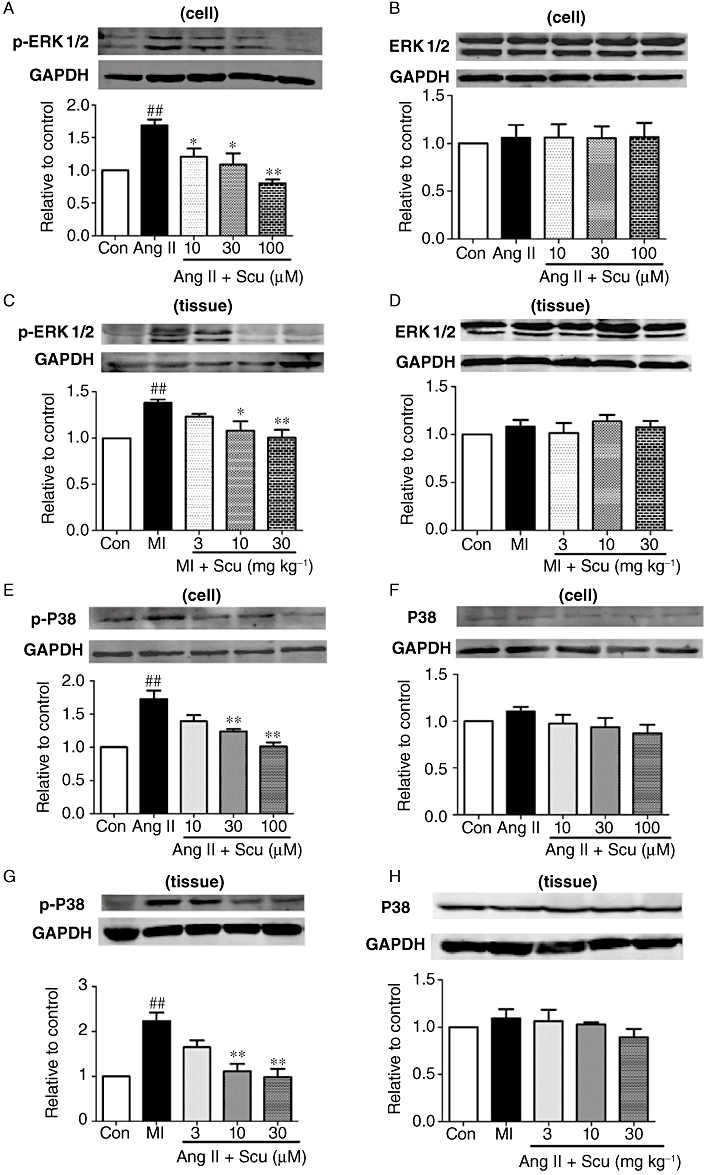
Effects of scutellarin (Scu) on the protein expression of ERK1/2, p-ERK1/2 and P38-MAPK, p-P38-MAPK in cultured cardiac fibroblasts and peri-infarct cardiac tissues. (A and B) p-ERK1/2 and ERK1/2 in cardiac fibroblasts; (C and D) p-ERK1/2 and ERK1/2 in cardiac tissues; (E and F) p-P38-MAPK and P38-MAPK in cardiac fibroblasts; (G and H) p-P38-MAPK and P38-MAPK in cardiac tissues. Ang II, angiotensin II (100 nM); MI, myocardial infarction. All values are expressed as mean ± SEM (n = 4). ##P < 0.01 versus sham; *P < 0.05, **P < 0.01 compared with Ang II or MI group.
Effect of Scu on p38MAPK and p-p38MAPK expression
The enhanced activation of p38MAPK has been reported to participate in cardiac fibrosis (See et al., 2004). We found that in cultured CFs, AngII (100 nM) caused a significant increase in the phosphorylation of p38MAPK, with p-p38MAPK increased by 72% in the Ang II group compared with the control group. Co-application of Scu significantly inhibited the activation of p38MAPK (Figure 7E). The expression of total p38MAPK did not change (Figure 7F). In infarct hearts of rats, the expression of p-p38MAPK was also increased by about 120%; this was inhibited by the 8 week application of Scu (Figure 7G). Similarly, the expression of total p38MAPK was not altered (Figure 7H).
Discussion and conclusions
At the early stage, fibrosis after MI is an integral component of the reparative process, maintaining structural integrity of the necrotic area by the formation of a connective tissue scar. However, in the long run, the continuous remodelling of the infarct left ventricle will lead to progressive ventricular dilatation, decreased cardiac performance and eventually chronic heart failure (Weber, 1997; Kurrelmeyer et al., 1998). In this process, the accumulation of collagenous material remote from the site of infarction is deeply involved; this contributes greatly to the reduced myocardial elasticity and impaired contractility (Boluyt et al., 1994; Hein et al., 2003). The extent of the detrimental remodelling predicts morbidity and mortality (Pfeffer and Braunwald, 1990; White and Braunwald, 1998). Despite the use of agents such as angiotensin converting enzyme-inhibitors, angiotensin II type 1 receptor blockers and β-adrenoceptor blocking agents, heart failure can still occur. Strategies that inhibit cardiac remodelling such as fibrosis following MI represent one of the major therapeutic goals of modern cardiology (Vanhoutte et al., 2006). Strikingly, herbal compounds such as sodium tanshinone IIA (Yang et al., 2009) and resveratrol (Lin et al., 2008) were demonstrated to produce beneficial effects on the infarct heart when applied for a long period. In this study, we found that Scu, a compound extracted from the traditional Chinese medicine E. breviscapus Hand Mazz, can improve the impaired cardiac function of chronic MI rats.
Other studies (Li et al., 2004; Lin et al., 2007; Jia et al., 2008) have demonstrated that Scu protects rats from cardiac ischaemic injury, and the mechanism may involve the ability of Scu to attenuate the increment of intracellular free calcium in cultured neonatal ventricular myocytes (Li et al., 2004). However, although these studies clarified the protective effects of Scu against acute cardiac injury, it was still unclear whether post-infarct administration of Scu could hinder the progressive deterioration of cardiac function. Our study confirmed that the treatment of MI rats with Scu for either 4 or 8 weeks significantly improved left ventricular function, as manifested by changes in the haemodynamic parameters, increased dP/dtmax and decreased LVEDP, and echocardiographic parameters, increased FS and EF, in comparision with sham rats.
Reactive fibrosis in the non-infarcted region, which adversely affects myocardial stiffness, is one of the major pathological alterations that participate in the development of heart failure after MI. In this study, we found that treatment with Scu apparently attenuated interstitial fibrosis, indicating a possible mechanism for its ability to improve cardiac function. The rapid proliferation of cardiac fibroblasts is responsible for the deposition of ECM and fibrosis, thus we examined the effects of Scu on the proliferation of cardiac fibroblasts and collagen production. We found that Scu significantly inhibited the increase in the number of cardiac fibroblasts induced by Ang II and the production of collagen, which explains the inhibitory effects of Scu on interstitial fibrosis after MI.
The molecular mechanism of interstitial fibrosis is very complex, involving the participation of a cascade of biochemical intracellular signalling processes. TGFβ1 is a key pro-fibrotic cytokine that is markedly elevated in experimental MI, and anti-TGF gene therapy mitigates cardiac remodelling by affecting cardiac fibrosis and infarct tissue dynamics (Okada et al., 2005). The finding that Scu inhibited the expression of TGFβ1 in this study indicates that the suppression of the TGFβ1 pathway underlies the anti-fibrotic action of Scu on post-infarct hearts. The p38 mitogen activated protein kinase (p38 MAPK) cascade has also been shown to play a critical role in the pathogenesis of cardiac fibrosis (Kompa et al., 2008). In a recent study, Ren et al. (2005) demonstrated that long-term (12 weeks) daily treatment of rats with the p38 MAPK inhibitor RWJ67657 and the ACE inhibitor ramipril resulted in a reduction in cardiac fibrosis and hypertrophy accompanied by a beneficial outcome on cardiac function after MI, while brief (1 week) inhibition of p38 MAPK activity after MI seemed to have no ameliorating effect on the progressive ventricular remodelling process (Kompa et al., 2008). Moreover, CFs, which rapidly proliferate after MI, are responsible for the main deposition of ECM, and activation of p38 MAPK underlies the proliferation and collagen production of CFs induced by Ang II (Chen and Mehta, 2006; Wu et al., 2009). Therefore, we hypothesize that the suppression of p38 MAPK cascade may be one underlying mechanism of Scu's beneficial effects on MI hearts. Our data indicate that long-term (8 weeks) treatment of MI rats with Scu significantly inhibited the up-regulation of the active form of p38 MAPK, phospho-p38 MAPK, while having no effect on the total p38 MAPK, the expression of which did not alter after MI. Scu also repressed Ang II-induced activation of p38 MAPK in CFs. These results strongly indicate that the inhibition of p38 MAPK accounts for the anti-fibrotic action of Scu.
Another MAPK cascade, extracellular signal regulated kinase (ERK1/2), was also implicated in the proliferation of CFs and interstitial fibrosis (Thum et al., 2008; Yeh et al., 2010). The activation of ERK1/2 by the up-regulation of miR-21 enhances cardiac fibroblast proliferation and thereby the interstitial fibrosis and cardiac dysfunction (Thum et al., 2008). The ERK1/2 signalling pathway accounts for Ang II-induced collagen I synthesis in the CFs (Olson et al., 2008; Gao et al., 2009). Treatment with resveratrol has been shown to inhibit the activation of ERK1/2 and the proliferation of CFs induced by Ang II (Olson et al., 2005). Our results showed that the activation of ERK1/2 induced by Ang II in CFs was significantly inhibited by Scu, as indicated by a decreased phospho-ERK1/2 expression. Moreover, we found that the expression of phospho-ERK1/2 in the heart of MI rats was significantly up-regulated and this was attenuated by long-term treatment with Scu. These data suggest that the suppression of ERK1/2 activation by Scu represents one of the possible mechanisms accounting for its beneficial effects on impaired cardiac function after MI. However, in terms of the role of ERK1/2 in interstitial fibrosis after MI in vivo, the findings so far are controversial. Some researchers have shown that the activity of ERK1/2 is increased after MI in mice (van Deel et al., 2008; Yeh et al., 2010) and rats (Shimizu et al., 1998), while others reported no change after MI in rabbits (Kobayashi et al., 2008) and mice (Takenaka et al., 2009). These disparities may be due to the different animal species used, or MI duration employed. However, the exact mechanism of Scu is currently unclear. Other studies have indicated that the action of Scu may be pleiotropic, as different pathways such as NFKB (Tan et al., 2010), caspase-6 (Chan et al., 2009), PKC (Yan et al., 2010), Ca2+ (Pan et al., 2008; Pan et al., 2010) have been shown to be involved in its action. In this study, long-term suppression of p38MAPK and ERK1/2 by Scu was demonstrated to be beneficial for the post-infarction heart. However, as the activation of p38MAPK and ERK1/2 is critical for cardioprotection (Fan et al., 2010), care should be taken when interpreting our results. Long-term suppression of the kinases by Scu may compromise the capacity of the heart to adapt to detrimental stimuli.
Taken together, the results of this study indicate that long-term application of Scu improves the cardiac function of MI rats by inhibiting interstitial fibrosis, irrespective of its other cardiovascular actions. The mechanisms may involve the suppression of TGFβ1 expression and inhibition of p38 MAPK and ERK1/2 phosphorylation. These data highlight the potential of Scu as an anti-fibrotic agent in the future.
Acknowledgments
This work was supported by the Ph.D. Programs Foundation of the Ministry of Education of China: Young Teacher, Opening Projects of Bio-Pharmaceutical Key Laboratory of Heilongjiang Province-Incubator of State Key Laboratory (to ZP), and by the National Basic Research Program of China (973 Program; 2007CB512000/2007CB512006; to BY).
Glossary
Abbreviations
- Ang II
angiotensin II
- CFs
cardiac fibroblasts
- CVF
collagen volume fraction
- ERK1/2
extracellular signal-regulated kinase
- FN1
fibronectin 1
- FS
fractional shortening
- IL-1β
interleukin 1β
- IL-6
interleukin 6
- LVDd
left ventricular diastolic diameter
- LVDP
left ventricular distolic pressure
- LVEF
left ventricular ejection fraction
- LVSd
left ventricular systolic diameter
- LVSP
left ventricular systolic pressure
- p38MAPK
p38 mitogen-activated protein kinase
- pERK1/2
phosphorylated extracellular signal–regulated kinase
- p-P38MAPK
phosphorylated p38 mitogen-activated protein kinase
- Scu
scutellarin
- TGFβ1
transforming growth factor β1
- TNFα
tumour necrosis factor α
Conflict of interest
None
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Boluyt MO, O'Neill L, Meredith AL, Bing OH, Brooks WW, Conrad CH, et al. Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components. Circ Res. 1994;75:23–32. doi: 10.1161/01.res.75.1.23. [DOI] [PubMed] [Google Scholar]
- Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY, Tan BK, Lee SC. Scutellarin sensitizes drug-evoked colon cancer cell apoptosis through enhanced caspase-6 activation. Anticancer Res. 2009;29:3043–3047. [PubMed] [Google Scholar]
- Chen J, Mehta JL. Angiotensin II-mediated oxidative stress and procollagen-1 expression in cardiac fibroblasts: blockade by pravastatin and pioglitazone. Am J Physiol Heart Circ Physiol. 2006;291:H1738–H1745. doi: 10.1152/ajpheart.00341.2006. [DOI] [PubMed] [Google Scholar]
- van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, et al. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic Biol Med. 2008;44:1305–1313. doi: 10.1016/j.freeradbiomed.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW. Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol. 2009;6:283–291. doi: 10.1038/nrcardio.2009.12. [DOI] [PubMed] [Google Scholar]
- Fan WJ, van Vuuren D, Genade S, Lochner A. Kinases and phosphatases in ischaemic preconditioning: a re-evaluation. Basic Res Cardiol. 2010;105:495–511. doi: 10.1007/s00395-010-0086-3. [DOI] [PubMed] [Google Scholar]
- Gao X, He X, Luo B, Peng L, Lin J, Zuo Z. Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur J Pharmacol. 2009;606:115–120. doi: 10.1016/j.ejphar.2008.12.049. [DOI] [PubMed] [Google Scholar]
- Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- Jia JH, Chen KP, Chen SX, Liu KZ, Fan TL, Chen YC. Breviscapine, a traditional Chinese medicine, alleviates myocardial ischaemia reperfusion injury in diabetic rats. Acta Cardiol. 2008;63:757–762. doi: 10.2143/AC.63.6.2033394. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Minatoguchi S, Yasuda S, Bao N, Kawamura I, Iwasa M, et al. Post-infarct treatment with an erythropoietin-gelatin hydrogel drug delivery system for cardiac repair. Cardiovasc Res. 2008;79:611–620. doi: 10.1093/cvr/cvn154. [DOI] [PubMed] [Google Scholar]
- Kompa AR, See F, Lewis DA, Adrahtas A, Cantwell DM, Wang BH, et al. Long-term but not short-term p38 mitogen-activated protein kinase inhibition improves cardiac function and reduces cardiac remodeling post-myocardial infarction. J Pharmacol Exp Ther. 2008;325:741–750. doi: 10.1124/jpet.107.133546. [DOI] [PubMed] [Google Scholar]
- Kurrelmeyer K, Kalra D, Bozkurt B, Wang F, Dibbs Z, Seta Y, et al. Cardiac remodeling as a consequence and cause of progressive heart failure. Clin Cardiol. 1998;21(Suppl 1):I14–I19. doi: 10.1002/clc.4960211304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XL, Li YQ, Yan WM, Li HY, Xu H, Zheng XX, et al. A study of the cardioprotective effect of breviscapine during hypoxia of cardiomyocytes. Planta Med. 2004;70:1039–1044. doi: 10.1055/s-2004-832644. [DOI] [PubMed] [Google Scholar]
- Lin JF, Lin SM, Chih CL, Nien MW, Su HH, Hu BR, et al. Resveratrol reduces infarct size and improves ventricular function after myocardial ischemia in rats. Life Sci. 2008;83:313–317. doi: 10.1016/j.lfs.2008.06.016. [DOI] [PubMed] [Google Scholar]
- Lin LL, Liu AJ, Liu JG, Yu XH, Qin LP, Su DF. Protective effects of scutellarin and breviscapine on brain and heart ischemia in rats. J Cardiovasc Pharmacol. 2007;50:327–332. doi: 10.1097/FJC.0b013e3180cbd0e7. [DOI] [PubMed] [Google Scholar]
- Muslin AJ. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci (Lond) 2008;115:203–218. doi: 10.1042/CS20070430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–2437. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- Olson ER, Naugle JE, Zhang X, Bomser JA, Meszaros JG. Inhibition of cardiac fibroblast proliferation and myofibroblast differentiation by resveratrol. Am J Physiol Heart Circ Physiol. 2005;288:H1131–H1138. doi: 10.1152/ajpheart.00763.2004. [DOI] [PubMed] [Google Scholar]
- Olson ER, Shamhart PE, Naugle JE, Meszaros JG. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension. 2008;51:704–711. doi: 10.1161/HYPERTENSIONAHA.107.098459. [DOI] [PubMed] [Google Scholar]
- Pan Z, Feng T, Shan L, Cai B, Chu W, Niu H, et al. Scutellarin-induced endothelium-independent relaxation in rat aorta. Phytother Res. 2008;22:1428–1433. doi: 10.1002/ptr.2364. [DOI] [PubMed] [Google Scholar]
- Pan ZW, Zhang Y, Mei DH, Zhang R, Wang JH, Zhang XY, et al. Scutellarin exerts its anti-hypertrophic effects via suppressing the Ca2+-mediated calcineurin and CaMKII signaling pathways. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:137–145. doi: 10.1007/s00210-009-0484-y. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA. Left ventricular remodeling after acute myocardial infarction. Annu Rev Med. 1995;46:455–466. doi: 10.1146/annurev.med.46.1.455. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38:617–623. doi: 10.1016/j.yjmcc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Schorb W, Conrad KM, Singer HA, Dostal DE, Baker KM. Angiotensin II is a potent stimulator of MAP-kinase activity in neonatal rat cardiac fibroblasts. J Mol Cell Cardiol. 1995;27:1151–1160. doi: 10.1016/0022-2828(95)90051-9. [DOI] [PubMed] [Google Scholar]
- See F, Thomas W, Way K, Tzanidis A, Kompa A, Lewis D, et al. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J Am Coll Cardiol. 2004;44:1679–1689. doi: 10.1016/j.jacc.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Yoshiyama M, Omura T, Hanatani A, Kim S, Takeuchi K, et al. Activation of mitogen-activated protein kinases and activator protein-1 in myocardial infarction in rats. Cardiovasc Res. 1998;38:116–124. doi: 10.1016/s0008-6363(97)00327-1. [DOI] [PubMed] [Google Scholar]
- Stockand JD, Meszaros JG. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol. 2003;284:H176–H184. doi: 10.1152/ajpheart.00421.2002. [DOI] [PubMed] [Google Scholar]
- Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- Takenaka H, Horiba M, Ishiguro H, Sumida A, Hojo M, Usui A, et al. Midkine prevents ventricular remodeling and improves long-term survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;296:H462–H469. doi: 10.1152/ajpheart.00733.2008. [DOI] [PubMed] [Google Scholar]
- Tan ZH, Yu LH, Wei HL, Liu GT. Scutellarin protects against lipopolysaccharide-induced acute lung injury via inhibition of NF-kappaB activation in mice. J Asian Nat Prod Res. 2010;12:175–184. doi: 10.1080/10286020903347906. [DOI] [PubMed] [Google Scholar]
- Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- Vanhoutte D, Schellings M, Pinto Y, Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res. 2006;69:604–613. doi: 10.1016/j.cardiores.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Wang LX, Zeng JP, Wei XB, Wang FW, Liu ZP, Zhang XM. Effects of scutellarin on apoptosis induced by cobalt chloride in PC12 cells. Chin J Physiol. 2007;50:301–307. [PubMed] [Google Scholar]
- Weber KT. Extracellular matrix remodeling in heart failure: a role for de novo angiotensin II generation. Circulation. 1997;96:4065–4082. doi: 10.1161/01.cir.96.11.4065. [DOI] [PubMed] [Google Scholar]
- White HD, Braunwald E. Applying the open artery theory: use of predictive survival markers. Eur Heart J. 1998;19:1132–1139. doi: 10.1053/euhj.1998.1017. [DOI] [PubMed] [Google Scholar]
- Wu M, Han M, Li J, Xu X, Li T, Que L, et al. 17beta-estradiol inhibits angiotensin II-induced cardiac myofibroblast differentiation. Eur J Pharmacol. 2009;616:155–159. doi: 10.1016/j.ejphar.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Yan L, Huang H, Tang QZ, Zhu LH, Wang L, Liu C, et al. Breviscapine protects against cardiac hypertrophy through blocking PKC-alpha-dependent signaling. J Cell Biochem. 2010;109:1158–1171. doi: 10.1002/jcb.22495. [DOI] [PubMed] [Google Scholar]
- Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- Yang L, Zou XJ, Gao X, Chen H, Luo JL, Wang ZH, et al. Sodium tanshinone IIA sulfonate attenuates angiotensin II-induced collagen type I expression in cardiac fibroblasts in vitro. Exp Mol Med. 2009;41:508–516. doi: 10.3858/emm.2009.41.7.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CC, Li H, Malhotra D, Turcato S, Nicholas S, Tu R, et al. Distinctive ERK and p38 signaling in remote and infarcted myocardium during post-MI remodeling in the mouse. J Cell Biochem. 2010;109:1185–1191. doi: 10.1002/jcb.22498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef ZR, Redwood SR, Marber MS. Postinfarction left ventricular remodelling: where are the theories and trials leading us? Heart. 2000;83:76–80. doi: 10.1136/heart.83.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.