

Abstract
Objective
Matrix metalloproteinase (MMP)-12 has been implicated in plaque progression and instability and is also amenable to selective inhibition. In this study, we investigated the influence of a greater than 10-fold selective synthetic MMP-12 inhibitor on plaque progression in the apolipoprotein E knockout mouse model of atherosclerosis.
Methods and Results
A phosphinic peptide (RXP470.1) that is a potent, selective murine MMP-12 inhibitor significantly reduced atherosclerotic plaque cross-sectional area by approximately 50% at 4 different vascular sites in male and female apolipoprotein E knockout mice fed a Western diet. Furthermore, RXP470.1 treatment resulted in less complex plaques with increased smooth muscle cell:macrophage ratio, less macrophage apoptosis, increased cap thickness, smaller necrotic cores, and decreased incidence of calcification. Additional in vitro and in vivo findings indicate that attenuated monocyte/macrophage invasion and reduced macrophage apoptosis probably underlie the beneficial effects observed on atherosclerotic plaque progression with MMP-12 inhibitor treatment.
Conclusion
Our data demonstrate that a selective MMP-12 inhibitor retards atherosclerosis development and results in a more fibrous plaque phenotype in mice. Our study provides proof of principle to motivate translational work on MMP-12 inhibitor therapy in humans.
Keywords: apoptosis, atherosclerosis, macrophages, metalloproteinases, pharmacology
Atherosclerotic plaque instability underlies most myocardial infarctions, most commonly through rupture of the fibrous cap.1 Plaque rupture results from excess hydrodynamic stress on a cap weakened by breakdown of the extracellular matrix (ECM). ECM integrity is tightly regulated through a balance between proteinases and their inhibitors. Among these, MMPs and tissue inhibitors of MMPs play an important role.2 In atherosclerotic plaques that are vulnerable to rupture matrix degradation prevails, particularly at the macrophage-rich shoulder regions where secretion of several matrix metalloproteinases (MMPs) is elevated and tissue inhibitors of MMP-3 in particular may be decreased.3 MMPs may therefore be suitable targets for therapeutic intervention against acute coronary syndromes.
However, recent findings reveal a more complex relationship between MMP activity and plaque stability. For example, we have demonstrated in hypercholesterolemic mice that MMP-3 and MMP-9 appear to have plaque-stabilizing roles, whereas MMP-12 is detrimental.4 Other murine studies using either gene ablation or protein overexpression have also shown that some MMPs support the growth of stable plaques through smooth muscle cell migration or matrix deposition or have no effect, whereas others, as expected, promote an unstable plaque phenotype via several mechanisms, including ECM destruction (see review5). Possibly because of the divergent roles MMPs play, broad-spectrum MMP inhibitors have failed to show clear benefits against atherosclerotic plaque growth or morphology in either animal or clinical studies.6-11 Manipulating expression of tissue inhibitors of MMPs in a mouse model of atherosclerosis has also yielded inconsistent results.12
These findings suggest that selectively targeting MMPs with a clearly detrimental role could be more effective for inhibiting atherosclerotic plaque progression and instability. The close structural similarity of MMPs around their active sites has made developing highly selective MMP inhibitors a difficult task in general.13 However, by exploiting phosphinic peptide chemistry, a strategy shown previously to provide more selective inhibitors of zinc-proteases,14 has led to the identification of a novel potent and highly selective MMP-12 inhibitor, RXP470.1, which has a Ki against human MMP-12 of 0.2 nmol/L but is 2 to 4 orders of magnitude less potent against other MMPs.15 Given the consistent evidence from several studies indicating a detrimental role for MMP-12 in plaque progression and instability, in both mouse4 and rabbit16,17 atherosclerosis models, and the association between MMP-12 expression and advanced human atherosclerotic lesions,18,19 we investigated the effect of RXP470.1 on plaque progression and morphology in apolipoprotein E (apoE) knockout mice. We treated mice with existing atherosclerosis with RXP470.1 for 4 weeks and tested its effects on plaques at 4 arterial sites.
Materials and Methods
An expanded Methods section is available in the Supplemental Material, available online at http://atvb.ahajournals.org.
Animals
All experiments were conducted according to the Animals (Scientific Procedures) Act 1986 (United Kingdom) and our institutional guidelines. Male and female mice homozygous null for the apoE gene on a 71% C57BL/6, 29% 129 background, were derived from a closed outbred colony housed in the Animal Unit of the University of Bristol.
MMP-12 Activity and Dose Ranging
MMP inhibitory activity was measured using a fluorogenic substrate in RXP470.1-treated animal plasmas, allowing determination of the inhibitor concentration, using a calibration inhibition curve. Furthermore, the presence of intact RXP470.1 was confirmed in plasma extraction mixtures with μ-high-performance liquid chromatography coupled to mass spectrometry.
Drug Administration
A total of 64 apoE-knockout mice were fed a high-fat rodent diet for 8 weeks to develop mature atherosclerotic plaques in the brachiocephalic artery. At this stage, 14 mice (10 male and 4 female) were terminated as a satellite group to evaluate atherosclerosis size and composition at the commencement of RXP-470.1 infusion (control, 8 weeks). Osmotic minipumps were implanted subcutaneously in the interscapular region of remaining mice (n=50). The reservoir of each pump was preloaded with 200 μL of either sterile phosphate-buffered saline (control, 12 weeks; n=27, 20 male and 7 female) or the MMP-12 selective inhibitor RXP470.1 solution at a concentration of 4.6 mg/kg of body weight per day in sterile phosphate-buffered saline (RXP470.1; n=23, 17 male and 6 female), and returned on a high-fat diet for a further 4 weeks (Supplemental Figure I).
ApoE/MMP-12 Double Knockout Mice
We generated both apoE/MMP-12 double knockout mice (apoE/MMP-12DKO) and apoE KO/MMP-12 wild-type (WT) controls (apoEKO/MMP-12WT) as described previously.4 Eight-week-old males of both types of mouse were fed a high-fat diet for 8 weeks (n=27 per group).
Characterization of Atherosclerotic Lesions
The brachiocephalic artery, proximal aorta, descending aorta, and heart underwent histological analysis, as previously described. The content of smooth muscle cells, macrophages, MMP-12, apoptosis, fibrillar collagens, and calcification was examined by immunohistochemical analyses. Necrotic core size was measured using the method described by Stoneman et al.20 Briefly, necrotic cores were defined as matrix-poor, acellular regions below a fibrous cap (SM actin-rich) region, and they were quantified by planimetry and expressed as percentage of total plaque area.
In Situ Zymography
Elastinolytic activity was assessed using a modification of the previously described in situ zymography method12 and is described in full in the supplemental information.
In Vivo Monocyte/Macrophage Invasion Assay
Multiple subcutaneous sponges were retrieved from male C57Bl/6;Sv129 mice infused by osmotic minipump with the MMP-12 specific inhibitor RXP470.1 (RXP470.1; n=7) or phosphate-buffered saline (control; n=7) and either monocytes/macrophages isolated as described previously3 or sponges fixed in formalin and subjected to histological examination for proliferation and apoptosis.
In Vitro Studies on Smooth Muscle Cells, Macrophages, and Macrophage-Derived Foam Cells
Proliferation and apoptosis frequencies were determined in mouse smooth muscle cells, monocyte-derived macrophages, and rabbit experimental foam cells with and without 100 nmol/L RXP470 coincubation, as described previously.21 In addition, apoptotic frequencies were also assessed in macrophages from both apoE/MMP-12DKO mice (n=7) and apoEKO/MMP-12WT control mice (n=7). N-cadherin expression was detected by Western blotting.
Statistical Analysis
The statistical analyses performed in this study are described in the supplemental information.
Results
Dose Ranging
The Ki values of RXP470.1 toward human and mouse MMP-12 are 0.2 and 4 nmol/L, respectively. According to Devel et al,15 the plasma concentration of RXP470.1 required to completely block MMP-12 but spare all other MMPs is 100 nmol/L. Infusing RXP470.1 from minipumps at a concentration of 4.6 mg/kg of body weight per day produced a plasma concentration of 100±10 nmol/L in apoE-knockout mice receiving high-fat diet (n=6). Mass spectroscopy fingerprint analysis demonstrated that RXP470.1 was observed in the intact form in the plasma of infused mice (Supplemental Figure II).
Effect of RXP470.1 on Body Weight and Plasma Lipids
Body weights were comparable between control (35.7±1.2 g) and RXP470.1-treated mice (36.2±0.9 g), indicating that drug treatment was well tolerated. RXP470.1 treatment for 4 weeks had no significant effect on total plasma cholesterol concentration when compared with controls (31.3±2.9 vs 28.2±1.8 mmol/L, respectively). There were also no effects on low-density lipoprotein cholesterol (29.4±2.8 vs 25.9±1.7 mmol/L, respectively), high-density lipoprotein cholesterol (1.3±0.1 vs 1.6±0.1 mmol/L, respectively), or triglyceride concentration (2.0±0.3 vs 2.4±0.4 mmol/L, respectively).
Effect of RXP470.1 on Vessel Morphometry
These data are summarized in Figure 1. Within the brachiocephalic artery, plaque cross-sectional area was reduced by 44% (P<0.05) in RXP470.1-treated animals compared with controls after 12 weeks of high-fat feeding. In addition, lesion size in the brachiocephalic artery of treated mice did not differ from that observed in mice at the commencement of drug administration (153±31 vs 147±32×103 μm2), suggesting that plaque progression was halted. Similarly, significant reductions in plaque area were detected at the aortic sinus (37%; P<0.05) and proximal aorta (61%; P<0.05). The most pronounced effect on plaque size was observed in the thoracic aorta (88%; P<0.05), largely because of a 70% reduction in the number of RXP470.1-treated mice with any detectable atherosclerotic lesion at this site. No significant differences were observed between males and females in either group (see Supplemental Table I).
Figure 1.
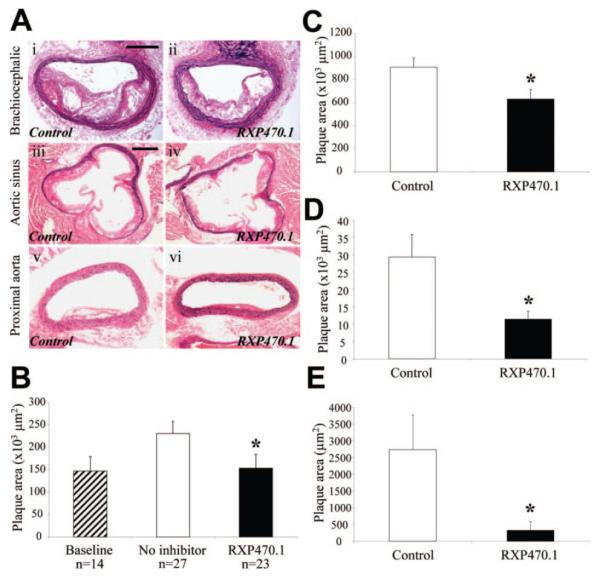
Effect of MMP-12 inhibition on atherosclerotic plaque area. A, Elastic van Gieson–stained brachiocephalic artery, aortic root, and proximal aorta atherosclerotic lesions. B to E, Quantification of brachiocephalic artery (B), aortic root (C), proximal aorta (D), and thoracic aorta (E) plaque cross-sectional area, from control and RXP470.1-treated mice (*P<0.05). Scale bar in panel i (A) represents 100 μm and is applicable to i, ii, v, and vi. Scale bar in iii represents 200 μm and is applicable to iii and iv.
Effect of RXP470.1 on Plaque Composition and Measures of Apoptosis
We focused on atherosclerotic plaques from the brachiocephalic artery to evaluate effects of MMP-12 inhibition on plaque composition.4,9,12 These data are summarized in Figure 2 and illustrate that RXP470.1 treatment significantly increased the percentage of smooth muscle cells as a proportion of all plaque cells (41±3% vs 31±3%; P<0.05) while reducing the percentage of macrophages (36±2% vs 42±1%; P<0.01). These results showed that RXP470.1 reduced plaque inflammation, resulting in a heightened smooth muscle cell:macrophage ratio by 33% (P<0.05). We used additional in vitro and in vivo studies of macrophage function to probe into the likely mechanisms for these effects. Addition of exogenous active MMP-12 to mouse monocyte-derived macrophages in vitro did not affect proliferative rates compared with controls (46±2% vs 36±5%). Furthermore, addition of RXP470.1 did not alter macrophage proliferation in either the absence (46±13%) or presence (39±12%) of added MMP-12. Accordingly, in brachiocephalic plaques from control and RXP470.1-treated mice, the percentage of proliferating macrophages as assessed by immunohistochemistry for Ki67 (14.8±3.1% and 13.6±2.2% respectively) and proliferating cell nuclear antigen (27.2±3% and 23.7±4% respectively) was unchanged. By contrast, RXP470.1 treatment significantly retarded the invasive capacity of monocyte/macrophages by 48% (P<0.01) in an in vivo subcutaneous sponge invasion assay compared with controls (1.7±0.3 vs 3.3±0.4×105 cells/sponge), even though the number of circulating monocytes was not significantly altered between RXP470.1-treated mice and controls (1.3±0.2 vs 1.9±0.6×106 monocytes/mL of blood). Proliferative rates of sponge macrophages were unchanged between RXP470.1-treated (5.6±1.6%) and control (4.0±1.1%) mice (Supplemental Figure III), and no apoptotic cells were detected in sponges from either group. However, it is possible that the effect of the inhibitor treatment could be different between invasion into a foreign body and an atherosclerotic plaque. We assessed endothelial coverage over plaques but observed no differences between control and RXP-470.1-treated mice (Supplemental Figure IV). In addition, we tested expression of vascular cell adhesion molecule-1 and MCP-1 and -2 molecules that determine leukocyte ingress into mouse plaques and detected no difference between the groups (Supplemental Figure IV). We therefore concluded that the effects of RXP470.1 on plaque inflammation were most likely explained by reduced monocyte/macrophage invasion rather than altered endothelial function.
Figure 2.

Effect of MMP-12 inhibition on brachiocephalic atherosclerotic plaque cellular composition. A to D, Immunohistochemical labeling of brachiocephalic artery lesions. Total numbers of lesional macrophages and smooth muscle cells were counted. The ratio was computed as summarized in the adjoining graph and depicts mean±SEM (*P<0.05, n≥23 per group). Scale bar in A represents 100 μm and is applicable to A to D. SMC indicates smooth muscle cell.
We next focused on effects on of RXP470.1 treatment on ECM integrity. Although a 26% increase in collagen content was observed in lesions from RXP470.1-treated mice, this did not reach statistical significance (Supplemental Table I and Supplemental Figure V). Similarly, no difference in total elastin content of plaques was observed between groups (Supplemental Table I). However, the number of lesions showing elastin fragmentation was decreased in RXP470.1-treated mice compared with controls (8% vs 37%; P<0.05, Supplemental Figure VI). Consistent with this, in situ zymography using a fluorescently quenched elastin substrate detected elastinolytic activity in brachiocephalic atherosclerotic lesions from 12-week high-fat-fed apoE-knockout mice (Figure 3A). However, the addition of 100 nmol/L RXP470.1 significantly reduced elastinolytic activity by 74% (4.1±0.5% vs 1.1±0.3% fluorescence of total plaque area; P<0.05; Figure 3B). The broad-spectrum MMP inhibitors BB94 and EDTA further reduced elastinolytic activity (95±4% and 94±5% respectively; Figure 3C and 3D), demonstrating the specificity of this assay.
Figure 3.
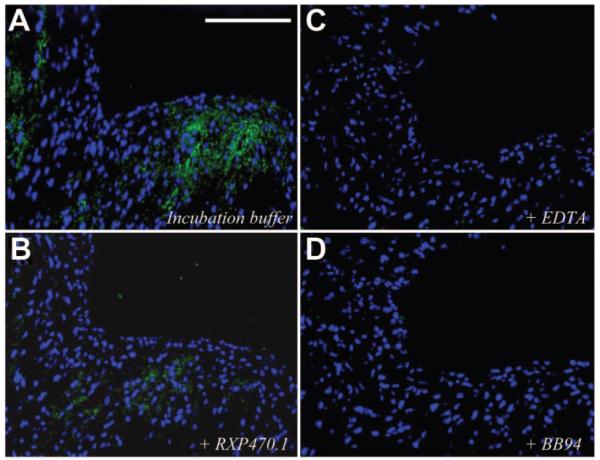
Effect of MMP-12 inhibition on brachiocephalic atherosclerotic plaque elastinolytic activity. Elastinolytic in situ zymography of brachiocephalic artery lesions with incubation buffer alone (A) or plus 100 nmol/L RXP470.1 (B), 20 mmol/L EDTA (C), or 1μmol/L BB94 (D). Green fluorescence represents elastinolytic activity. Scale bar in A represents 50 μm and is applicable to all panels.
We also observed a significant 65% reduction in the number of plaques containing buried fibrous layers from RXP470.1-treated mice compared with controls (Figure 4A). We further analyzed for changes in the complexity of lesions including fibrous cap thickness and necrotic core size. RXP470 treatment significantly increased minimum cap thickness (16.4±2.2 μm) compared with both no inhibitor (8.7±1.0 μm, P<0.001) and baseline (10.2±1.5 μm, P<0.05) mice. Necrotic core size was measured using the method described by Stoneman et al.20 RXP470.1-treated mice showed a significant 46% reduction in lesion necrotic core size compared with controls (13±3% and 24±2% respectively, P<0.05). Consistent with this, as shown in Figure 5A, RXP470.1 treatment significantly reduced the number of ISEL-positive macrophages by 42% (P<0.05) in brachiocephalic artery plaques compared with controls. This finding was corroborated by immunohistochemistry for cleaved PARP-1 and also assessing the numbers of pyknotic/fragmented nuclei (Supplemental Figures VII and VIII), both showing a 57% and a 43% reduction in apoptotic frequencies, respectively. Indeed, the frequencies of intraplaque macrophage apoptosis were similar between the baseline (15.3±3.0%) and RXP470.1-treated mice (18.1±5.8%), suggesting that MMP-12 inhibition retards increased macrophage apoptosis associated with plaque progression. Furthermore, the rate of in vitro serum starvation-induced apoptosis, as detected by immunocytochemistry for the apoptotic marker cleaved caspase-3 and changes in nuclear morphology, was significantly increased in both mouse monocyte-derived macrophages and rabbit granuloma foam-cell macrophages by treatment with exogenous active MMP-12 compared with untreated cells (Figure 5B and 5C). Conversely, apoptosis was significantly inhibited in both macrophages (91%) and foam cell macrophages (91%) after RXP470.1 addition compared with active MMP-12 alone (Figure 5B and 5C). Furthermore, RXP470.1 treatment significantly reduced apoptotic rates in control macrophages and foam-cell macrophages relative to control cells (Figure 5B and 5C).
Figure 4.

Effect of MMP-12 inhibition on brachiocephalic atherosclerotic plaque composition. A, Identification of buried fibrous layers (arrows) from control and RXP470.1-treated mice. SMC indicates smooth muscle cell. B, Presence of calcification (brown/black, arrows) in brachiocephalic artery lesions from control and RXP470.1-treated mice. Quantification is summarized in the adjoining graphs and depicts mean±SEM (*P<0.05, n≥23 per group). Scale bars represent 100 μm and are applicable to all panels.
Figure 5.
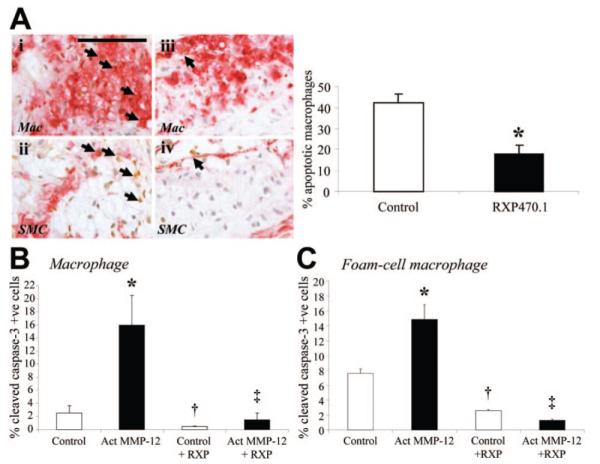
Effect of MMP-12 inhibition on apoptosis. A, Immunohistochemical labeling of brachiocephalic artery lesions for macrophages, smooth muscle cells (both red), and apoptosis (brown, arrows), from control and RXP470.1-treated mice. Quantification is summarized in the adjoining graph and depicts mean±SEM (*P<0.05, n≥23 per group). Scale bar in i represents 100 μm and is applicable to all panels. SMC indicates smooth muscle cell. B and C, Quantification of apoptosis in macrophages (B) and foam-cell macrophages (C), as assessed by cleaved caspase-3 immunocytochemistry. *P<0.05 vs control, †P<0.05 vs control, ‡P<0.05 vs active MMP-12. All values are expressed as mean±SEM. All groups, n=3.
To further investigate the effects of MMP-12 on macrophage apoptosis, macrophages from apoE/MMP-12 double-deficient mice and apoE deficient/MMP-12 WT controls were subjected to serum-starvation induced apoptosis. In accordance with the pharmacological findings, the absence of MMP-12 reduced macrophage apoptosis by 60% (20±4% vs 50±3%; P<0.01). In contrast to the results for macrophages, the rate of FasL-induced apoptosis was unchanged in smooth muscle cells treated with RXP470.1 relative to untreated cells (70±29% vs 78±29×104 ALU). Accordingly, the frequency of apoptotic vascular smooth muscle cells detected within atherosclerotic plaques was not altered in mice treated with RXP470.1 (0.7±0.3% vs 1.0±0.2% apoptotic vascular smooth muscle cell). Western blotting for the prosurvival protein N-cadherin was conducted to gain mechanistic insight into the antiapoptotic effect of RXP470.1. We demonstrate that active MMP-12 reduces full-length N-cadherin expression in macrophages, but this is reversed by coincubation with RXP470.1. In addition, RXP470.1 treatment negated the formation of the cleaved fragment of N-cadherin observed in the presence of active MMP-12 (Supplemental Figure IX).
In addition to the changes in cap thickness and necrotic core size, there was a 72% (P<0.05) reduction in the incidence of calcification in lesions from RXP470.1-treated mice compared with controls (Figure 4B). Moreover, when normalized to lesion area, an 86% (P<0.01) decrease in the area of calcification was detected in plaques from treated animals (0.8±0.6%) versus plaques from control mice (5.9±2.2%). On closer examination, it was observed that calcification, macrophage apoptosis, and MMP-12 expression colocalized in atherosclerotic lesions from control mice (Supplemental Figure X). In accordance with this observation, there were significant correlations between the extent of calcification and necrotic core area (P<0.05), macrophage content (P<0.01), and apoptosis (P<0.001) in all atherosclerotic lesions. These observations suggest that calcification may have been a secondary consequence of macrophage apoptosis and or increased plaque size.
Effect of RXP470.1 Compared With Those of MMP-12 Knockout
Effects of MMP-12 knockout on necrotic core size, macrophage apoptosis and calcification have not been reported previously. We therefore conducted new analyses of archive material of brachiocephalic atherosclerotic plaques from apoE/MMP-12DKO (n=27) and apoEKO/MMP-12WT (n=25) mice from a previous study.4 As shown in Figure 6, apoE/MMP-12DKO mice significantly reduced necrotic core size (50%; P<0.05) and the frequency of macrophage apoptosis (39%; P<0.05) compared with controls. Moreover, the percentage area of calcification normalized to lesion area was decreased by 88% (P<0.01) in plaques from apoE/MMP-12DKO animals (0.4±0.4%) versus plaques from apoEKO/MMP-12WT mice (3.5±1.0%). Also shown in Figure 6, MMP-12 knockout reduced the number of calcified lesions (90%; P<0.05). These above findings parallel those observed in RXP470.1-treated mice.
Figure 6.

Effect of MMP-12 gene deletion on apoptosis and calcification. A, Necrotic core area of brachiocephalic artery lesions from apoEKO/MMP-12WT and apoE/MMP-12DKO mice. SMC indicates smooth muscle cell. B, Immunohistochemical labeling of brachiocephalic artery lesions for macrophages, smooth muscle cells (both red) and apoptosis (brown, arrows). C, Presence of calcification (brown/black, arrows) in brachiocephalic artery lesions from apoEKO/MMP-12WT and apoE/MMP-12DKO mice. Quantification is summarized in the adjoining graphs and depicts mean±SEM (*P<0.05, n≥23 per group). Scale bars represent 100 μm and are applicable to adjoining panels.
Discussion
Our present study provides strong new evidence supporting the therapeutic potential of highly selective intervention against MMP-12 using RXP470.1. Administration of RXP470.1 to mice with already established atherosclerosis significantly reduced plaque growth at 4 differing vascular sites and, in fact, totally blocked lesion progression in the brachiocephalic artery. We also assessed the effects of RXP470.1 on morphological surrogates that have been widely used in previous mouse studies to infer beneficial changes in plaque phenotype. The proportion of macrophages to smooth muscle cells, the frequency of macrophage apoptosis, loss of fibrous cap thickness, the necrotic core area, the frequency of plaques exhibiting a layered phenotype, and the number of plaques exhibiting calcification were all significantly reduced by RXP470.1 treatment. If translated to humans, the dramatic reduction in all of these parameters support the potential of RXP470.1 to promote plaque stability.1 These effects of RXP470.1 were obtained at circulating concentrations at which MMP-12 is powerfully inhibited—as shown directly by elastinolytic in situ zymography— but other MMPs are spared.15 Furthermore, RXP470.1 was well tolerated by apoE-knockout mice and had no effect on plasma lipid concentrations or circulating monocyte numbers.
Substantial data support the importance of MMPs in atherosclerotic plaque development and destabilization.22 However, neither animal studies6-9 nor clinical trials10,11 have so far demonstrated conclusive benefits from broad-spectrum MMP inhibitors, perhaps because of a balance of positive and negative effects. MMP-12 expression, in particular, has been correlated with advanced18,19 and ruptured23 human atherosclerotic plaques, and a common functional MMP-12 promoter polymorphism that increases expression is associated with increased coronary artery stenosis in diabetic patients.24 Furthermore, macrophage-specific overexpression of active MMP-12 in atherosclerotic rabbits accelerates plaque progression.16,17 Conversely, we have shown previously4 that MMP-12 knockout decreases plaque size in the brachiocephalic artery of fat-fed apoE-knockout mice by 52%, increases smooth muscle to macrophage ratio by 224%, and reduces the number of plaques with a layered phenotype by 59%. Here we report very similar values after RXP470.1 treatment. In another study, Luttun et al25 demonstrated a decrease in the number of medial elastin breaks in the aortic arch of MMP-12 knockout mice, which we also saw here after RXP470.1 treatment. New data on the effects of RXP470.1 treatment and MMP-12 knockout on necrotic core size, macrophage apoptosis in plaques and in vitro and calcification reported here were also very similar. Hence, selective MMP-12 inhibition with RXP470.1 proved as effective against atherosclerosis as MMP-12 knockout and more effective than global MMP inhibition, consistent with our arguments in the Introduction.
Three underlying mechanisms seem to be of key importance: inhibition of macrophage invasion, elastinolysis, and foam cell apoptosis. Consistent with this, we demonstrated directly that RXP470.1 treatment reduced macrophage accumulation into subcutaneous sponges without changing proliferative or apoptotic rates, implying a reduction in macrophage invasion. A lack of any changes in endothelial coverage and expression of molecules associated with recruitment and adhesion imply a direct effect of MMP-12 inhibition on invasion. Furthermore RXP470.1 dramatically affected early plaque formation in the thoracic aorta, a process reliant primarily on monocyte/macrophage recruitment. Reduced macrophage invasion was also shown in MMP-12 knockout mice by Shipley et al26 and in an allergen-induced lung inflammation mouse model.27 Conversely, macrophage-specific overexpression of MMP-12 in rabbits augmented macrophage recruitment to already inflamed lesions.28,29 MMP-12 contributes to monocyte/macrophage invasion in part through ECM proteolysis. For example, macrophages from MMP-12 knockout mice have a diminished capacity to degrade ECM components, fail to digest soluble elastin, and are unable to penetrate reconstituted basement membranes in vitro and in vivo.26 In addition, elastin peptides generated by MMP-12 mediated hydrolysis are also potent chemoattractants for macrophages.30
MMP-12 activity is associated with apoptosis in several pathologies.31,32 Apoptosis is readily detected in advanced atherosclerotic plaques and is postulated to contribute to plaque vulnerability and rupture,33 mainly through formation and expansion of the acellular necrotic core,34 in part because of ineffective clearance.35 Interestingly, MMP-12 expression is prominently localized in advanced human atherosclerotic lesions where the rupture-prone shoulder regions and acellular necrotic core are juxtaposed.18 Our current in vitro results demonstrate that active MMP-12 supports apoptosis in both macrophages and foam-cell macrophages and that RXP470.1 therefore promotes survival of these cells. Although a detailed analysis of the mechanism by which MMP-12 promotes macrophage apoptosis will require a separate study, we have identified cleavage of N-cadherin as one potential mechanism. We have previously shown21 an association of N-cadherin expression and macrophage survival and that N-cadherin is cleaved by MMPs including MMP-12.36,37 We observed, furthermore, that exogenous N-cadherin reduced macrophage and foam-cell macrophage apoptosis in vitro and that systemic adenoviral overexpression of N-cadherin retarded macrophage apoptotic frequencies in brachiocephalic plaques of apoE-deficient mice, which displayed a less complex plaque phenotype compared with controls. Consequently, we examined here N-cadherin protein levels in foam-cell macrophages treated with active MMP-12 and RXP470.1. We observed a higher turnover rate of N-cadherin in foam-cell macrophages treated with active MMP-12 which was retarded by addition of RXP470.1. Taken together, these observations suggest that inhibition of MMP-12-dependent cleavage of macrophage N-cadherin may be one mechanism by which RXP470.1 inhibits macrophage apoptosis. Although it is beyond the scope of the present study, we aim to further validate this novel and intriguing potential mechanistic link in future studies.
Reduced calcification observed in RXP470.1-treated and MMP-12 knockout mice is probably a secondary consequence of reduced plaque size and inflammation. For example, both in vitro and in vivo studies have linked inflammation and cardiovascular calcification in atherosclerosis.38-40 Reduced calcification might also be a secondary consequence of reduced macrophage apoptosis. Consistent with this, calcification correlates with the extent of lipid-rich necrotic core formation.40,41
Among the limitations of our study, it must be noted that the background strain of mice influences atherosclerosis and hence we cannot rule out the possibility that the effects of RXP470.1 we observed are strain dependent. Another limitation is that we were unable to directly assess the in vivo specificity of the inhibitor in the present study and therefore cannot exclude an effect of RXP470.1 on other MMPs or indeed other proteases, but we feel our data and those previously published strongly imply that beneficial effects observed with RXP470.1 are through inhibition of MMP-12 activity. Given the very large in vitro selectivity of RXP470.1 for MMP-12 over other MMPs (greater than 100-fold) it is highly unlikely that any other MMP is inhibited in vivo. Consistent with this, RXP470.1 retarded elastinolytic activity within atherosclerotic plaques by in situ zymography but did not increase collagen content, in contrast to the previously published MMP-13 knockout.42 Moreover, our data showing that the effects of 100 nmol/L RXP470.1 closely mirror those of MMP-12 gene deletion in plaques strengthen the conclusion that the effects are due largely to MMP-12 inhibition. The pharmacokinetics of peptides is an acknowledged limitation to translation of our inhibitor strategy into humans. However, previous in vivo studies performed with phosphinic peptides show relatively good in vivo stability.43,44 In addition, in vivo studies using phosphinic peptides reported an encouraging lack of toxicity for phosphinic peptides.43,44 Nevertheless, translation into humans will require a full toxicological screen. Given that RXP470.1 inhibited monocyte recruitment into both plaques and subcutaneous sponges, MMP-12 inhibition might have effects on other inflammatory processes.
In summary, our study demonstrates that a selective MMP-12 inhibitor, RXP470.1, can reduce atherosclerosis progression and alter plaque phenotype. Our results provide proof of principle in mice to motivate translation of selective MMP-12 inhibitor treatment to human atherosclerosis.
Supplementary Material
Acknowledgments
The authors thank Michelle Somerville for excellent technical assistance.
Sources of Funding
This work was supported by grants from the British Heart Foundation to JLJ (FS/07/053/24069) and ACN (CH95/001), grants by Commissariat à l’Energie Atomique, and the National Institute for Health Research Bristol Biomedical Research Unit in Cardiovascular Medicine.
Footnotes
Drs Newby and Dive contributed equally to this work.
Disclosures
None.
References
- 1.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 2.Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–2114. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- 3.Johnson JL, Sala-Newby GB, Ismail Y, Aguilera CNM, Newby AC. Low tissue inhibitor of metalloproteinases 3 and high matrix metalloproteinase 14 levels defines a subpopulation of highly invasive foam-cell macrophages. Arterioscler Thromb Vasc Biol. 2008;28:1647–1653. doi: 10.1161/ATVBAHA.108.170548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases -3, -7, -9 and -12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci U S A. 2005;102:15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovas Med. 2007;17:253–258. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prescott MF, Sawyer WK, Von Linden-Reed J, Jeune M, Chou M, Caplan SL, Jeng AY. Effect of matrix metalloproteinase inhibition on progression of atherosclerosis and aneurysm in LDL receptor-deficient mice overexpressing MMP-3, MMP-12, and MMP-13 and on restenosis in rats after balloon injury. Ann NY Acad Sci. 1999;878:179–190. doi: 10.1111/j.1749-6632.1999.tb07683.x. [DOI] [PubMed] [Google Scholar]
- 7.Cherr GS, Motew SJ, Travis JA, Fingerle J, Fisher L, Brandl M, Williams JK, Geary RL. Metalloproteinase inhibition and the response to angioplasty and stenting in atherosclerotic primates. Arterioscler Thromb Vasc Biol. 2002;22:161–166. doi: 10.1161/hq0102.101129. [DOI] [PubMed] [Google Scholar]
- 8.Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2003;23:483–488. doi: 10.1161/01.ATV.0000058404.92759.32. [DOI] [PubMed] [Google Scholar]
- 9.Johnson JL, Fritsche-Danielson R, Behrendt M, Westin-Eriksson A, Wennbo H, Herslof M, Elebring M, George SJ, McPheat W, Jackson CL. Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc Res. 2006;71:586–595. doi: 10.1016/j.cardiores.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Axisa B, Loftus IM, Naylor AR, Goodall S, Jones L, Bell PRF, Thompson MM. Prospective, randomized, double-blind trial investigating the effect of doxycycline on matrix metalloproteinase expression within atherosclerotic carotid plaques. Stroke. 2002;33:2858–2864. doi: 10.1161/01.str.0000038098.04291.f6. [DOI] [PubMed] [Google Scholar]
- 11.Brown DL, Desai KK, Vakili BA, Nouneh C, Lee H-M, Golub LM. Clinical and biochemical results of the Metalloproteinase Inhibition with Subantimicrobial Doses of Doxycycline to Prevent Acute Coronary Syndromes (MIDAS) pilot trial. Arterioscler Thromb Vasc Biol. 2004;24:733–738. doi: 10.1161/01.ATV.0000121571.78696.dc. [DOI] [PubMed] [Google Scholar]
- 12.Johnson JL, Baker AH, Oka K, Chan L, Newby AC, Jackson CL, George SJ. Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: involvement of macrophage migration and apoptosis. Circulation. 2006;113:2435–2444. doi: 10.1161/CIRCULATIONAHA.106.613281. [DOI] [PubMed] [Google Scholar]
- 13.Yiotakis A, Dive V. Synthetic active site-directed inhibitors of metzincins: achievement and perspectives. Mol Aspects Med. 2008;29:329–338. doi: 10.1016/j.mam.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Dive V, Cotton JL, Yiotakis A, Michaud A, Vassiliou S, Jiracek J, Vazeux G, Chauvet M-Trs, Cuniasse P, Corvol P. RXP 407, a phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. P Natl Acad Sci USA. 1999;96:4330–4335. doi: 10.1073/pnas.96.8.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devel L, Rogakos V, David A, Makaritis A, Beau F, Cuniasse P, Yiotakis A, Dive V. Development of selective inhibitors and substrate of matrix metalloproteinase-12. J Biol Chem. 2006;281:11152–11160. doi: 10.1074/jbc.M600222200. [DOI] [PubMed] [Google Scholar]
- 16.Liang J, Liu E, Yu Y, Kitajima S, Koike T, Jin Y, Morimoto M, Hatakeyama K, Asada Y, Watanabe T, Sasaguri Y, Watanabe S, Fan J. Macrophage metalloelastase accelerates the progression of atherosclerosis in transgenic rabbits. Circulation. 2006;113:1993–2001. doi: 10.1161/CIRCULATIONAHA.105.596031. [DOI] [PubMed] [Google Scholar]
- 17.Yamada S, Wang K-Y, Tanimoto A, Fan J, Shimajiri S, Kitajima S, Morimoto M, Tsutsui M, Watanabe T, Yasumoto K, Sasaguri Y. Matrix metalloproteinase 12 accelerates the initiation of atherosclerosis and stimulates the progression of fatty streaks to fibrous plaques in transgenic rabbits. Am J Pathol. 2008;172:1419–1429. doi: 10.2353/ajpath.2008.070604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halpert I, Sires UI, Roby JD, PotterPerigo S, Wight TN, Shapiro SD, Welgus HG, Wickline SA, Parks WC. Matrilysin is expressed by lipid-laden macrophages at sites of potential rupture in atherosclerotic lesions and localizes to areas of versican deposition, a proteoglycan substrate for the enzyme. Proc Natl Acad Sci U S A. 1996;93:9748–9753. doi: 10.1073/pnas.93.18.9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas AC, Sala-Newby GB, Ismail Y, Johnson JL, Pasterkamp G, Newby AC. Genomics of foam cells and nonfoamy macrophages from rabbits identifies arginase-1 as a differential regulator of nitric oxide production. Arterioscler Thromb Vasc Biol. 2007;27:571–577. doi: 10.1161/01.ATV.0000256470.23842.94. [DOI] [PubMed] [Google Scholar]
- 20.Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, Bennett M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100:884–893. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyon CA, Johnson JL, Williams H, Sala-Newby GB, George SJ. Soluble N-cadherin overexpression reduces features of atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2009;29:195–201. doi: 10.1161/ATVBAHA.108.178087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newby AC. Dual role of matrix metalloproteinases (matrixins) in neointima formation and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 23.Morgan AR, Rerkasem K, Gallagher PJ, Zhang B, Morris GE, Calder PC, Grimble RF, Eriksson P, McPheat WL, Shearman CP, Ye S. Differences in matrix metalloproteinase-1 and matrix metalloproteinase-12 transcript levels among carotid atherosclerotic plaques with different histopathological characteristics. Stroke. 2004;35:1310–1315. doi: 10.1161/01.STR.0000126822.01756.99. [DOI] [PubMed] [Google Scholar]
- 24.Jormsjo S, Ye S, Moritz J, Walter DH, Dimmeler S, Zeiher AM, Henney A, Hamsten A, Eriksson P. Allele-specific regulation of matrix metalloproteinase-12 gene activity is associated with coronary artery luminal dimensions in diabetic patients with manifest coronary artery disease. Circ Res. 2000;86:998–1003. doi: 10.1161/01.res.86.9.998. [DOI] [PubMed] [Google Scholar]
- 25.Luttun A, Lutgens E, Manderveld A, Maris K, Collen D, Carmeliet P, Moons L. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109:1408–1414. doi: 10.1161/01.CIR.0000121728.14930.DE. [DOI] [PubMed] [Google Scholar]
- 26.Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A. 1996;93:3942–3946. doi: 10.1073/pnas.93.9.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warner RL, Lukacs NW, Shapiro SD, Bhagarvathula N, Nerusu KC, Varani J, Johnson KJ. Role of metalloelastase in a model of allergic lung responses induced by cockroach allergen. Am J Pathol. 2004;165:1921–1930. doi: 10.1016/S0002-9440(10)63244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Liang J, Koike T, Sun H, Ichikawa T, Kitajima S, Morimoto M, Shikama H, Watanabe T, Sasaguri Y, Fan J. Overexpression of human matrix metalloproteinase-12 enhances the development of inflammatory arthritis in transgenic rabbits. Am J Pathol. 2004;165:1375–1383. doi: 10.1016/S0002-9440(10)63395-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan J, Wang X, Wu L, Matsumoto S-I, Liang J, Koike T, Ichikawa T, Sun H, Shikama H, Sasaguri Y, Watanabe T. Macrophage-specific overexpression of human matrix metalloproteinase-12 in transgenic rabbits. Transgenic Res. 2004;13:261–269. doi: 10.1023/b:trag.0000034717.70729.61. [DOI] [PubMed] [Google Scholar]
- 30.Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest. 1980;66:859–862. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorrin-Rivas MJ, Arii S, Mori A, Kaneda Y, Imamura M. Mouse macrophage metalloelastase gene delivery by HVJ-cationic liposomes in experimental antiangiogenic gene therapy for murine CT-26 colon cancer. Int J Cancer. 2001;93:731–735. doi: 10.1002/ijc.1389. [DOI] [PubMed] [Google Scholar]
- 32.Matute-Bello G, Wurfel MM, Lee JS, Park DR, Frevert CW, Madtes DK, Shapiro SD, Martin TR. Essential role of MMP-12 in Fas-induced lung fibrosis. Am J Respir Cell Mol Biol. 2007;37:210–221. doi: 10.1165/rcmb.2006-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolodgie FD, Narula J, Burke AP, Haider N, Farb A, You HL, Smialek J, Virmani R. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol. 2000;157:1259–1268. doi: 10.1016/S0002-9440(10)64641-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartung D, Sarai M, Petrov A, Kolodgie F, Narula N, Verjans J, Virmani R, Reutelingsperger C, Hofstra L, Narula J. Resolution of apoptosis in atherosclerotic plaque by dietary modification and statin therapy. J Nuclear Med. 2005;46:2051–2056. [PubMed] [Google Scholar]
- 35.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk Receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoE−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–1428. doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uglow EB, Slater S, Sala-Newby GB, Aguilera-Garcia CM, Angelini GD, Newby AC, George SJ. Dismantling of cadherin-mediated cell-cell contacts modulates smooth muscle cell proliferation. Circ Res. 2003;92:1314–1321. doi: 10.1161/01.RES.0000079027.44309.53. [DOI] [PubMed] [Google Scholar]
- 37.Dwivedi A, Slater SC, George SJ. MMP-9 and -12 cause N-cadherin shedding and thereby β-catenin signalling and vascular smooth muscle cell proliferation. Cardiovasc Res. 2009;81:178–186. doi: 10.1093/cvr/cvn278. [DOI] [PubMed] [Google Scholar]
- 38.Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–655. doi: 10.1161/hc0502.102969. [DOI] [PubMed] [Google Scholar]
- 39.Aikawa E, Nahrendorf M, Figueiredo J-L, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 40.Jeziorska M, McCollum C, Woolley DE. Calcification in atherosclerotic plaque of human carotid arteries: associations with mast cells and macrophages. J Pathol. 1998;185:10–17. doi: 10.1002/(SICI)1096-9896(199805)185:1<10::AID-PATH71>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 41.Shanahan CM, Cary NRB, Metcalfe JC, Weissberg PL. High expression of genes for calcification-regulating proteins in human atherosclerotic plaques. J Clin Invest. 1994;93:2393–2402. doi: 10.1172/JCI117246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- 43.Dive V, Andarawewa KL, Boulay A, Matziari M, Beau F, Guerin E, Rousseau B, Yiotakis A, Rio MC. Dosing and scheduling influence the antitumor efficacy of a phosphinic peptide inhibitor of matrix metalloproteinases. Int J Cancer. 2005;113:775–781. doi: 10.1002/ijc.20459. [DOI] [PubMed] [Google Scholar]
- 44.Georgiadis D, Beau F, Czarny B, Cotton J, Yiotakis A, Dive V. Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin: insights from selective inhibitors. Circ Res. 2003;93:148–154. doi: 10.1161/01.RES.0000081593.33848.FC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.