

Abstract
This review focuses on thiol/disulfide redox switches that regulate heme binding to proteins and modulate their activities. The importance of redox switches in metabolic regulation and the general mechanism by which redox switches modulate activity are discussed. Methods are described to characterize heme-binding sites and to assess their physiological relevance. For thiol/disulfide interconversion to regulate activity of a system, the redox process must be reversible at the ambient redox potentials found within the cell; thus, methods (and their limitations) are discussed that can address the physiological relevance of a redox switch. We review recent results that define a mechanism for how thiol/disulfide redox switches that control heme binding can regulate the activities of an enzyme, heme oxygenase-2, and an ion channel, the BK potassium channel. The redox switches on these proteins are composed of different types of Cys-containing motifs that have opposite effects on heme affinity, yet have complementary effects on hypoxia sensing. Finally, a model is proposed to describe how the redox switches on heme oxygenase-2 and the BK channel form an interconnected system that is poised to sense oxygen levels in the bloodstream and to elicit the hypoxic response when oxygen levels drop below a threshold value. Antioxid. Redox Signal. 14, 1039–1047.
Introduction
Redox homeostasis is essential for growth and survival. Like cellular buffers that maintain the pH in a suitable range, the ambient intracellular redox potential is buffered and regulated by the ratio of several thiol/disulfide systems, including thioredoxin, glutathione, and cysteine. Cysteine residues within proteins also play an important role as thiol/disulfide redox defense systems against oxidative stress (25). Thus, the ratios of the intracellular concentrations of the oxidized versus reduced states of these redox buffers poise the ambient redox potential. Yet, what is ambient can be ambiguous, because intracellular redox buffers are not fully in equilibrium and seem to regulate the redox status of different subsets of proteins (33, 43). Further, the intracellular ambient redox poise varies as the metabolic state of the cell changes. On the basis of the intracellular glutathione/glutathione disulfide ratio, the redox poise ranges from ∼−250 mV when cells are rapidly dividing to ∼−200 mV when they are differentiating to ∼−160 mV when undergoing apoptosis (43). Different compartments within the cell also maintain different ambient potentials; for example, based on the thioredoxin redox poise, the cytoplasm, nucleus, and mitochondria exhibit redox potentials of −280, −300, and −340 mV, respectively (33). In addition, thiol/disulfide redox systems are also important in altering (as well as maintaining) the redox potential of the extracellular environment; for example, extracellular redox remodeling is important in controlling the activation and proliferation of T cells within the immune system (65). Thus, because redox poise is spatially and temporally dynamic, systems are required to sense and to respond to changes in ambient redox potential as well as to coordinate events that occur under discrete redox conditions.
Redox switches and sensors are poised to respond to the dynamic redox environment within the cell. A switch is an electrical component that opens or closes a circuit, thus regulating the flow of current to a light bulb, a machine, etc. By analogy, a redox switch is a biochemical component that undergoes a reversible redox change that controls activity by promoting activation/inhibition of an enzyme, enhancement/reduction of transcriptional activity, alteration in ligand binding affinity, etc. A redox sensor is defined as a switch that additionally measures the ambient redox potential. Any biological molecule that is redox active could potentially serve as a redox switch or redox sensor. As well as cysteine thiol/disulfide (Cys-SS-Cys/CysSH), which is the focus of this review, the list of potential redox switches or sensors includes, CysSH/Cys-sulfonate, transition metal ions (Fe3+/2+, Ni2+/1+, Cu2+/1+, iron-sulfur clusters, etc.); cofactors like flavins (FAD/FADH°/FADH2), folate (folate/H2folate/H4folate), NAD(P)+/NAD(P)H, and quinones (Q/QH°/QH2); and redox equilibrium between the oxidized and reduced states of small molecules like glutathione, CoA, and mycothiol (20).
This review focuses on thiol/disulfide redox switches that interconvert between the oxidized disulfide and the reduced thiol(ate) states (Eq. 1) and, as a result of this redox change, bind their ligand with higher or lower affinity. A related redox switch involves interconversion between the sulfenic acid and thiol states (Eq. 2). As described in Poole's article in this volume (36), when there is a nearby reactive Cys residue, that is, a resolving Cys, the sulfenate can undergo conversion to the disulfide. Thus, as indicated by summing Equations 2 and 3, interconversion between the thiol and disulfide states can involve a sulfenate intermediate. Because the sulfenate can undergo facile irreversible oxidation eventually to the sulfonate, the presence of a resolving Cys residue helps to maintain a reversible two-electron redox interconversion.
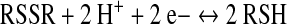 |
(1) |
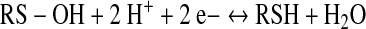 |
(2) |
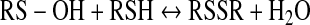 |
(3) |
Switches and Redox Coupling
Figure 1 illustrates how a redox switch can regulate activity (ligand binding, a chemical reaction, etc), or, similarly, how activity can alter the apparent redox potential of the switch. The fraction of the reduced component of the redox switch has been plotted versus the ambient redox potential of the solution in which the reaction occurs. The two parameters obtained from this Nernst plot are the number of electrons (n) transferred and the midpoint redox potential (E0) for the reaction. E0 represents the potential at which the concentrations of oxidized and reduced species are equal. The plot is described by the Nernst equation (Eq. 4), where R is the universal gas constant (8.31 J K−1 mol−1), T is the absolute temperature, F is the Faraday constant (9.65 × 104 C mol−1), and n is the number of electrons transferred in the reaction. Thus, the curve on the left would represent any two-electron reaction, for example, Equation 1, with an E0 of ∼−185 mV.
FIG. 1.

Comparison of standard redox reactions (left) and an EC reaction (right) in which a redox process is coupled to a chemical reaction. B binds to (or reacts with) only the Ared state. Because only the reduced form of the protein can bind ligand, the apparent redox potential of the Eox/Ered couple shifts to a more positive value.
Any redox-active biological molecule can potentially serve as a redox switch; however, for the redox interconversion to be physiologically relevant, the midpoint redox potential of the Aox/Ared couple must be near the range of the ambient intracellular redox potential, which ranges from −170 to −325 mV (17, 32). Thus, the redox switch shown in Figure 1 would appear to be physiologically relevant, because the midpoint potential (∼−185 mV) is within the range of the ambient intracellular redox potential.
Figure 1 also describes what happens when a redox process is coupled to a chemical process, which might be a redox-dependent conformational change, a binding event, or a chemical reaction, which is classed by an electrochemist as an EC system. In this example, only the reduced form of the enzyme can bind its ligand (B), thus lowering the concentration of Ared and shifting the apparent E0 to more positive values. The magnitude of the shift relates to how favorable the coupled reaction is. For example, reduction of Co2+ is a very unfavorable reaction; however, in methionine synthase this reaction can be accomplished at relatively mild redox potentials because it is coupled to the binding of methyltetrahydrofolate and conversion of Co1+ to methyl-Co3+ (4).
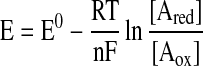 |
(4) |
Various processes in proteins have been shown to respond to redox changes. Redox-dependent transcriptional activation or repression is observed in several systems (3). OxyR, SoxR, and CprK are redox switches that control transcription in response to oxidative stress in bacteria (3, 14, 23, 26, 49). Exposure of cells to H2O2 leads to the formation of a disulfide bond in OxyR, which activates the transcription of genes that are part of a peroxide regulon and are involved in defense against oxidative stress. In SoxR, the oxidation or nitrosylation of a [2Fe-2S] cluster is coupled to a conformational change that is linked to transcriptional activation of oxidative stress defense genes. In the case of CprK, only the reduced state of a thiol/disulfide switch can promote a conformational change that activates transcription of genes involved in dehalogenation of haloaromatic environmental pollutants, a process that requires two oxygen-sensitive cofactors: vitamin B12 and an iron–sulfur cluster (13).
The same types of redox changes and redox-sensitive modules that modulate transcriptional activity can also elicit changes in catalytic activity of an enzyme. As described in the article by Becker and coworkers, profound redox-dependent changes occur in PutA (8). In its reduced state, PutA is a membrane-bound proline dehydrogenase; however, upon oxidation, the enzyme is released from the membrane to become a transcriptional repressor of genes involved in proline utilization (8, 74). As described in more detail below, a thiol-disulfide redox switch in mammalian heme oxygenase-2 (HO2) regulates the binding of its substrate (Fe3+-heme). Similarly, for the mammalian voltage- and Ca++-activated large conductance potassium channel (BK channel), the thiol-disulfide redox switch controls the affinity of a regulatory site for heme, which is an allosteric inhibitor of K+ transport.
Thus, redox switches can couple redox processes occurring in redox-responsive modules to events in other functional domains, allowing the regulation of transcription, enzymatic activity, ion channel activity, membrane binding, etc. This coupling allows the cell to maintain metabolic homeostasis and effect metabolic changes under varying redox conditions, as exemplified by the various systems described in this forum issue on redox switches.
Measurement of the Thiol/Disulfide Midpoint Redox Potential
The midpoint redox potential of the thiol/disulfide couple can be measured by several methods. Regardless of the technique, it is important to ensure that the Cys residues are in redox equilibrium with the solution, because thiols and disulfides usually are sluggish to equilibrate. Redox mediators with midpoint potentials in the range of the couple to be determined are used to establish and maintain the ambient potential of the solution (the x-axis of Fig. 1). Common mediators are dithiothreitol (E0 = −327 mV) (39) and glutathione (E0 = −240 mV) (54). For example, the reduced form of glutathione/oxidized disulfide state of glutathione ratio can be varied to establish a gradient of ambient redox potentials between −130 and −250 mV.
Because only minor changes in the UV–visible spectrum are associated with the thiol/disulfide interconversion, continuous UV–visible spectroelectrochemical measurements are usually too insensitive to monitor the redox titration. On the other hand, if a Trp residue is near the targeted Cys residues, fluorescence methods can often be used to continuously monitor the redox status of the thiol/disulfide couple, because the disulfide generally quenches the intrinsic fluorescence of a nearby excited Trp residue by an electron transfer mechanism (52). Thus, with this type of redox titration, the y-axis of Figure 1 would represent the extent of fluorescence quenching. Direct electrochemistry, performed by attaching the proteins directly to electrodes, is another option for continuous measurements of the extent of oxidation/reduction at varied redox potentials (15).
Discontinuous methods are also available to measure the midpoint potential of a thiol/disulfide redox switch. The ambient potential is set by incubation of the sample with a redox buffer as in the continuous methods described above; however, it is important to ensure that the redox state of the Cys residue(s) established by the redox buffer system is trapped and does not change during preparation of the samples for the redox measurement. Treatment with ice-cold trichloroacetic acid is often used for thiol trapping (40) before the samples are reacted with a thiol modification reagent, for example, maleimide, iodoacetate, and vinylpyridine. Then, the extent of modification can be monitored by a polyacrylamide gel-based method, such as the maleimide-polyethylene glycol 5000 thiol-trapping method, which places a 5 kDa adduct on each free thiol on the protein (68).
In Vivo Measurements of the Thiol/Disulfide Ratio
Besides the above-mentioned in vitro methods, one can also measure the ratio of oxidized/reduced Cys by an in vivo or ex vivo method. This measurement is an important test of the hypothesis that the particular redox switch is physiologically relevant. This is a more demanding experiment than the in vitro measurement with a purified protein because the protein of interest is only one component of all other proteins in the cell and because the desire is to trap the thiol status under a particular metabolic condition, for example, hypoxia, normoxia, and peroxide stress. The isotope coded affinity tag (ICAT) method is a novel mass spectrometric technique that combines thiol trapping with the ICAT technique to quantify oxidative thiol modifications within the growing cell (40). ICAT reagents consist of an iodoacetamide group linked to a cleavable biotin affinity tag via an isotopically light (12C) or heavy (13C) nine-carbon linker (24). Briefly, cells are lysed at low pH to trap the thiol status and then reacted with the light ICAT reagent. After washing to remove the first reagent, the precipitated protein sample is reduced with tris(2-carboxyethyl)phosphine and all newly reduced thiols are alkylated with heavy ICAT reagent. The alkylated proteins are then digested with typsin, and the cysteine containing peptides are enriched on a cation-exchange cartridge followed by an avidin affinity cartridge to enrich the tagged peptides. Finally, the samples are analyzed by liquid chromatography followed by tandem mass spectrometry to quantify the amounts of reduced (dithiol) protein conjugating the light ICAT reagent and oxidized (disulfide) protein conjugating the heavy ICAT reagent.
Thiol/Disulfide Redox Regulation of Heme Binding and Activity of HO2 and the BK Channel
The methods and principles described above have been used to characterize the thiol/disulfide redox switches in HO2 and the BK channel, which form an integrated system to sense oxygen and hypoxia. The switches in both proteins modulate activity by regulating ligand (heme) binding.
Catalyzing the rate-limiting step in heme catabolism to generate CO, biliverdin, and free iron, heme oxygenase (HO, EC 1:14:99:3) is a key regulator of heme and iron homeostasis. HO is linked to various signaling pathways and is important in the response to oxidative stress (1, 7). In cyanobacteria, algae, and plants, HO and biliverdin reductase are involved in generating phytochromes, which modulate growth and photosynthesis (53, 63). The HO-catalyzed degradation of heme requires three oxygenation cycles and seven electrons provided by cytochrome P450 reductase (Eq. 5). Then, biliverdin is transformed to bilirubin by biliverdin reductase. In animals, HO is the only known significant source of the signaling molecule CO (35).
 |
(5) |
Mammals (in fact, all amniotes) contain two HO isoforms: inducible HO1 and constitutively expressed HO2. HO1 and HO2 share similar physical and kinetic properties; however, they have different tissue distributions, with HO2 being highly expressed in the brain, testes, and carotid body (41). HO1 and HO2 share a high level of sequence (55% identity and 76% similarity, as indicated by the middle shaded region in Fig. 2) and structural homology within their core catalytic domains. The heme is sandwiched between the distal and proximal helices (37, 55), the latter of which donates a histidine ligand to the heme iron (His25 in HO1 and His45 in HO2) (Fig. 2). HO1 (33 kDa) and HO2 (36 kDa) have similar molecular masses, comparable enzymatic activities, and share a stretch of 20 hydrophobic residues at their C-terminus that forms a transmembrane helix that anchors them to the microsomal membrane (41). There are two divergent regions between HO1 and HO2. The first is an ∼20 residue extension at the N-terminus of HO2. The other is a region near the C-terminus (between residues 240–295, HO2 numbering), which contains two heme responsive motifs (HRMs) (Fig. 2). Early studies on HO2 indicated that heme binds to each of the HRMs (42); however, more recent studies clearly demonstrate that the HRMs in HO2 do not bind heme per se, but instead form a thiol/disulfide redox switch that is involved in regulating the affinity of HO2 for a single heme that binds to the catalytic domain (70). HO2 also contains a third HRM; however, its role (if any) is unknown. In contrast, there are no Cys residues in HO1.
FIG. 2.

Comparison of HO1 and HO2. (A) Organization of HO1 and HO2. Conserved regions are shaded. (B) Overlay of the core structures of HO1 (PDB 1N45) and HO2 (PDB 2QPP). The C-terminal region containing the HRMs, which was not observed in the structure of HO2 is depicted as a rectangle. The “?” indicates that the HRM region could not be located in the electron density due to disorder HO2, heme oxygenase-2; HRM, heme responsive motif. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
HRMs are found in various proteins and are known to control processes related to iron and oxidative metabolism in organisms ranging from bacteria to humans. The HRM consists of a conserved Cys-Pro core sequence that is usually flanked at the N-terminus by basic amino acids and at the C-terminus by a hydrophobic residue (Table 1). There are three HRMs in both isozymes of aminolevulinate synthase (the housekeeping enzyme aminolevulinate synthase-1 and the erythroid tissue-specific enzyme aminolevulinate synthase-2), which catalyzes the first and rate-limiting enzymatic reaction in the mammalian heme biosynthetic pathway. In the transcriptional regulator, Bach1, HRMs are reported to bind heme and thus inhibit its DNA-binding activity (11, 45). Three HRMs, which have been proposed to control its activity and stability, exist in iron responsive regulator (Irr) (50, 66, 67). The yeast transcriptional activator Hap1 contains seven HRMs that are proposed to mediate heme-dependent transcriptional activation of genes that mediate the effects of oxygen on transcription (27, 38). In HO2, the two C-terminal HRMs constitute a thiol/disulfide redox switch that responds to the intracellular redox potential (68) and regulates the heme affinity of the enzyme (21, 70) (Fig. 3). Thus, HRM-heme interactions regulate the activity and/or stability of proteins that play central roles in respiration and oxidative damage (27, 38), coordinate protein synthesis, and heme availability in reticulocytes (13, 60) and control iron and heme homeostasis (19, 42, 44, 67, 71).
Table 1.
Heme Responsive Motifs in Various Proteins
| Protein | Species | HRM sequence |
|---|---|---|
| Aminolevulinate synthase-1 | Human | RCPFLS, NCPKMM, KCPFLA |
| Aminolevulinate synthase-2 | Human | RCPVLA, RCPILA, HCPFML |
| Bach1–transcriptional regulator of H01 expression | Human | ECPRKK, DCPLSF, PCPYAC, NCPFIS, ECPWLG, QCPEK, LCPKYR |
| eIF2 (eukaryotic Initiation Factor 2) Kinase | Rabbit | ACPYVM, RCPAQA |
| Hap1 | Yeast | KCPIMH, KCPVDH, RCPVDH, RCPVDH, KCPVDH, RCPIDH, KCPVYQ |
| Heme oxygenase-2 | Human | KC265PFYA, SC282PFRT |
| IRP-2 | Human | LCPFHL |
| IRR (Iron Responsive Regulator) | Gm negative bacteria | GCPWHD |
| Per2 (Period-2 circadian regulator) | Human | SCPAVPF |
This table lists the species from which the HRMs have been identified and does not imply that the CP (shown in bold face) motifs are not conserved. For example, the HRMs in heme oxygenase-2 are conserved in all amniotes.
HRM, heme responsive motif.
FIG. 3.

Regulation of heme binding by the HRMs in HO2. Conversion from the thiol(ate) to the disulfide state appears to involve a sulfenate intermediate (33). The oxidized disulfide state has much higher affinity for heme than the reduced state. The midpoint redox potential for the SS/2RSH couple is −200 mV (22). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
We began to explore the role of the HRMs in HO2 because they represent the major obvious difference between HO1 and HO2. The properties of HO2 variants containing single, double, and triple Cys-to-Ala substitutions of the HRMs, along with a truncated variant that lacks the entire HRM region, were compared with those of the wild-type protein in both the oxidized and reduced states (70). In addition, thiol modification experiments were performed to characterize the redox state of each Cys residue under oxidizing and reducing conditions. As summarized in Figure 3, under oxidizing conditions, the two C-terminal HRMs were shown to form a disulfide link that is reducible with glutathione or dithiothreitol. A Cys sulfenate was characterized in the C127A/C282A variant, indicating this as a possible intermediate in oxidation of the dithiol to the disulfide. The redox state of the HRMs and the Cys-to-Ala substitutions were shown to not affect the stoichiometry of heme binding (one heme per mol protein), stability, spectral properties of the heme (in the ferric, ferrous, and ferrous-CO states), or activity at saturating heme concentrations; however, the oxidized disulfide state was shown to bind heme with ∼10-fold higher affinity than that of the reduced dithiol state (70). Because heme binds within the alpha helical core catalytic domain of HO2, which is nearly identical to that of HO1, the HRM region must regulate heme affinity through allosteric effects.
Because HO2 is constituitively expressed, it is important to understand how the levels of CO, heme, and bilirubin are regulated in neurons and in other tissues in which HO2 is the dominant HO isoform. We have proposed that the HRMs at the C-terminus of HO2 play an important mode in regulating HO2 activity and, in turn, cellular functions linked to HO2 and in integrating redox regulation with heme and CO metabolism (68).
In accord with the hypothesis, the HRMs in HO2 do not independently bind heme, but form a thiol/disulfide redox switch that regulates affinity of the active site for heme (70). The Kd value for the HO2-heme complex was found to be around 350 nM when the cysteines of the HRMs are in the reduced dithiol state; however, this value drops significantly to ∼33 nM, similar to the intracellular free heme level, when the HRMs switch to the oxidized disulfide state, indicating a much stronger affinity of oxidized HO2 for its substrate. Recent in vitro and ex vivo studies indicated the physiological relevance of this redox switch (68). Using fluorescence quenching and thiol trapping methods, the midpoint potential of the thiol/disulfide redox couple in HO2 (−200 mV) (68) was shown to be within the −170 to −325 mV range of the ambient cellular redox potential. Then, by the OxICAT method (described above), the redox state of the HRMs in growing cells exposed to different redox conditions was assessed. These studies demonstrated that the HRMs in HO2 respond to the cellular redox state with the disulfide state predominating under oxidizing or oxidative stress conditions, the dithiolate under reducing conditions, and an approximately equal mixture of the dithiol and disulfide states under normoxic conditions (68).
A key function of HO2 that is linked to redox, heme, and CO metabolism is to regulate the activity of the BK channel, which functions in the carotid body to regulate oxygen sensing and the hypoxic response (58). BK channels have the largest single-channel conductance of all K+- selective channels and control neural firing patterns, modulate blood vessel tone, and are an element of the electrical resonator in the inner hair cells of the cochlea (16). BK channels are activated by membrane depolarization and increases of intracellular Ca2+ levels; therefore, activation of the BK channel repolarizes the cell membrane and leads to closing of voltage-gated Ca2+ channels, allowing it to serve as a key negative feedback regulator of both membrane potential and intracellular Ca2+ levels (16). Because membrane potential and Ca2+ levels control so many physiological processes in various tissues, dysfunction of the BK channel can lead to hypertension, hearing loss, motor impairment, urinary problems, and asthma (16).
As in other voltage-gated K+ channels (depicted in Fig. 4), the BK channel is composed of four pore-forming α subunits, each of which contains a seven-helix transmembrane segment, a voltage-sensing domain, one-fourth of the ion conduction pore (2), and, at the C-terminal end, a large 700-residue cytoplasmic region containing two regulators of conductance of potassium (RCK1 and RCK2) domains that are required for Ca2+ activation of the channel (64, 72). RCK1 also serves as a H+ sensor (29, 72) and appears to be involved in inhibition of the BK channel by heme and activation by CO (58). It appears that heme, CO, and HO2 bind to a linker region between the two RCK domains, which is called the heme-binding domain (HBD) (69). BK channel activity is also regulated by accessory β subunits, which can modulate voltage, Ca2+, and Mg2+ sensitivity and are important for the different functions of BK channels in various tissues (46, 59).
FIG. 4.

Model for redox regulation of HO2 and BK channel activity. The membrane-spanning, HBD, RCK1, RCK2, and C-terminal domains of the BK channel (one of the four pore-forming α subunits is removed to show the internal channel) are shown in interaction with HO2, which contains a core catalytic domain that binds and metabolizes heme and a C-terminal regulatory region that contains the thiol/disulfide switch. The surface representation of HO2 is based on its crystal structure, which lacks the redox switch, here represented by two rectangles (9a). The cartoon of the BK channel is loosely derived from its EM structure (61a). Under normoxic conditions, the channel opens because inhibitor heme has dissociated from the HBD (due to its low heme affinity in the SS state) or because CO (generated by HO2) is bound. Under hypoxic conditions, the channel is closed because inhibitor heme is bound (due to the high affinity of HBD for heme in its reduced RSH state) and because CO levels are relatively low (due to low affinity of HO2 for heme and to low O2 levels). Thus, heme is bound to HO2 under normoxic conditions and to the HBD of the BK channel under hypoxia. HBD, heme-binding domain; RCK, regulator of conductance of potassium. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Recent results indicate that the BK channel is regulated by a thiol/disulfide-mediated redox switch, within the HBD, which is similar in principle to that just described for HO2 (69). This HBD contains a characteristic CXXCH thiol/disulfide regulatory motif in which the histidine residue serves as the axial heme ligand. The CXXC forms a thiol/disulfide redox switch that regulates the affinity of the HBD for heme and CO. This is a rather robust switch in which the dithiol state binds heme with a Kd (210 nM) that is similar to the free heme pool, whereas the disulfide state has 14-fold poorer affinity. The redox switches in these two proteins are composed of different Cys-containing motifs—in HO2, two C-terminal HRMs form the thiol/disulfide switch (above), whereas a CXXC motif in the BK channel comprises its redox switch. Further, the heme in the HBD tightly binds CO (Kd = 50 nM) (69), which is significant because CO binding to the BK channel has been shown to activate its potassium transport activity (58, 62).
Figure 4 presents a two-tier model for regulation of the activities of HO2 and the BK channel. It is proposed that, at one level, the activities of HO2 and the BK channel are modulated by the redox state of the cell. An important component of this model is that the thiol/disulfide redox switches in these proteins exhibit similar midpoint redox potentials: −200 mV for HO2 (68) and −185 mV for the BK channel (69). Another key component of the model is that the thiol-disulfide redox switches in HO2 and the BK channel exert opposite (but complementary) effects on the affinity for heme. Thus, the oxidized (disulfide) state of HO2 and the reduced (dithiol) state of the BK channel would bind heme at the low concentrations found within the cell; however, heme would be released from reduced HO2 or oxidized BK channel. Figure 4 also proposes a second tier of regulation involving modulation of the rate at which HO2 generates CO and the consequent effect of this variation in local concentrations of CO on BK channel activity.
As proposed in Figure 4, under normoxic conditions, there is sufficient O2 present to poise the thiol/disulfide switches of HO2 and the BK channel in the disulfide state, where HO2 has high affinity and the BK channel has low affinity for heme. Thus, under normoxic conditions, release of heme from the BK channel would promote the open state and high K+ transport activity. Because O2 is a required substrate for HO2, these O2-rich conditions favor production of CO, which would react with any remaining heme bound to the BK channel and activate K+ transport. Conversely, under hypoxic conditions, the low O2 levels and low affinity of HO2 for heme would result in low heme degradation and CO production rates, thus increasing local heme levels and decreasing CO concentrations. These conditions, coupled with the high affinity of the BK channel for heme, would favor the inhibited heme-bound state of the BK channel, poising the channel in the closed state (represented by a cork stopper).
The second tier of regulation involves CO binding to the BK channel. Figure 4 describes two recently proposed mechanisms by which CO could mediate activation of the BK channel. The first mechanism involves CO binding to heme (31) in the HBD (69), whereas the other is a heme-independent mechanism in which CO was proposed to activate the BK channel by directly interacting with specific His and Asp residues in the RCK1 domain (30). This mechanism was proposed because mutation of these residues leads to loss of CO-dependent activation. Because it is difficult to imagine metal-independent CO binding, we favor the heme-dependent mechanism and have suggested that the His/Asp residues in the RCK1 domain either bind a metal ion (that can bind CO) or are involved in allosteric control of heme-dependent CO binding (69).
Ultimately, inhibition of the BK channel results in a depolarization wave from the glomus cells of the carotid body to the respiratory system, causing increased ventilation to restore circulating blood O2 levels. Although, given all the inputs to which the BK channel is sensitive, Figure 4 is undoubtedly a simplistic explanation for how the BK channel is regulated by redox, heme, CO, and HO2, it provides a working and testable hypothesis that redox poise and the levels of heme and CO converge with other known regulators, for example, Ca++ and pH, to modulate BK channel activity.
Significance of HO2-Mediated Redox Regulation of Heme and CO Homeostasis
CO, one of the three products of the HO-catalyzed reaction, acts as a signaling molecule in various physiological processes, including circadian modulation of heme biosynthesis, regulating T cell function, modulating caveolin-1 status in growth control (35), activating guanylate cyclase (5), and mediating O2 sensing and the hypoxic response through regulation of the BK channel (62), which enables the O2-sensing function of the human carotid body (28, 34), as described above. Although both HO1 and HO2 catalyze CO formation, the specific functions of HO2 are revealed in studies of HO2 knockout mice, which are more susceptible to neurotoxicity, cerebral ischemia, stroke damage, and traumatic brain injury, indicating an important role for HO2 in neural signaling, neuroprotection (against oxidative stress in brain injury), and in regulating cerebral blood flow (10, 12, 18, 51, 61, 73). A role of HO2 in traumatic brain injury is in reducing lipid peroxidation (10). Cerebral microvascular endothelial cells from HO2 knockout mice exhibit higher sensitivity to TNF-α induced apoptosis and glutamate toxicity, whereas bilirubin and CO rescue this defect (6, 47). Because HO1 is not induced under these conditions, it appears that HO2 is the key enzyme responding to TNF-α-induced oxidative stress in this cell type (48). Similarly, HO2-null corneas exhibit chronic inflammation that is attenuated by supplementation with biliverdin (48, 56). Surprisingly, even though HO1 is relatively more highly expressed in the kidney than HO2, CO and biliverdin production decreases by more than twofold in HO2−/− versus wild-type mice (9, 10, 57). Further, HO2 has been shown to protect against renal pathology induced by oxidative stress related to diabetes (22).
Conclusion and Future Directions
This review covers a system composed of two interacting proteins: HO2 and the BK channel. Thiol/disulfide redox switches allosterically regulate both proteins by controlling the binding of heme, which is a substrate for HO2 and an allosteric inhibitor for the channel. Although the redox switches in the two proteins are evolutionarily unrelated, a CXXCH motif in the BK channel and HRMs in HO2, their midpoint redox potentials are similar, −185 to −200 mV, which is well within the range of ambient intracellular redox potential.
The scope of thiol/disulfide regulation of heme binding is not known and it is expected to reach to other systems. One approach is to combine experimental methods, like OxICAT, and in silico methods to identify all proteins that contain thiol/disulfide switches with midpoint potentials within the range of the intracellular ambient redox potential. Similarly, methods might be developed to identify all proteins that allosterically and reversibly bind heme at physiologically relevant concentrations. Thus, such heme-regulated proteins should have Kd values for heme in the 20–150 nM range of the free heme poise within cells, allowing them to bind and release heme as a function of an external signal (redox, etc.). It is also important to identify the thiol/disulfide exchange factors or oxidoreductases that can partner with the redox switches. Another layer of regulation is observed in the BK channel, which although inhibited by heme, undergoes activation by CO. It will be interesting to examine how many heme- and redox-regulated systems are also subject to another tier of regulation by binding gaseous messengers like CO, NO, and H2S.
Abbreviations Used
- HBD
heme binding domain
- HO2
heme oxygenase-2
- HRM
heme responsive motif
- ICAT
isotope coded affinity tag
- RCK
regulator of conductance of potassium
Acknowledgments
We thank members of the Ragsdale laboratory (especially those in the present and past HO2 group: Ireena Bagai, Andrea Morris, Jeff Morgan, Chris Walters, and Ryan Kunz) for their work and valuable discussions related to redox switches. We also thank our collaborators for discussions and for their creative involvement in the work described, especially George Phillips, Jr., Thomas Brunold, and Chris Bianchotti at the University of Wisconsin–Madison, and Ursula Jakob, Jeff Martens, Erik Zuiderweg, and Lars Leichert at the University of Michigan. The work in the Ragsdale laboratory on HO2 and the BK channel has been supported in part by National Institutes of Health grants R21 HL089837 and R01 HL102662.
References
- 1.Abraham NG. Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 2.Adelman JP. Shen KZ. Kavanaugh MP. Warren RA. Wu YN. Lagrutta A. Bond CT. North RA. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- 3.Antelmann H. Helmann JD. Thiol-based redox switches and gene regulation. Antioxid Redox Signal. 2011;14:1049–1063. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee RV. Harder SR. Ragsdale SW. Matthews RG. Mechanism of reductive activation of cobalamin-dependent methionine synthase: an electron paramagnetic resonance spectroelectrochemical study. Biochemistry. 1990;29:1129–1135. doi: 10.1021/bi00457a005. [DOI] [PubMed] [Google Scholar]
- 5.Baranano DE. Snyder SH. Neural roles for heme oxygenase: contrasts to nitric oxide synthase. Proc Natl Acad Sci U S A. 2001;98:10996–11002. doi: 10.1073/pnas.191351298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basuroy S. Bhattacharya S. Tcheranova D. Qu Y. Regan RF. Leffler CW. Parfenova H. HO-2 provides endogenous protection against oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am J Physiol Cell Physiol. 2006;291:C897–C908. doi: 10.1152/ajpcell.00032.2006. [DOI] [PubMed] [Google Scholar]
- 7.Bauer M. Huse K. Settmacher U. Claus RA. The heme oxygenase-carbon monoxide system: regulation and role in stress response and organ failure. Intensive Care Med. 2008;34:640–648. doi: 10.1007/s00134-008-1010-2. [DOI] [PubMed] [Google Scholar]
- 8.Becker DF. Zhu W. Moxley MA. Flavin redox switching of protein functions. Antioxid Redox Signal. 2011;14:1079–1091. doi: 10.1089/ars.2010.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellner L. Martinelli L. Halilovic A. Patil KA. Puri N. Dunn MW. Regan RF. Schwartzman ML. HO-2 deletion causes endothelial cell activation marked by oxidative stress, inflammation and angiogenesis. J Pharmacol Exp Ther. 2009;331:925–932. doi: 10.1124/jpet.109.158352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Bianchetti CM. Li Y. Ragsdale SW. Phillips GN., Jr Comparison of Apo and heme-bound crystal structures of a truncated human heme oxygenase-2. J Biol Chem. 2007;282:37624–37631. doi: 10.1074/jbc.M707396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang EF. Wong RJ. Vreman HJ. Igarashi T. Galo E. Sharp FR. Stevenson DK. Noble-Haeusslein LJ. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci. 2003;23:3689–3696. doi: 10.1523/JNEUROSCI.23-09-03689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chefalo PJ. Oh J. Rafie-Kolpin M. Kan B. Chen JJ. Heme-regulated eIF-2alpha kinase purifies as a hemoprotein. Eur J Biochem. 1998;258:820–830. doi: 10.1046/j.1432-1327.1998.2580820.x. [DOI] [PubMed] [Google Scholar]
- 12.Chen J. Tu Y. Connolly EC. Ronnett GV. Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr Neurovasc Res. 2005;2:121–131. doi: 10.2174/1567202053586767. [DOI] [PubMed] [Google Scholar]
- 13.Chen JJ. London IM. Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem Sci. 1995;20:105–108. doi: 10.1016/s0968-0004(00)88975-6. [DOI] [PubMed] [Google Scholar]
- 14.Chen PR. Brugarolas P. He C. Redox Signaling in human pathogens. Antioxid Redox Signal. 2011;14:1107–1118. doi: 10.1089/ars.2010.3374. [DOI] [PubMed] [Google Scholar]
- 15.Chobot SE. Hernandez HH. Drennan CL. Elliott SJ. Direct electrochemical characterization of archaeal thioredoxins. Angew Chem Int Ed Engl. 2007;46:4145–4147. doi: 10.1002/anie.200604620. [DOI] [PubMed] [Google Scholar]
- 16.Cui J. Yang H. Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66:852–875. doi: 10.1007/s00018-008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dooley CT. Dore TM. Hanson GT. Jackson WC. Remington SJ. Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 18.Dore S. Goto S. Sampei K. Blackshaw S. Hester LD. Ingi T. Sawa A. Traystman RJ. Koehler RC. Snyder SH. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience. 2000;99:587–592. doi: 10.1016/s0306-4522(00)00216-5. [DOI] [PubMed] [Google Scholar]
- 19.Eisenstein RS. Blemings KP. Iron regulatory proteins, iron responsive elements and iron homeostasis. J Nutr. 1998;128:2295–2298. doi: 10.1093/jn/128.12.2295. [DOI] [PubMed] [Google Scholar]
- 20.Fahey RC. Novel thiols of prokaryotes. Annu Rev Microbiol. 2001;55:333–356. doi: 10.1146/annurev.micro.55.1.333. [DOI] [PubMed] [Google Scholar]
- 21.Gardner JD. Yi L. Ragsdale SW. Brunold TC. Spectroscopic insights into axial ligation and active-site H-bonding in substrate-bound human heme oxygenase-2. J Biol Inorg Chem. 2010;15:1117–1127. doi: 10.1007/s00775-010-0672-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodman AI. Chander PN. Rezzani R. Schwartzman ML. Regan RF. Rodella L. Turkseven S. Lianos EA. Dennery PA. Abraham NG. Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J Am Soc Nephrol. 2006;17:1073–1081. doi: 10.1681/ASN.2004121082. [DOI] [PubMed] [Google Scholar]
- 23.Gupta N. Ragsdale SW. Dual roles of an essential cysteine residue in activity of a redox-regulated bacterial transcriptional activator. J Biol Chem. 2008;283:28721–28728. doi: 10.1074/jbc.M800630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gygi SP. Rist B. Gerber SA. Turecek F. Gelb MH. Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 25.Hansen RE. Roth D. Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassett DJ. Cohen MS. Bacterial adaptation to oxidative stress: implications for pathogenesis and interaction with phagocytic cells. FASEB J. 1989;3:2574–2582. doi: 10.1096/fasebj.3.14.2556311. [DOI] [PubMed] [Google Scholar]
- 27.Hon T. Hach A. Lee HC. Cheng T. Zhang L. Functional analysis of heme regulatory elements of the transcriptional activator Hap1. Biochem Biophys Res Commun. 2000;273:584–591. doi: 10.1006/bbrc.2000.2995. [DOI] [PubMed] [Google Scholar]
- 28.Hou S. Heinemann SH. Hoshi T. Modulation of BKCa channel gating by endogenous signaling molecules. Physiology (Bethesda) 2009;24:26–35. doi: 10.1152/physiol.00032.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou S. Xu R. Heinemann SH. Hoshi T. Reciprocal regulation of the Ca2+ and H+ sensitivity in the SLO1 BK channel conferred by the RCK1 domain. Nat Struct Mol Biol. 2008;15:403–410. doi: 10.1038/nsmb.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hou S. Xu R. Heinemann SH. Hoshi T. The RCK1 high-affinity Ca2+ sensor confers carbon monoxide sensitivity to Slo1 BK channels. Proc Natl Acad Sci U S A. 2008;105:4039–4043. doi: 10.1073/pnas.0800304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaggar JH. Li A. Parfenova H. Liu J. Umstot ES. Dopico AM. Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+ -activated K+ channels. Circ Res. 2005;97:805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 33.Kemp M. Go YM. Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kemp PJ. Hemeoxygenase-2 as an O2 sensor in K+ channel-dependent chemotransduction. Biochem Biophys Res Commun. 2005;338:648–652. doi: 10.1016/j.bbrc.2005.08.110. [DOI] [PubMed] [Google Scholar]
- 35.Kim HP. Ryter SW. Choi AM. CO as a cellular signaling molecule. Annu Rev Pharmacol Toxicol. 2006;46:411–449. doi: 10.1146/annurev.pharmtox.46.120604.141053. [DOI] [PubMed] [Google Scholar]
- 36.Klomsiri C. Karplus PA. Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lad L. Schuller DJ. Shimizu H. Friedman J. Li H. Ortiz de Montellano PR. Poulos TL. Comparison of the heme-free and -bound crystal structures of human heme oxygenase-1. J Biol Chem. 2003;278:7834–7843. doi: 10.1074/jbc.M211450200. [DOI] [PubMed] [Google Scholar]
- 38.Lee HC. Hon T. Lan C. Zhang L. Structural environment dictates the biological significance of heme-responsive motifs and the role of Hsp90 in the activation of the heme activator protein Hap1. Mol Cell Biol. 2003;23:5857–5866. doi: 10.1128/MCB.23.16.5857-5866.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lees WJ. Whitesides GM. Equilibrium constants for thiol- disulfide interchange reactions: a coherent, corrected set. J Org Chem. 1993;58:642–647. [Google Scholar]
- 40.Leichert LI. Gehrke F. Gudiseva HV. Blackwell T. Ilbert M. Walker AK. Strahler JR. Andrews PC. Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci U S A. 2008;105:8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 42.McCoubrey WK., Jr Huang TJ. Maines MD. Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J Biol Chem. 1997;272:12568–12574. doi: 10.1074/jbc.272.19.12568. [DOI] [PubMed] [Google Scholar]
- 43.Moriarty-Craige SE. Jones DP. Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr. 2004;24:481–509. doi: 10.1146/annurev.nutr.24.012003.132208. [DOI] [PubMed] [Google Scholar]
- 44.Munakata H. Sun JY. Yoshida K. Nakatani T. Honda E. Hayakawa S. Furuyama K. Hayashi N. Role of the heme regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J Biochem (Tokyo) 2004;136:233–238. doi: 10.1093/jb/mvh112. [DOI] [PubMed] [Google Scholar]
- 45.Ogawa K. Sun J. Taketani S. Nakajima O. Nishitani C. Sassa S. Hayashi N. Yamamoto M. Shibahara S. Fujita H, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001;20:2835–2843. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orio P. Rojas P. Ferreira G. Latorre R. New disguises for an old channel: MaxiK channel beta-subunits. News Physiol Sci. 2002;17:156–161. doi: 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- 47.Parfenova H. Basuroy S. Bhattacharya S. Tcheranova D. Qu Y. Regan RF. Leffler CW. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: contributions of HO-1 and HO-2 to cytoprotection. Am J Physiol Cell Physiol. 2006;290:C1399–C1410. doi: 10.1152/ajpcell.00386.2005. [DOI] [PubMed] [Google Scholar]
- 48.Parfenova H. Neff RA., 3rd Alonso JS. Shlopov BV. Jamal CN. Sarkisova SA. Leffler CW. Cerebral vascular endothelial heme oxygenase: expression, localization, and activation by glutamate. Am J Physiol Cell Physiol. 2001;281:C1954–C1963. doi: 10.1152/ajpcell.2001.281.6.C1954. [DOI] [PubMed] [Google Scholar]
- 49.Pomposiello PJ. Demple B. Redox-operated genetic switches: the SoxR and OxyR transcription factors. Trends Biotechnol. 2001;19:109–114. doi: 10.1016/s0167-7799(00)01542-0. [DOI] [PubMed] [Google Scholar]
- 50.Qi Z. Hamza I. O'Brian MR. Heme is an effector molecule for iron-dependent degradation of the bacterial iron response regulator (Irr) protein. Proc Natl Acad Sci U S A. 1999;96:13056–13061. doi: 10.1073/pnas.96.23.13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin X. Kwansa H. Bucci E. Dore S. Boehning D. Shugar D. Koehler RC. Role of heme oxygenase-2 in pial arteriolar response to acetylcholine in mice with and without transfusion of cell-free hemoglobin polymers. Am J Physiol Regul Integr Comp Physiol. 2008;295:R498–R504. doi: 10.1152/ajpregu.00188.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu W. Wang L. Lu W. Boechler A. Sanders DA. Zhong D. Dissection of complex protein dynamics in human thioredoxin. Proc Natl Acad Sci U S A. 2007;104:5366–5371. doi: 10.1073/pnas.0608498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhie G. Beale SI. Phycobilin biosynthesis: reductant requirements and product identification for heme oxygenase from Cyanidium caldarium. Arch Biochem Biophys. 1995;320:182–194. doi: 10.1006/abbi.1995.1358. [DOI] [PubMed] [Google Scholar]
- 54.Schafer FQ. Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 55.Schuller DJ. Wilks A. Ortiz de Montellano PR. Poulos TL. Crystal structure of human heme oxygenase-1. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 56.Seta F. Bellner L. Rezzani R. Regan RF. Dunn MW. Abraham NG. Gronert K. Laniado-Schwartzman M. Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am J Pathol. 2006;169:1612–1623. doi: 10.2353/ajpath.2006.060555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sodhi K. Inoue K. Gotlinger K. Canestraro M. Vanella L. Kim DH. Manthati VL. Koduru SR. Falck JR. Schwartzman ML, et al. EET agonist rescues the metabolic syndrome phenotype of HO-2 null mice. J Pharmacol Exp Ther. 2009;331:906–916. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang XD. Xu R. Reynolds MF. Garcia ML. Heinemann SH. Hoshi T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature. 2003;425:531–535. doi: 10.1038/nature02003. [DOI] [PubMed] [Google Scholar]
- 59.Torres YP. Morera FJ. Carvacho I. Latorre R. A marriage of convenience: beta-subunits and voltage-dependent K+ channels. J Biol Chem. 2007;282:24485–24489. doi: 10.1074/jbc.R700022200. [DOI] [PubMed] [Google Scholar]
- 60.Uma S. Matts RL. Guo Y. White S. Chen JJ. The N-terminal region of the heme-regulated eIF2alpha kinase is an autonomous heme binding domain. Eur J Biochem. 2000;267:498–506. doi: 10.1046/j.1432-1327.2000.01021.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang J. Zhuang H. Dore S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol Dis. 2006;22:473–476. doi: 10.1016/j.nbd.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 61a.Wang L. Sigworth FJ. Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature. 2009;461:292–295. doi: 10.1038/nature08291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams SE. Wootton P. Mason HS. Bould J. Iles DE. Riccardi D. Peers C. Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 63.Willows RD. Mayer SM. Foulk MS. DeLong A. Hanson K. Chory J. Beale SI. Phytobilin biosynthesis: the Synechocystis sp. PCC 6803 heme oxygenase-encoding ho1 gene complements a phytochrome-deficient Arabidopsis thalianna hy1 mutant. Plant Mol Biol. 2000;43:113–120. doi: 10.1023/a:1006489129449. [DOI] [PubMed] [Google Scholar]
- 64.Xia XM. Zeng X. Lingle CJ. Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature. 2002;418:880–884. doi: 10.1038/nature00956. [DOI] [PubMed] [Google Scholar]
- 65.Yan Z. Banerjee R. Redox remodeling as an immunoregulatory strategy. Biochem. 2010;49:1059–1066. doi: 10.1021/bi902022n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang J. Ishimori K. O'Brian MR. Two heme binding sites are involved in the regulated degradation of the bacterial iron response regulator (Irr) protein. J Biol Chem. 2005;280:7671–7676. doi: 10.1074/jbc.M411664200. [DOI] [PubMed] [Google Scholar]
- 67.Yang J. Panek HR. O'Brian MR. Oxidative stress promotes degradation of the Irr protein to regulate haem biosynthesis in Bradyrhizobium japonicum. Mol Microbiol. 2006;60:209–218. doi: 10.1111/j.1365-2958.2006.05087.x. [DOI] [PubMed] [Google Scholar]
- 68.Yi L. Jenkins PM. Leichert LI. Jakob U. Martens JR. Ragsdale SW. The heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J Biol Chem. 2009;284:20556–20561. doi: 10.1074/jbc.M109.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yi L. Morgan JT. Ragsdale SW. Identification of a thiol/disulfide redox switch in the human BK channel that controls its affinity for heme and CO. J Biol Chem. 2010;285:20117–20127. doi: 10.1074/jbc.M110.116483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yi L. Ragsdale SW. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J Biol Chem. 2007;282:20156–21067. doi: 10.1074/jbc.M700664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshino K. Munakata H. Kuge O. Ito A. Ogishima T. Haeme-regulated degradation of delta-aminolevulinate synthase 1 in rat liver mitochondria. J Biochem. 2007;142:453–458. doi: 10.1093/jb/mvm159. [DOI] [PubMed] [Google Scholar]
- 72.Yusifov T. Savalli N. Gandhi CS. Ottolia M. Olcese R. The RCK2 domain of the human BKCa channel is a calcium sensor. Proc Natl Acad Sci U S A. 2008;105:376–381. doi: 10.1073/pnas.0705261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zakhary R. Poss KD. Jaffrey SR. Ferris CD. Tonegawa S. Snyder SH. Targeted gene deletion of heme oxygenase 2 reveals neural role for carbon monoxide. Proc Natl Acad Sci USA. 1997;94:14848–14853. doi: 10.1073/pnas.94.26.14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang W. Zhang M. Zhu W. Zhou Y. Wanduragala S. Rewinkel D. Tanner JJ. Becker DF. Redox-induced changes in flavin structure and roles of flavin N(5) and the ribityl 2′-OH group in regulating PutA—membrane binding. Biochem. 2007;46:483–491. doi: 10.1021/bi061935g. [DOI] [PMC free article] [PubMed] [Google Scholar]