

Abstract
BACKGROUND AND PURPOSE
Recent evidence suggests that corticotropin-releasing factor (CRF) receptor signalling is involved in modulating the negative symptoms of opiate withdrawal. In this study, a series of experiments were performed to further characterize the role of CRF-type 2 receptor (CRF2) signalling in opiate withdrawal-induced physical signs of dependence, hypothalamus-pituitary-adrenal (HPA) axis activation, enhanced noradrenaline (NA) turnover in the hypothalamic paraventricular nucleus (PVN) and tyrosine hydroxylase (TH) phosphorylation (activation), as well as CRF2 expression in the nucleus of the solitary tract-A2 noradrenergic cell group (NTS-A2).
EXPERIMENTAL APPROACH
The contribution of CRF2 signalling in opiate withdrawal was assessed by i.c.v. infusion of the selective CRF2 antagonist, antisauvagine-30 (AS-30). Rats were implanted with two morphine (or placebo) pellets. Six days later, rats were pretreated with AS-30 or saline 10 min before naloxone and the physical signs of abstinence, the HPA axis activity, NA turnover, TH activation and CRF2 expression were measured using immunoblotting, RIA, HPLC and immunohistochemistry.
KEY RESULTS
Rats pretreated with AS-30 showed decreased levels of somatic signs of naloxone-induced opiate withdrawal, but the corticosterone response was not modified. AS-30 attenuated the increased production of the NA metabolite, 3-methoxy-4-hydroxyphenylglycol, as well as the enhanced NA turnover observed in morphine-withdrawn rats. Finally, AS-30 antagonized the TH phosphorylation at Serine40 induced by morphine withdrawal.
CONCLUSIONS AND IMPLICATIONS
These results suggest that physical signs of opiate withdrawal, TH activation and stimulation of noradrenergic pathways innervating the PVN are modulated by CRF2 signalling. Furthermore, they indicate a marginal role for the HPA axis in CRF2-mediation of opiate withdrawal.
Keywords: morphine withdrawal, corticotropin-releasing factor-2 receptor, hypothalamic paraventricular nucleus, noradrenergic pathways activity
Introduction
Corticotropin-releasing factor (CRF) and noradrenaline (NA) are two neurochemicals released following stress, which have been strongly implicated in different aspects of drug dependence, including craving and relapse to drug use (Sinha, 2007; 2008;). CRF is widely distributed throughout the brain and plays a major role in coordinating the behavioural and autonomic responses to stress (Owens and Nemeroff, 1991). In addition, CRF has been reported to contribute to the anxiogenic and adverse symptoms of withdrawal from exposure to several drugs of abuse, including cocaine and opioids (Koob, 2008). Two G protein-coupled receptors have been identified that bind CRF with high affinity: CRF receptor 1 (CRF1) and CRF receptor 2 (CRF2). Many lines of evidence indicate that the central CRF system is involved in the expression of morphine withdrawal signs. Thus, CRF receptor antagonists have been shown to attenuate several behavioural signs of morphine withdrawal (Iredale et al., 2000; Lu et al., 2000).
NA is also a key player in the mammalian stress response as well as in stress-induced relapse of drug-seeking for opiates, cocaine, nicotine and ethanol (for review, see Smith and Aston-Jones, 2008). In addition, NA has also been implicated in addiction and in particular in the adverse effects of acute opiate withdrawal (Maldonado, 1997; Aston-Jones and Kalivas, 2008; Núñez et al., 2009). Thus, opiate withdrawal results in marked activity of central noradrenergic neurones and it has been proposed that noradrenergic afferent neurones to the extended amygdala and the hypothalamic paraventricular nucleus (PVN) are critically involved in the adverse effects of opiate withdrawal. These noradrenergic afferent neurones originate in the nucleus of the solitary tract (NTS) and ventrolateral medulla noradrenergic A2 and A1 cell groups (Delfs et al., 2000).
Increased noradrenergic activity in the PVN during acute opiate withdrawal is also accompanied by increased signalling of CRF activity and NA turnover in the PVN (Fuertes et al., 2000; Laorden et al., 2000; Núñez et al., 2007a). Rat tyrosine hydroxylase (TH) activity is directly regulated by the phosphorylation at Ser31 and Ser40 (Haycock, 1993; Dunkley et al., 2004) and recently, we have shown that naloxone-induced morphine withdrawal stimulates TH activity and accelerates NA turnover in the PVN via a mechanism involving phosphorylation of TH at Ser31 (Núñez et al., 2007b).
The possible involvement of CRF1 and CRF2 subtypes in the interaction between morphine withdrawal and the noradrenergic system innervating the PVN has not been well documented and it may be possible that CRF could act to alter NA transmission. Recent data from our laboratory have suggested that the CRF1 subtype may not contribute to the functional interaction between NA and the CRF systems in mediating morphine withdrawal-activation of the brain stress neurocircuitry (Navarro-Zaragoza et al., 2010). On the other hand, conflicting findings have been reported with regard to the role of CRF1 and/or CRF2 in drug withdrawal. The stimulation of CRF2 decreases alcohol intake in alcohol-dependent animals and decreases alcohol withdrawal-induced anxiety-like behaviour (Valdez et al., 2004). In contrast, other studies have shown that the negative emotional state associated with precipitated nicotine withdrawal is partially mediated by an increase in the release of CRF in the extended amygdala (Marcinkiewcz et al., 2009). Furthermore, CRF2 knockout mice display decreased somatic morphine withdrawal signs, suggesting that the activation of CRF2 contributes to drug withdrawal (Papaleo et al., 2008). So, additional studies are needed in order to elucidate the role of CRF2 in drug withdrawal.
The aim of the present series of experiments was to investigate (i) the role of CRF2 in mediating somatic and behavioural states produced during precipitated morphine withdrawal; (ii) the response of noradrenergic pathways innervating the PVN and the activation of the hypothalamus-pituitary-adrenocortical (HPA) axis induced by morphine withdrawal in morphine-dependent rats pretreated with the selective CRF2 antagonist antisauvagine-30; and (iii) the effect of morphine withdrawal on CRF2 expression within the NTS-A2.
Methods
Materials
Pellets of morphine base (Alcaliber Laboratories., Madrid, Spain) or lactose (control) were prepared in the Department of Pharmacy and Pharmaceutics Technology (School of Pharmacy, Granada, Spain); naloxone HCl (Sigma Chemical Co., St Louis, MO, USA); Antisauvagine-30 (AS-30; Tocris, Bristol, UK); Protease inhibitors (Boehringer Mannheim, Mannheim, Germany); phosphatase inhibitor Cocktail Set (Calbiochem, Darmstadt, Germany); HPLC reagents were purchased from Sigma Chemical Co. Naloxone and AS-30 were prepared fresh each day by reconstitution in sterile 0.9% NaCl (saline; Laboratorios ERN, Barcelona, Spain). Ketamine and xylazine were purchased from Labs. Merial and Labs Calier, respectively (Barcelona, España).
Animals
Male Wistar rats (220–240 g; Harlan, Barcelona, Spain; n= 33 at the beginning of the experiment) were housed in pairs in cages (length, 45 cm; width, 24 cm; height, 20 cm) on arrival in a room with controlled temperature (22 ± 2°C) and humidity (50 ± 10%), with free access to water and food (Harlan Teklad standard rodent chow; Harlan Interfauna Ibérica, Barcelona, Spain). Animals were adapted to a standard 12 h light-dark cycle (lights on: 08 h 00 min – 20 h 00 min) for 7 days before the beginning of the experiments. All surgical and experimental procedures were performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC), and were approved by the local Committees for animal research (REGA ES300305440012).
Surgery
Rats were anaesthetized with 150 mg·kg−1 ketamine chlorhydrate and 8 mg·kg−1 xylazin (i.p.) before being implanted with a chronic guide cannula aimed at the right lateral ventricle, with the incisor bar set at −2.7 mm below the interaural line and according to coordinates from the stereotaxic atlas of Paxinos and Watson (Paxinos and Watson, 2007): anteroposterior =−1.0 mm from bregma, L =+2.0 mm (right hemisphere) and V =−3.0 mm, with the cranium surface as dorsal reference. Each guide cannula consisted of a threaded cylindrical pedestal molded around a piece of stainless steel tubing (26-gauge size) that extends 3.0 mm below the pedestal (Plastics One®, Bilaney Consultants GMBH, Düsseldorf, Germany). The cannula was fixed with dental cement (Vertex self-curing, Dentimex, Zeist, the Netherlands) and stainless-steel screws, which held the cannula in place. Sterile dummy stylets (Plastics One®) were placed into the cannula to prevent occlusion. After surgery, the skin was sutured and antiseptic (Topionic, Almirall Prodesfarma, Barcelona, Spain) was applied to the sutured area. After surgery, rats were individually housed.
Drug treatment and experimental procedure
One week after stereotaxic surgery, rats were implanted subcutaneously (s.c.) with two 75 mg morphine pellets under light ether anaesthesia. Control rats received placebo pellets containing the excipient without morphine. This procedure has been shown to produce consistent plasma morphine concentrations beginning a few hours after the implantation of the pellets and a full withdrawal syndrome after acute injection of opioid antagonists (Frenois et al., 2002). Dependence on morphine is achieved 24 h after implantation of pellets and remained constant for 15 days (Gold et al., 1994). Six days after the implantation of morphine or placebo pellets, precipitated withdrawal was induced by s.c. injection of naloxone (1 mg·kg−1; in a volume of 1 mL·kg−1 body weight). The four experimental conditions investigated for opiate withdrawal-induced physical signs of dependence, corticosterone, adrenocorticotropic hormone (ACTH), NA and 3-methoxy-4-hydroxyphenylethylen glycol (MHPG), TH phosphorylated at Ser31 and at Ser40, and CRF2 determination were: (i) placebo-vehicle-naloxone; (ii) placebo-AS-30-naloxone; (iii) morphine-vehicle-naloxone; (iv) morphine-AS-30-naloxone (Table 1).
Table 1.
Number of animals used in each experiment
| Parameters | Plac + Veh + Nx | Plac + AS30 + Nx | Morph + Veh + Nx | Morph + AS30 + Nx | Total n |
|---|---|---|---|---|---|
| Number of rats at the beginning of the experiment = 33 | 7 | 7 | 9 | 10 | |
| Somatic signs of opiate withdrawal | 6 | 5 | 8 | 9 | 28 |
| ACTH levels | 5 | 6 | 9 | 10 | 30 |
| Corticosterone levels | 5 | 6 | 8 | 8 | 27 |
| MHPG, NA and NA turnover | 4 | 6 | 5 | 10 | 25 |
| TH pSer31-IR | 6 | 6 | 5 | 5 | 22 |
| TH pSer40-IR | 5 | 6 | 4 | 6 | 21 |
| CRF2-IR | 7 | 6 | 13 |
ACTH, adrenocorticotropic hormone; NA, noradrenaline; MHPG, 3-methoxy-4-hydroxyphenylethylen glycol; TH, tyrosine hydroxylase; Veh, vehicle; Nx, naloxone; Plac, placebo; Morph, morphine.
Measurement of the withdrawal syndrome
Experiments were carried out in a quiet room. The observer was unaware of the drug combination used. Rats were individually placed into transparent plastic cages 15 min before the naloxone injection and observed continuously for the occurrence of somatic signs of opiate withdrawal up to 30 min after the naloxone injection. Subsequently, previously identified behavioural characteristics of the rat opiate abstinence syndrome (Lu et al., 2000) were evaluated, including: wet dog shakes, jumping, paw tremour, teeth chattering, mastication, ptosis, piloerection, sniffing, writhing, tremour and diarrhoea. The number of wet dog shakes, jumping, sniffing, and paw tremour was counted as the number of events occurring during the total test time period (graded signs). Teeth chattering, body tremour, mastication, ptosis, piloerection and diarrhoea were scored 1 for appearance or 0 for non-appearance within each 5 min time. To obtain a comprehensive index of the severity of somatic opioid withdrawal including all the signs examined, a global withdrawal score was calculated for each animal by giving each individual sign a relative weight as previously reported (Maldonado et al., 1996): jumping ×0.8; wet dog shakes ×1; paw tremour ×0.35; sniffing ×0.5; writhing ×0.5; ptosis ×1.5; teeth chattering ×1.5; body tremour ×1.5; diarrhoea ×1.5 and piloerection ×1.5. Body weight loss was determined as the difference between the weight determined immediately before naloxone injection and a second determination made 30 min later. The weight gain of the rats was checked during treatment to ensure that the morphine was liberated correctly from the pellets because it is known that chronic morphine treatment induces a decrease in body weight gain due to lower caloric intake (Houshyar et al., 2004; Núñez et al., 2009).
In order to investigate the effect of CRF2 blockade on the physical symptoms of morphine withdrawal, rats were infused with the selective CRF2 antagonist, AS-30 [AS-30; (Rühmann et al., 1998] or saline (control) administered i.c.v. Briefly, rats were gently restrained while the dummy stylets were removed and replaced with a 33-gauge stainless-steel injector (Plastics One®) extending 1 mm below the cannula tip. The injector was connected by polyethylene tubing (Plastics One®) to one 10 µL syringe (Hamilton Microliter Syringes, Bonaduz, GR, Switzerland) mounted to an infusion pump (11 Plus Syringe Pump, Harvard Apparatus Inc., Holliston, MA, USA). AS-30 was dissolved in 0.9% sterile saline (pH 7.4) and a dose of 20 µg was administered to rats in the AS-30 groups. Rats in the control vehicle group received a vehicle injection (saline). The solution was infused unilaterally in a volume of 3 µL into the right hemisphere for 2 min. The injector was left in place for 1 min after the infusion was completed to allow for diffusion.
Thirty minutes after naloxone injection, rats were decapitated (between 11 h 00 min and 12 h 00 min to avoid circadian variations in plasma levels of the hormones), the brains were rapidly removed, and stored immediately at −80°C until use for Western blot analysis of TH Ser31, TH Ser40 and CRF2. A second set of animals from each treatment group was used for NA and MHPG determination. One set of each treatment group was randomly assigned for plasma ACTH and corticosterone determination.
In order to determine the effect of inhibiting CRF2 on the morphine withdrawal-induced activation of the axis, morphine-dependent and control rats were treated with vehicle (saline; 3 µL i.c.v.) or AS-30 (20 µg 3 µL−1 i.c.v.) 10 min before the administration of naloxone. The dose of AS-30 was selected on the basis of published reports (Maruyama et al., 2007). After treatment, the following parameters were determined: NA and MHPG content and NA turnover in the PVN (HPLC); TH phosphorylation (activation) at Serine 31 (pSer31) and/or Serine 40 (pSer40) in the NTS-A2 noradrenergic cell group (Western blot); CRF2 expression in the NTS-A2 (Western blot); and plasma corticosterone and ACTH concentrations (RIA).
Western blotting
Brainstem tissue corresponding to NTS-A2 cell group was dissected between the area postrema (AP), rostrally, to the pyramidal decussation caudally (plane of sections relative to bregma: −13.68 to −14.60; Paxinos and Watson, 2007). Samples were placed in homogenization buffer (Núñez et al., 2007b), homogenized and sonicated for 30 s prior to centrifugation at 6000×g for 5 min at 4°C. Samples containing equal quantities of total proteins (40 µg) were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). Western analysis was performed with the following primary antibodies: 1:500 goat polyclonal anti-CRF2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA); 1:500 rabbit polyclonal anti-tyrosine-hydroxylase phosphorylated at ser31 (pSer31; Millipore, Temecula, CA, USA); 1:500 rabbit polyclonal anti-tyrosine-hydroxylase phosphoSer40 (pSer40; Millipore); and 1:1000 anti-beta actin (rabbit polyclonal antibody, Cell Signaling Technology Inc., Danvers, MA, USA). We used β-actin as our loading control for all the experiments. Before re-probing, blots were stripped by incubation with stripping buffer (glycine 25 mM and SDS 1%, pH 2) for 1 h at 37°C. Blots were subsequently reblocked and probed with anti β-actin (1:1000, overnight at room temperature). The ratios of CRF2/β-actin, pSer31-TH/β-actin and TH pSer40-TH/β-actin were plotted and analysed. Protein levels were corrected for individual levels.
Estimation of noradrenaline and its metabolite MHPG in the PVN
NA and its metabolite in the central nervous system, MHPG, were determined by HPLC with electrochemical detection as described previously (Navarro-Zaragoza et al., 2010). Bilateral tissue samples of the PVN were dissected according to the technique of Palkovits (1973), frozen in liquid nitrogen, weighed, placed in perchloric acid (0.1 M), homogenized and centrifuged and the supernatants taken for analysis and filtered through 0.22 µm GV (Millipore). Ten µL of each sample were injected into a 5-µm C18 reversed-phase column (Waters, Milford, MA, USA) through a Rheodyne syringe-loading injector (Waters). Electrochemical detection was accomplished with an electrochemical detector (Waters 2465). NA and MHPG were quantified by reference to calibration curves run at the beginning and the end of each series of assays. The content of NA and MHPG in the PVN was expressed as ng·g−1 wet weight of tissue. The NA turnover was determined as the NA ratio, which was calculated as: NA ratio = MHPG/NA.
Radioimmunoassay
After the rats had been decapitated, trunk blood was collected into ice-cooled tubes containing 5% EDTA and was then centrifuged (500×g; 4°C; 15 min). Plasma was separated, divided into two aliquots and stored at −80°C until assayed for corticosterone or ACTH. Plasma levels of corticosterone and ACTH were quantified using specific corticosterone and ACTH antibodies for rats ([125I]-CORT and [125I]-hACTH RIA; MP Biomedicals, Orangeburg, NY, USA). The sensitivity of the assay was 7.7 ng·mL−1 for corticosterone and 5.7 pg·mL−1 for ACTH.
Statistical analysis
Data are presented as mean ± SEM and were analysed using the SPSS 15.0 statistical package (SPSS Inc., Chicago, IL, USA). Somatic signs of withdrawal, body weight loss, and hormonal and biochemical parameters were analysed by two-way analysis of variance (anova) with pretreatment (placebo, morphine) and acute treatment (vehicle, AS-30) as independent variables. The Newman–Keuls post hoc test was used for individual group comparisons. To compare two groups, Student's t-test was used. Differences with a P < 0.05 were considered significant.
Results
In accordance with previous findings, Student's t-test showed that rats receiving long-term morphine treatment had significantly lower weight gain (−0.68 ± 2.31 g; t29= 5.74; P < 0.001; n= 12) than the placebo control group (19.75 ± 2.56 g; n= 19), which might be due to the reduced food intake observed during chronic morphine treatment (Núñez et al., 2009).
AS-30 attenuates the somatic expression of naloxone-precipitated morphine withdrawal
Six days after the implantation of morphine or placebo pellets, rats were challenged with naloxone (1 mg·kg−1 s.c.) and immediately tested for the occurrence of somatic signs of opiate withdrawal. The following somatic signs were significantly present in morphine-treated groups when compared with placebo-treated groups: paw tremour (P < 0.001), wet dog shakes (P < 0.001), body tremour (P < 0.001), writhing (P < 0.010), sniffing (P < 0.010), teeth chattering (P < 0.001), ptosis (P < 0.001), mastication (P < 0.001), diarrhoea (P < 0.001), piloerection (P < 0.001) and weight loss (P < 0.001). The analysis of the global withdrawal score confirmed these differences between morphine- and placebo-treated rats (P < 0.001). The results for two-way anova analysis are shown in Table 2.
Table 2.
AS-30 attenuates the somatic expression of naloxone-precipitated morphine withdrawal
| Chronic treatment (morphine vs. placebo) | Two-way anova Pretreatment (AS-30 vs. vehicle) | Interaction | ||||
|---|---|---|---|---|---|---|
| Signs | F (1, 27) | P < | F (1,27) | P < | F (1,27) | P < |
| Jumping | 0.553 | n.s | 0.553 | n.s | 0.553 | n.s. |
| Wet dog shakes | 180.477 | 0.001 | 4.677 | 0.050 | 4.677 | 0.050 |
| Paw tremour | 14.766 | 0.001 | 5.203 | 0.050 | 5.203 | 0.050 |
| Sniffing | 8.105 | 0.010 | 1.269 | n.s | 1.269 | n.s. |
| Writhing | 7.727 | 0.010 | 0.003 | n.s | 0.003 | n.s. |
| Tremour | 16.086 | 0.001 | 2.380 | n.s | 2.380 | n.s. |
| Ptosis | 21.407 | 0.001 | 0.114 | n.s | 0.114 | n.s. |
| Mastication | 48.882 | 0.001 | 4.846 | 0.050 | 4.846 | n.s. |
| Teeth chattering | 30.156 | 0.001 | 4.106 | 0.050 | 4.106 | 0.050 |
| Piloerection | 233.107 | 0.001 | 92.537 | 0.001 | 92.537 | 0.050 |
| Diarrhoea | 22.605 | 0.001 | 0.560 | n.s | 0.560 | n.s. |
| Weight loss | 78.523 | 0.001 | 1.418 | n.s | 5.092 | 0.050 |
| Global score | 212.377 | 0.001 | 14.874 | 0.001 | 14.874 | 0.001 |
Two-way anova with chronic treatment (morphine vs. placebo) and pretreatment before naloxone (AS-30 vs. vehicle) as between-subjects factors. When significant interactions in pretreatment or between these two factors were observed, a subsequent post hoc test was applied.
AS-30, Antisauvagine-30.
In the CRF2 blockade study after naloxone-precipitated morphine withdrawal, comparisons between morphine groups showed that wet dog shakes (P < 0.05), paw tremour (P < 0.05), mastication (P < 0.05), teeth chattering (P < 0.05), piloerection (P < 0.001) and weight loss (P < 0.05) were significantly decreased in rats receiving AS-30 microinfusion (Table 2, Figure 1A–F). The analysis of the global withdrawal score confirmed that AS-30 significantly reduced the somatic expression of withdrawal in morphine-treated rats (P < 0.001; Table 2, Figure 1G). Thus, the blockade of CRF2 overall decreased the expression of naloxone-precipitated somatic signs of opiate withdrawal, reducing global scores of morphine-dependent AS-30-treated rats.
Figure 1.
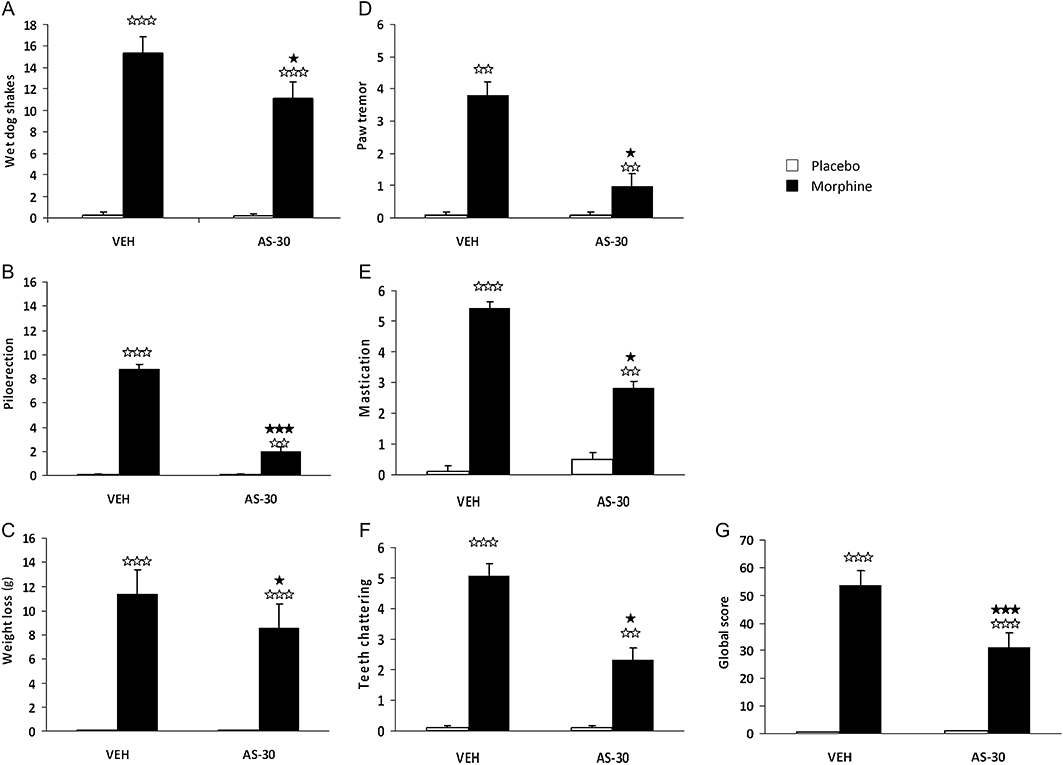
Attenuation of the severity of somatic signs of naloxone-precipitated morphine withdrawal by antisauvagine-30 microinfusion. The following variables were counted: (A) wet dog shakes; (C) body weight loss; (D) paw tremour and checked: (B) piloerection; (E) mastication; (F) teeth chattering. Somatic signs of withdrawal were observed during 30 min immediately after naloxone injection (1 mg·kg−1 s.c.). A global withdrawal score (G) was calculated for each animal as described in the Methods. Data are expressed as mean ± SEM. ⋆P < 0.050; ⋆⋆⋆P < 0.001, versus morphine + vehicle (VEH) + naloxone; ⋆⋆P < 0.01; ⋆⋆⋆P < 0.001 versus similar groups receiving chronic placebo.
Effects of CRF2 blockade on morphine withdrawal-induced HPA axis activation
We measured plasma ACTH and corticosterone concentrations (as HPA axis activation markers) in blood samples obtained from morphine-dependent or control rats 30 min after injection of naloxone. Two-way anova for ACTH revealed a major effect of chronic morphine treatment [F(1,23)= 55.65; P < 0.0001]. Newman–Keuls post hoc test showed that naloxone-precipitated morphine withdrawal in morphine-dependent animals administered vehicle plus naloxone i.c.v. evoked a dramatic increase (P < 0.001) in ACTH secretion (Figure 2A) compared with placebo-treated rats administered i.c.v. vehicle plus naloxone.
Figure 2.
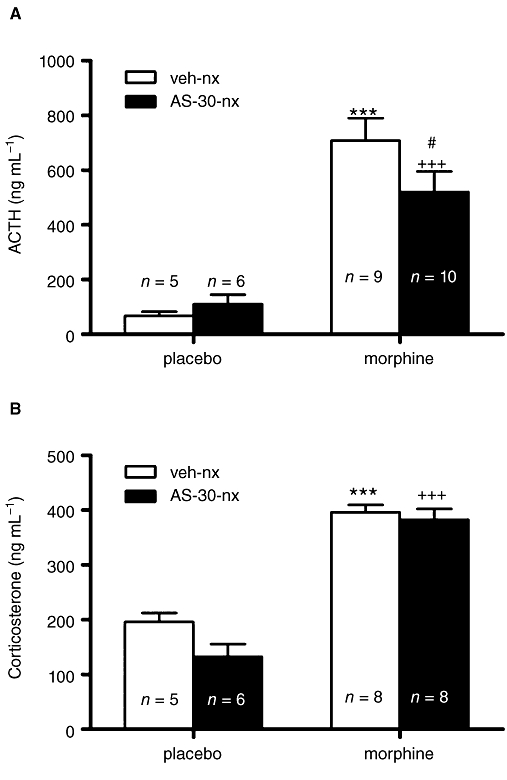
Antisauvagine-30 (AS-30) attenuated the plasma ACTH (A) but not the corticosterone (B) response to naloxone-induced morphine withdrawal. Data represent the mean ± SEM of plasma ACTH and corticosterone concentration 30 min after naloxone injection to control pellets- or morphine-treated rats administered vehicle, or AS-30 10 min before naloxone administration. ***P < 0.001 versus control pellets + vehicle + naloxone; +++P < 0.001 versus placebo + AS-30 + naloxone; #P < 0.05 versus morphine + vehicle + naloxone. ACTH, adrenocorticotropic hormone.
To evaluate whether a causal link exists between CRF2 activation and HPA axis hyperactivation during morphine withdrawal, we measured plasma ACTH concentrations in animals made dependent on morphine and pretreated with AS-30 10 min before naloxone administration. Newman–Keuls post hoc test showed that naloxone-precipitated morphine withdrawal evoked an increase in ACTH in animals treated with the CRF2 antagonist (P < 0.001) versus placebo plus AS-30 plus naloxone. However, levels of ACTH in rats pretreated with AS-30 10 min before naloxone injection were significantly (P < 0.05) lower than those seen in morphine-dependent rats administered vehicle instead of AS-30 (Figure 2A).
Two-way anova for corticosterone revealed a significant effect of chronic morphine treatment [F(1,26)= 131.14; P < 0.0001]. As shown in Figure 2B, in morphine-withdrawn rats administered vehicle i.c.v., the plasma corticosterone levels increased significantly (P < 0.001). Corticosterone levels in AS-30 plus naloxone-treated morphine-pelleted animals were significantly (P < 0.001) higher than those observed in the placebo group also administered AS-30 plus saline.
CRF2 blockade attenuates naloxone-induced MHPG production and NA turnover elevation in the hypothalamic PVN
Figure 3 summarizes the changes in NA content, MHPG production and NA turnover (as estimated by the ratio MHPG/NA) after injection of naloxone to control and morphine-dependent rats injected i.c.v. with vehicle or AS-30. The overall anova on NA content in the PVN revealed the main effects of morphine pretreatment [F(1,22)= 4.64; P= 0.0425], and significant effects for acute AS-30 administration [F(1,22)= 4.64; P= 0.0425]. Post hoc analysis indicated that groups rendered dependent on morphine and injected with vehicle or AS-30 on day 6 showed significantly (P < 0.05) lower levels of NA than the placebo-pelleted groups also injected with vehicle or AS-30 10 min before naloxone (Figure 3A).
Figure 3.
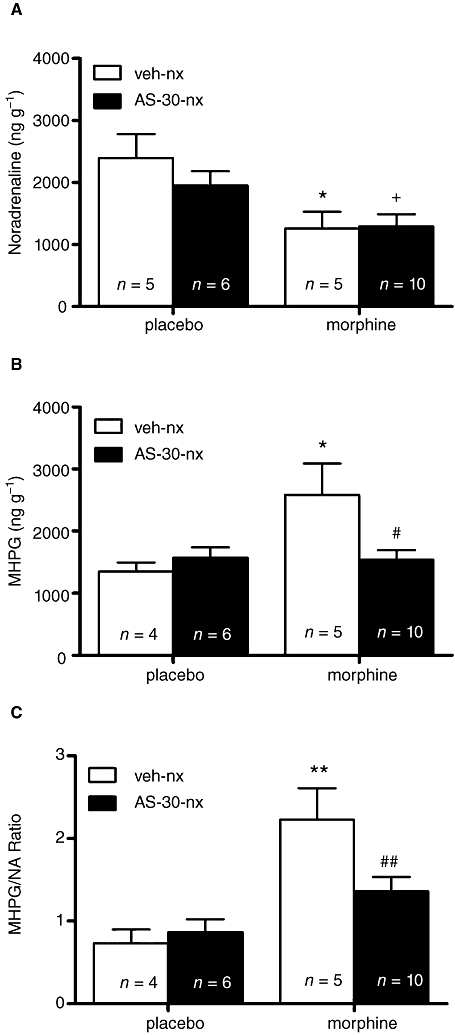
Effects of CRF2 blockade on NA (A) and MHPG (B) levels at the PVN and on the morphine withdrawal-induced increased NA turnover (as estimated by the MHPG/NA ratio; C) in control and in morphine-dependent rats after administration of naloxone. AS-30 attenuated morphine withdrawal-induced increase in MHPG levels and NA turnover. Data represent the mean ± SEM 30 min after naloxone injection to control pellets- or morphine-treated rats receiving vehicle or AS-30 10 min before naloxone (nx) administration. *P < 0.05; **P < 0.01 versus control pellets (placebo) + saline + naloxone; +P < 0.05 versus control pellets + AS-30 + nx; #P < 0.05; ## P < 0.01 versus morphine-treated rats + saline + naloxone. CRF2, corticotropin-releasing factor type-2 receptor; NA, noradrenaline; MHPG, 3-methoxy-4-hydroxyphenylethylen glycol; PVN, paraventricular nucleus; AS-30, antisauvagine-30; veh, vehicle.
The anova for MHPG production showed a significant effect of morphine pretreatment [F(1,21)= 5.19; P= 0.0332], and a significant interaction between morphine pretreatment and AS-30 administration [F(1,21)= 5.72; P= 0.0262]. Post hoc analysis showed that the MHPG levels increased significantly (P < 0.05) in the naloxone-precipitated morphine withdrawal group injected with vehicle i.c.v., as compared with the placebo-treated group injected with vehicle plus naloxone (Figure 3B). Post hoc analysis also showed that pretreatment with AS-30 10 min before naloxone injection significantly (P < 0.05) reduced morphine withdrawal-induced increases in MHPG levels compared with morphine-dependent rats administered vehicle instead of AS-30.
Results for the two-way anova for MHPG/NA ratio in the PVN revealed a significant effect of morphine pretreatment [F(1,21)= 17.87; P= 0.0004], and significant interaction between pretreatment and acute AS-30 administration [F(1,21)= 4.52; P= 0.0456]. As shown in Figure 3C, rats rendered dependent on morphine and injected with vehicle i.c.v. plus naloxone showed a significantly (P < 0.01) higher NA turnover in the PVN than the placebo group also injected with vehicle i.c.v. plus naloxone. An administration of AS-30 10 min before naloxone to morphine-dependent rats significantly (P < 0.01) antagonized the elevation in NA turnover (Figure 3C) compared with the morphine-pelleted group pretreated with vehicle before naloxone.
Effects of CRF2 blockade on morphine withdrawal-induced TH phosphorylation in the NTS
Additional experiments were performed in the NTS to determine whether naloxone-induced morphine withdrawal would activate phosphorylation of TH at Ser31 (pSer31) and/or Ser40 (pSer40). Next, we evaluated the effects of CRF2 blockade on the phosphorylation of TH after naloxone-induced morphine withdrawal. Two-way anova for pSer31-TH revealed a main effect of morphine pretreatment [F(1,18)= 14.94; P= 0.0011]. As shown in Figure 4A, rats dependent on morphine and given vehicle i.c.v. 10 min before naloxone (1 mg·kg−1 s.c.) injection showed a significant (P < 0.05) enhancement in pSer31-TH levels in the NTS compared with the placebo-pretreated group also injected with vehicle i.c.v. plus naloxone. An administration of AS-30 i.c.v. to morphine-dependent rats did not modify the increased phosphorylation of TH at Ser31 that was seen 30 min after naloxone injection in the vehicle-treated morphine-dependent rats (Figure 4A).
Figure 4.
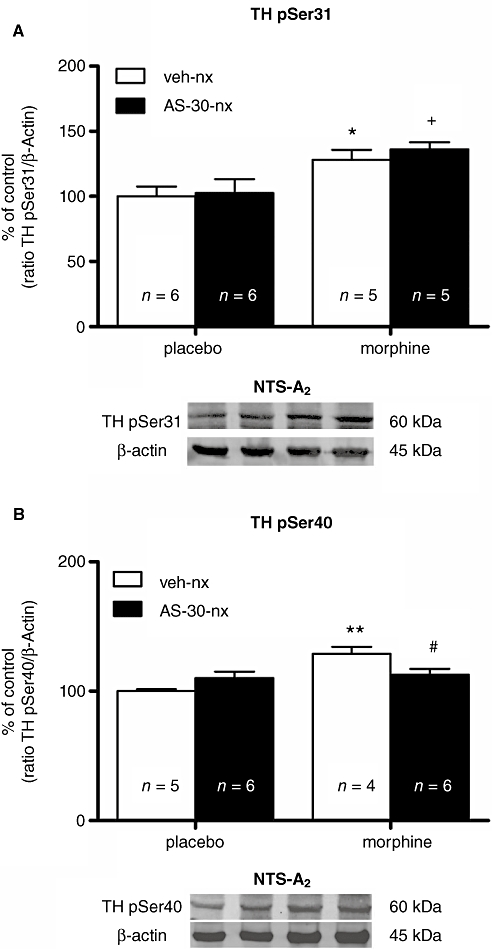
Morphine withdrawal-induced TH phosphorylation at Ser40 is dependent on the activation of CRF2 in the NTS-A2 noradrenergic cell group. Representative immunoblots of TH phosphorylated at Ser31 (A) and Ser40 (B) in NTS tissues isolated from placebo or morphine-dependent rats 30 min after the administration of naloxone in the absence or presence of an i.c.v. infusion of AS-30 (20 µg·2 µL−1) 10 min before naloxone administration. Data correspond to mean ± SEM. In morphine-dependent rats, a post hoc comparison test revealed a significant increase in TH phosphorylation at Ser31 during morphine withdrawal, which was not antagonized by AS-30. By contrast, the increase in TH phosphorylated at Ser40 after naloxone-precipitated morphine withdrawal was attenuated in rats pretreated with AS-30. *P < 0.05; **P < 0.01 versus control pellets (placebo) + saline + naloxone; +P < 0.05 versus control pellets + AS-30 + naloxone; #P < 0.05 morphine-treated rats + saline + naloxone. TH, tyrosine hydroxylase; CRF2, corticotropin-releasing factor type-2 receptor; NTS-A2, nucleus of the solitary tract-A2 noradrenergic cell group; AS-30, antisauvagine-30; veh, vehicle; nx, naloxone.
The anova for pSer40-TH showed a main effect of morphine pretreatment [F(1,17)= 11.95; P= 0.0030], and interaction between morphine pretreatment and AS-30 administration [F(1,17)= 8.15; P= 0.0110]. Figure 4B depicts that there was a significant (P < 0.01) increase in pSer40-TH levels in the NTS during naloxone-induced morphine withdrawal in rats administered vehicle i.c.v., compared with the corresponding placebo control group administered vehicle i.c.v. plus naloxone (1 mg·kg−1 s.c.). In addition, an administration of AS-30 i.c.v. to morphine-dependent rats significantly (P < 0.05) prevented the enhancement in pSer40-TH levels that was seen 30 min after naloxone injection.
Influence of morphine withdrawal on CRF2 immunoreactivity in NTS as determined by Western blot
Figure 5 shows that there was a significant (t11= 3.28; P < 0.001) increase in CRF2-IR levels in NTS during morphine withdrawal compared with the control placebo-treated group also administered naloxone. AS-30 pretreatment did not modify the enhanced CRF2-IR (data not shown).
Figure 5.
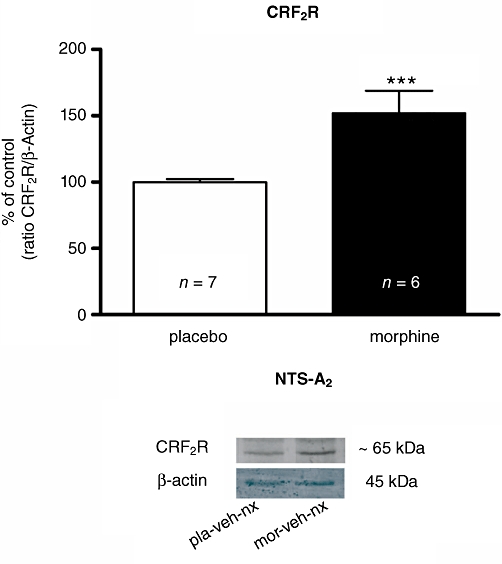
Representative immunoblots of CRF2 expression in NTS tissue isolated from control pellets-treated (placebo) or morphine-dependent rats 30 min after administration of naloxone. β-Actin was used as loading control. Data represent the optical density of immunoreactive bands expressed as a percentage (%) of the mean ± SEM of placebo control band. ***P < 0.001 versus control group. CRF2, corticotropin-releasing factor type-2 receptor; NTS, nucleus of the solitary tract; veh, vehicle; nx, naloxone; pla, placebo; mor, morphine.
Discussion
In this study, we demonstrated an important role for the CRF2 pathways in the somatic expression of morphine withdrawal. Thus, morphine-dependent rats infused with the CRF2 antagonist AS-30 before naloxone injection displayed lower levels of major somatic reactions to the stressful condition of naloxone-induced morphine withdrawal than rats infused with vehicle instead of AS-30. In addition, morphine-dependent rats infused with AS-30 also showed lower global morphine withdrawal scores than morphine-dependent animals infused with vehicle, suggesting that the activation of the CRF2 pathway might positively modulate the somatic expression of opiate withdrawal.
Conflicting findings have been reported with regard to the role of CRF receptors in opiate withdrawal. Thus, previous studies have proposed that CRF1 is the primary mediator of the action of the CRF system on the behavioural signs of morphine withdrawal (Iredale et al., 2000; Contarino and Papaleo, 2005). However, CRF2 knockout mice display decreased somatic morphine withdrawal signs, suggesting that the activation of the CRF2 subtype contributes to drug withdrawal (Papaleo et al., 2008). It should be pointed out that, in contrast to CRF1, the role for CRF2 pathways in stress-responsive circuitry is less clear. We have recently reported that specific pharmacological antagonism of CRF1 with antalarmin or CP-154526 decreased anxiety-like behaviour but not other characteristic symptoms of opiate withdrawal (Navarro-Zaragoza et al., 2010). AS-30 is a peptide antagonist exhibiting high affinities for the cloned human CRF2(a) (Ki 0.8 nM) and mouse CRF2(b) receptor (Kd 1.4 nM) (Rühmann et al., 1998). In the present study, AS-30 blocked the morphine withdrawal-increased NA turnover in the PVN, the increased MHPG production and attenuated most of the somatic signs of abstinence, whereas in a recent study from our group none of these parameters were attenuated by the selective CRF1 antagonists antalarmin or CP-154526 at different doses (Navarro-Zaragoza et al., 2010). In accordance with the present results, Papaleo et al. (2008) have reported that genetic disruption of the CRF2 pathway in mice reduced the severity of major somatic signs of morphine withdrawal. Together with our previous research, the present study provides initial evidence for opposite roles for the two CRF receptors in somatic opiate withdrawal.
All major drugs of abuse stimulate the HPA axis during acute withdrawal from the drug via the activation of CRF in the hypothalamic PVN, with a common response of elevated ACTH and corticosterone (Laorden et al., 2002a; Koob and Kreek, 2007; Núñez et al., 2007a; Koob, 2008). However, the relationship between HPA axis activity and alterations in drug withdrawal-induced behaviour has not been elucidated, and contradictory results have been obtained (Grakalic et al., 2006; Papaleo et al., 2007). The results of the present study show that morphine-dependent rats infused with AS-30, and the morphine-dependent group-receiving vehicle showed similar plasma corticosterone responses to naloxone-induced morphine withdrawal. In agreement with the present results, Papaleo et al. (2008) demonstrated that both wild-type mice and mice deficient in CRF2 showed increased plasma corticosterone responses to opiate withdrawal. Although it has been found that rat anterior pituitary corticotrophs express very high levels of CRF1 mRNA but no CRF2 mRNA (Kageyama et al., 2003), recent results from our laboratory showed that pretreatment with CP-154526, a selective CRF1 antagonist, did not block the corticosterone release that is produced as a consequence of morphine withdrawal (Navarro-Zaragoza et al., 2010). The present data do not support a role for the HPA axis in the decreased somatic expression of morphine withdrawal displayed by rats in which the CRF2 has been blocked. However, ACTH concentrations were found to be, slightly but significantly, decreased in rats infused with AS-30. A potential explanation for this finding is that, although CRF is thought to be the major secretagogue in stimulating ACTH secretion, arginine-vasopressin (AVP), changes in adrenal cortex sensitivity to the ACTH signal and other factors also play a role (Tilders et al., 1985). In addition, there is increasing evidence that AVP and CRF production and release from the parvocellular PVN neurones are under independent regulation. Thus, it is possible that AVP may play an important role in mediating the pituitary-adrenal response to drug withdrawal and stress (Deak et al., 1999; Núñez et al., 2007a).
NA has been implicated in addiction and in particular in acute opiate withdrawal (Delfs et al., 2000; for review, see Maldonado, 1997; Smith and Aston-Jones, 2008). The present findings demonstrated that the administration of naloxone to morphine-treated rats significantly elevated MHPG production and NA turnover in the PVN, which project from noradrenergic NTS-A2 cell group, which is in agreement with previous data from our laboratory showing that morphine withdrawal stimulates NA turnover in the PVN as well as the activity of NTS-A2 TH-positive neurones (as reflected by c-Fos expression) (Laorden et al., 2000; 2002a;). In addition, we have found that morphine withdrawal is associated with an increase in TH enzymatic activity in the PVN and TH phosphorylation (activation) at Ser31 and TH mRNA expression in the NTS-A2 (Núñez et al., 2007b; 2009;), which occurs 60–120 min after morphine withdrawal. We also found elevated hnRNA CRF and AVP expression in the PVN from morphine-withdrawn rats (Núñez et al., 2007a).
There are strong neurochemical and electrophysiological evidence suggesting an interaction between CRF and the catecholaminergic systems, and the existence of a NA-CRF loop has been proposed in which NA would modulate the release of CRF in the brain stress system, including the central amygdala, the bed nucleus of the stria terminalis (important components of the extended amygdala) and the PVN. CRF from these nuclei would induce the release of NA by the brain stem noradrenergic areas (Koob, 1999; Stinus et al., 2005). However, the possible involvement of CRFR subtypes in the interaction between morphine withdrawal and the noradrenergic system innervating the PVN has not been well documented. We recently demonstrated that the activation of CRF1 subtype is not responsible for the elevation of NA neurotransmission innervating the PVN (Navarro-Zaragoza et al., 2010). By contrast, the results of the present study demonstrated that pretreatment with the selective CRF2 antagonist AS-30 did block both increased MHPG production and NA turnover in the PVN during morphine withdrawal. These results indicate that CRF2 activation may contribute to the increase in brain stem noradrenergic response in opiate-withdrawn rats and suggest that the effects of the CRF2 antagonist were at the NTS, where a robust hybridization signal for CRF2 mRNA has been found (Van Pett et al., 2000; Hauger et al., 2006). We have previously shown a role for the noradrenergic system in the somatic signs of opiate withdrawal (Laorden et al., 2000; 2002b;). Support for this idea can be found in a number of studies that implicate the NTS-A2 noradrenergic cell group in the affective disorders associated with drug withdrawal (Delfs et al., 2000; Smith and Aston-Jones, 2008). In addition, the present results raise the possibility that the effect of CRF2 blockade may be mediated via its action at extrahypothalamic sites, independent of its effects on the HPA axis. Next, the expression of CRF2 in the NTS was determined. Morphine withdrawal resulted in the up-regulation of CRF2 immunoreactivity. Although the functional consequences of increased CRF2 expression in response to opiate withdrawal have not been identified, one possibility is that the up-regulation of CRF2 may contribute to the hyperactivation of noradrenergic transmission that was seen in morphine-withdrawn rats, which agrees with recent reports showing no modifications in CRF1 immunoreactivity during morphine withdrawal (Navarro-Zaragoza et al., 2010).
It is well known that changes in the state of phosphorylation of TH, the rate-limiting enzyme in the synthesis of catecholamines, are critically involved in the regulation of catecholamine synthesis and function. In particular, increases in the phosphorylation of Ser31 and Ser40 accelerate TH activity, thereby stimulating production of neurotransmitters in catecholamine terminals (for review, see Kumer and Vrana, 1996; Bobrovskaya et al., 2007). Information regarding the effect of CRF on TH activation is limited. Using phosphorylation state-specific antibodies directed towards Ser31 or Ser40, in the present study, we have shown that naloxone-induced morphine withdrawal greatly increased the levels of TH phosphorylated at Ser31 and Ser40 in the rat NTS concomitantly with the previously described enhanced NA turnover. Together, these data confirm that Ser31 and/or Ser40 phosphorylation of TH may be an important modulator of TH activity during opiate withdrawal and might be directly involved with increasing NA turnover in morphine-withdrawn rats. The present findings are in agreement with those from previous studies showing that TH phosphorylation at Ser 40 results in a marked increase in TH activity (Dunkley et al., 2004). In the present study, we used the CRF2 antagonist AS-30 to check the involvement of the CRF2 subtype in the TH phosphorylation at Ser31 and/or at Ser40 during morphine withdrawal and we found that pretreatment with AS-30 significantly attenuated the morphine withdrawal stimulation of Ser40 but not Ser31 phosphorylation in the NTS. These results suggest that morphine withdrawal would induce activation of the CRF2 at the NTS level, which would result in enhanced Ser40 phosphorylation, TH activity and catecholamine synthesis and release in the PVN.
In conclusion, the results of this study help to show that noradrenergic afferents to the PVN and the CRF/CRF2 pathways are critically involved in the physical symptoms of opiate withdrawal, supporting a therapeutic potential of CRF2 antagonists in addictive disorders. Present data would help to the identification of the mechanisms that underlie the complex interactions between CRF and noradrenergic pathways during opiate withdrawal. Given the known neuroanatomical and functional interactions between CRF and NA, it is possible that these systems might be important therapeutic targets that mediate the adverse effects of opiate dependence.
Acknowledgments
This work was supported by the Ministerio de Ciencia e Innovación (Grants SAF/FEDER 2007-62758 and 2009-07178), Spain; Red de Trastornos Adictivos (RD06/0001/1006 and RD06/0001/1001), Spain. JN-Z was supported by a pre-doctoral fellowship from the Ministerio de Ciencia e Innovación. CN was supported by a post-doctoral contract from the Ministerio de Ciencia e Innovación. J.R-M was supported by a post-doctoral fellowship from A4U. The authors wish to thank Juana María Hidalgo and Lucía Mora for their technical assistance.
Glossary
Abbreviations
- CRF2
corticotropin-releasing factor type-2 receptor
- HPA
hypothalamus-pituitary-adrenocortical
- NTS-A2
nucleus of the solitary tract-A2 noradrenergic cell group
- PVN
hypothalamic paraventricular nucleus
Conflict of interest
The authors have no conflict of interest.
Supplemental material
References
- Aston-Jones G, Kalivas PW. Brain norepinephrine rediscovered in addiction research. Biol Psychiatry. 2008;63:1005–1006. doi: 10.1016/j.biopsych.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovskaya L, Gilligan C, Bolster EK, Flaherty JJ, Dickson PW, Dunkley PR. Sustained phosphorylation of tyrosine hydroxylase at serine 40: a novel mechanism for maintenance of catecholamine synthesis. J Neurochem. 2007;100:479–489. doi: 10.1111/j.1471-4159.2006.04213.x. [DOI] [PubMed] [Google Scholar]
- Contarino A, Papaleo F. The corticotropin-releasing factor receptor-1 pathway mediates the negative affective states of opioid withdrawal. Proc Natl Acad Sci USA. 2005;102:18649–18654. doi: 10.1073/pnas.0506999102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak T, Nguyen KT, Ehrlich AL, Watkins LR, Spencer RL, Maier SF, et al. The impact of the nonpeptide corticotropin-releasing hormone antagonist antalarmin on behavioral and endocrine responses to stress. Endocrinology. 1999;140:79–86. doi: 10.1210/endo.140.1.6415. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Bobrovskaya L, Graham ME, von Nagy-Felsobuki EI, Dickson PW. Tyrosine hydroxylase phosphorylation: regulation and consequences. J Neurochem. 2004;91:1025–1043. doi: 10.1111/j.1471-4159.2004.02797.x. [DOI] [PubMed] [Google Scholar]
- Frenois F, Cador M, Caille S, Stinus L, Le Moine C. Neural correlates of the motivational and somatic components of naloxone-precipitated morphine withdrawal. Eur J Neurosci. 2002;16:1377–1389. doi: 10.1046/j.1460-9568.2002.02187.x. [DOI] [PubMed] [Google Scholar]
- Fuertes G, Laorden ML, Milanés MV. Noradrenergic and dopaminergic activity in the hypothalamic paraventricular nucleus after naloxone-induced morphine withdrawal. Neuroendocrinology. 2000;71:60–67. doi: 10.1159/000054521. [DOI] [PubMed] [Google Scholar]
- Gold LH, Stinus L, Inturrisi CE, Koob GF. Prolonged tolerance, dependence and abstinence following subcutaneous morphine pellets implantation in the rat. Eur J Pharmacol. 1994;253:45–51. doi: 10.1016/0014-2999(94)90755-2. [DOI] [PubMed] [Google Scholar]
- Grakalic I, Schindler C, Baumann M, Rice K, Riley A. Effects of stress modulation on morphine-induced conditioned place preferences and plasma corticosterone levels in Fischer, Lewis, and Sprague-Dawley rat strains. Psychopharmacology. 2006;189:277–286. doi: 10.1007/s00213-006-0562-5. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5:453–479. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycock JW. Multiple signaling pathways in bobine chromaffin cells regulate tyrosine hydroxylase phosphorylation at Ser19, Ser31 and Ser40. Neurochem Res. 1993;18:15–26. doi: 10.1007/BF00966919. [DOI] [PubMed] [Google Scholar]
- Houshyar H, Manalo S, Dallman MF. Time-dependent alterations in mRNA expression of brain neuropeptides regulating energy balance and hypothalamo-pituitary-adrenal activity after withdrawal from intermittent morphine treatment. J Neurosci. 2004;24:9414–9424. doi: 10.1523/JNEUROSCI.1641-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iredale PA, Alvaro JD, Lee Y, Terwilliger R, Chen YL, Duman RS. Role of corticotropin-releasing factor receptor-1 in opiate withdrawal. J Neurochem. 2000;74:199–208. doi: 10.1046/j.1471-4159.2000.0740199.x. [DOI] [PubMed] [Google Scholar]
- Kageyama K, Li C, Vale WW. Corticotropin-releasing factor receptor type 2 messenger ribonucleic acid in rat pituitary: localization and regulation by immune challenge, restraint stress, and glucocorticoids. Endocrinology. 2003;144:1524–1532. doi: 10.1210/en.2002-221046. [DOI] [PubMed] [Google Scholar]
- Koob GF. Stress, corticotropin-releasing factor, and drug addiction. Ann N Y Acad Sci. 1999;897:27–45. doi: 10.1111/j.1749-6632.1999.tb07876.x. [DOI] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress system in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumer SC, Vrana KE. Intricate regulation of tyrosine hydroxylase activity and gene expression. J Neurochem. 1996;67:443–462. doi: 10.1046/j.1471-4159.1996.67020443.x. [DOI] [PubMed] [Google Scholar]
- Laorden ML, Fuertes G, González-Cuello A, Milanés MV. Changes in catecholaminergic pathways innervating paraventricular nucleus and pituitary-adrenal axis response during morphine dependence: implication of a1- and a2-adrenoceptors. J Pharmacol Exp Ther. 2000;293:578–584. [PubMed] [Google Scholar]
- Laorden ML, Castells MT, Milanés MV. Effects of morphine and morphine withdrawal on brainstem neurons innervating hypothalamic nuclei that control the pituitary-adrenocortical axis in rats. Br J Pharmacol. 2002a;136:67–75. doi: 10.1038/sj.bjp.0704684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laorden ML, Núñez C, Almela P, Milanés MV. Morphine withdrawal-induced c-fos expression in the hypothalamic paraventricular nucleus is dependent on the activation of catecholaminergic neurones. J Neurochem. 2002b;83:132–140. doi: 10.1046/j.1471-4159.2002.01123.x. [DOI] [PubMed] [Google Scholar]
- Lu L, Liu D, Ceng X, Ma L. Differential role of corticotropin-releasing factor receptor subtypes 1 and 2 in opiate withdrawal and in relapse to opiate dependence. Eur J Neurosci. 2000;12:4398–4404. [PubMed] [Google Scholar]
- Maldonado R. Participation of noradrenergic pathways in the expression of opiate withdrawal: biochemical and pharmacological evidence. Neurosci Biobehav Rev. 1997;1:91–104. doi: 10.1016/0149-7634(95)00061-5. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Haoune J, et al. Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science. 1996;273:657–659. doi: 10.1126/science.273.5275.657. [DOI] [PubMed] [Google Scholar]
- Marcinkiewcz CA, Prado MM, Isaac SK, Marshall A, Rylkova D, Bruijnzeel AW. Corticotropin-releasing factor within the central nucleus of the amygdala and the nucleus accumbens shell mediates the negative affective state of nicotine withdrawal in rats. Neuropsychopharmacology. 2009;34:1743–1752. doi: 10.1038/npp.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Makino S, Noguchi T, Nishioka T. Central type 2 corticotropin-releasing hormone receptor mediates hypothalamic-pituitary-adrenocortical axis activation in the rat. Neuroendocrinology. 2007;86:1–16. doi: 10.1159/000103556. [DOI] [PubMed] [Google Scholar]
- Navarro-Zaragoza J, Núñez C, Laorden ML, Milanes MV. Effects of corticotropin-releasing factor receptor-1 (CRF1) antagonists on the brain stress system responses to morphine withdrawal. Mol Pharmacol. 2010;77:864–873. doi: 10.1124/mol.109.062463. [DOI] [PubMed] [Google Scholar]
- Núñez C, Foldes A, Laorden ML, Milanes MV, Kovács KJ. Activation of stress-related hypothalamic neuropeptide gene expression during morphine withdrawal. J Neurochem. 2007a;101:1060–1071. doi: 10.1111/j.1471-4159.2006.04421.x. [DOI] [PubMed] [Google Scholar]
- Núñez C, Laorden ML, Milanes MV. Regulation of serine (Ser)-31 and Ser40 tyrosine hydroxylase phosphorylation during morphine withdrawal in the hypothalamic paraventricular nucleus and nucleus tractus solitarius-A2 cell group. Role of ERK1/2. Endocrinology. 2007b;148:5780–5793. doi: 10.1210/en.2007-0510. [DOI] [PubMed] [Google Scholar]
- Núñez C, Földes A, Pérez-Flores D, García-Borrón JC, Laorden ML, Kovács KJ, et al. Elevated glucocorticoid levels are responsible for induction of tyrosine hydroxylase (TH) mRNA expression, phosphorylation and enzyme activity in the nucleus of the solitary tract (NTS-A2) during morphine withdrawal. Endocrinology. 2009;150:3118–3127. doi: 10.1210/en.2008-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin releasing factor. Pharmacol Rev. 1991;43:425–473. [PubMed] [Google Scholar]
- Palkovits M. Isolated removal of hypothalamic or other brain nuclei of the rat. Brain Res. 1973;59:449–450. doi: 10.1016/0006-8993(73)90290-4. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Kitchener P, Contarino A. Disruption of the CRF/CRF(1) receptor stress system exacerbates the somatic signs of opiate withdrawal. Neuron. 2007;53:577–589. doi: 10.1016/j.neuron.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Ghozland S, Ingallinesi M, Roberts AJ, Koob GF, Contarino A. Disruption of the CRF2 receptor pathway decreases the somatic expression of opiate withdrawal. Neuropsychopharmacology. 2008;33:2878–2887. doi: 10.1038/npp.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Amsterdam: Academic Press; 2007. [Google Scholar]
- Rühmann A, Bonk I, Lin CR, Rosenfeld MG, Spiess J. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): development of CRFR2beta-selective antisauvagine-30. Proc Natl Acad Sci USA. 1998;95:15264–15269. doi: 10.1073/pnas.95.26.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R. The role of stress in addiction relapse. Curr Psych Report. 2007;9:388–395. doi: 10.1007/s11920-007-0050-6. [DOI] [PubMed] [Google Scholar]
- Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann N Y Acad Sci. 2008;1141:105–130. doi: 10.1196/annals.1441.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Aston-Jones G. Noradrenergic transmission in the extended amygdala: role in increased drug-seeking and relapse during protracted drug abstinence. Brain Struct Funct. 2008;213:43–61. doi: 10.1007/s00429-008-0191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinus L, Cador M, Zorrilla EP, Koob GF. Buprenorphine and a CRF1 antagonists block the acquisition of opiate withdrawal-induced conditioned place aversion in rats. Neuropsychopharmacology. 2005;30:90–98. doi: 10.1038/sj.npp.1300487. [DOI] [PubMed] [Google Scholar]
- Tilders FJ, Berkenbosch F, Vermes I, Linton EA, Smelek PG. Role of epinephrine and vasopressin in the control of the pituitary-adrenal response to stress. Fed Proc. 1985;44:155–168. [PubMed] [Google Scholar]
- Valdez GR, Sabino V, Koob GF. Increased anxiety-like behavior and ethanol self-administration in dependent rats: reversal via corticotropin-releasing factor-2 receptor activation. Alcohol Clin Exp Res. 2004;28:865–872. doi: 10.1097/01.alc.0000128222.29875.40. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Li HY, Arias C, Prins GS, et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.