

Abstract
Polyketide and nonribosomal peptides constitute important classes of small molecule natural products. Due to the proven biological activities of these compounds, novel methods for discovery and study of the polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) enzymes responsible for their production remains an area of intense interest, and proteomic approaches represent a relatively unexplored avenue. While these enzymes may be distinguished from the proteomic milieu by their use of the 4′-phosphopantetheine (PPant) posttranslational modification, proteomic detection of PPant peptides is hindered by their low abundance and labile nature which leaves them unassigned using traditional database searching. Here we address key experimental and computational challenges to facilitate practical discovery of this important posttranslational modification during shotgun proteomics analysis using low-resolution ion-trap mass spectrometers. Activity-based enrichment maximizes MS input of PKS/NRPS peptides, while targeted fragmentation detects putative PPant active sites. An improved data analysis pipeline allows experimental identification and validation of these PPant peptides directly from MS2 data. Finally, a machine learning approach is developed to directly detect PPant peptides from only MS2 fragmentation data. By providing new methods for analysis of an often cryptic posttranslational modification, these methods represent a first step towards the study of natural product biosynthesis in proteomic settings.
Keywords: Natural products, polyketide synthase, nonribosomal peptide synthetase, posttranslational modification, LC-MS/MS, support vector machine, carrier protein domain, InsPecT
1 Introduction
Polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) biosynthetic enzymes are responsible for the production of a wide-range of biologically active natural products.1 To facilitate the biosynthesis of complex molecules, PKS and NRPS enzymes utilize carrier protein (CP) domains.2 These small proteins serve as points of covalent tethering for small molecules within the PKS/NRPS megasynthase, allowing biosynthetic intermediates to be channeled to cognate partner proteins for condensation and further biochemical elaboration. The site of covalent tethering is the terminal thiol of a posttranslationally introduced 4′-phosphopantetheine (PPant) arm, which modifies a conserved serine residue of the CP domain. The past decade has seen the development of a number of novel approaches to accelerate the discovery and characterization of new natural product biosynthetic enzymes, a majority of which are genetics-based.3-5 Recently, we introduced a proteomic approach to natural product enzyme discovery, demonstrating enrichment of PKS and NRPS enzymes from unfractionated proteomic samples by PKS/NRPS-directed active site probes, and subsequent identification by tandem mass spectrometry (MS/MS) using multidimensional protein identification technology (MudPIT).6 Despite its utility, one shortcoming of this identification strategy was its inability to identify 4′-PPant CP active site peptides, presumably due to inefficient fragmentation of the CP active site peptide during MS/MS after ejection of the 4′-PPant arm (Figure 1a).7 In contrast to phosphorylated serine residues which undergo a characteristic neutral loss that can be used to trigger an additional fragmentation event on the remaining peptide backbone for increased sequence coverage (MS3),8 tandem MS-based identification of CP peptides is extremely difficult using traditional ESI-MS/MS instruments as the PPant cofactor can undergo multiple pathways of elimination during collisionally induced dissociation (CID), many of which change the charge state of the peptide (Figure 2a and S1).9
Figure 1.
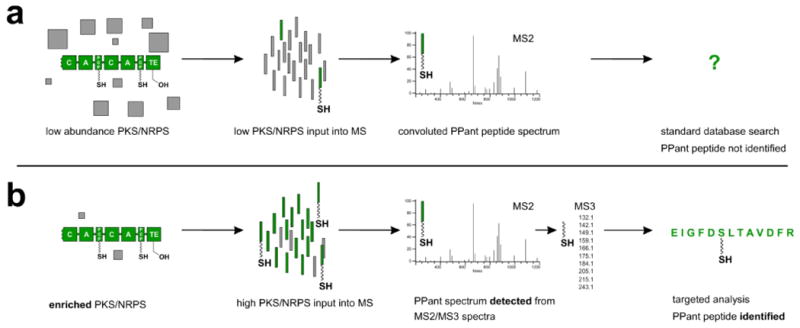
Approaches towards the proteomic identification of 4′-phosphopantetheinylated (PPant) peptides. (a) Traditional methods of proteomic analysis have difficulty in detecting PPant peptides due to their low abundance. In cases when PPant peptides are sampled by the spectrometer, spectra are often left unassigned due to weak fragmentation of the peptide backbone as well as convolution of MS2 spectra by PTM and parent mass ejection ions. (b) New approaches to the identification of PPant peptides investigated in the current study. Enrichment methods maximize the input of PPant peptides to the spectrometer. PPant peptides are initially detected by multistage fragmentation resulting in a characteristic MS3 pantetheine signature, or by a machine learning approach which detects PPant spectra based on their MS2 fragmentation patterns. Finally, PPant peptides are identified by a modified database search.
Figure 2.
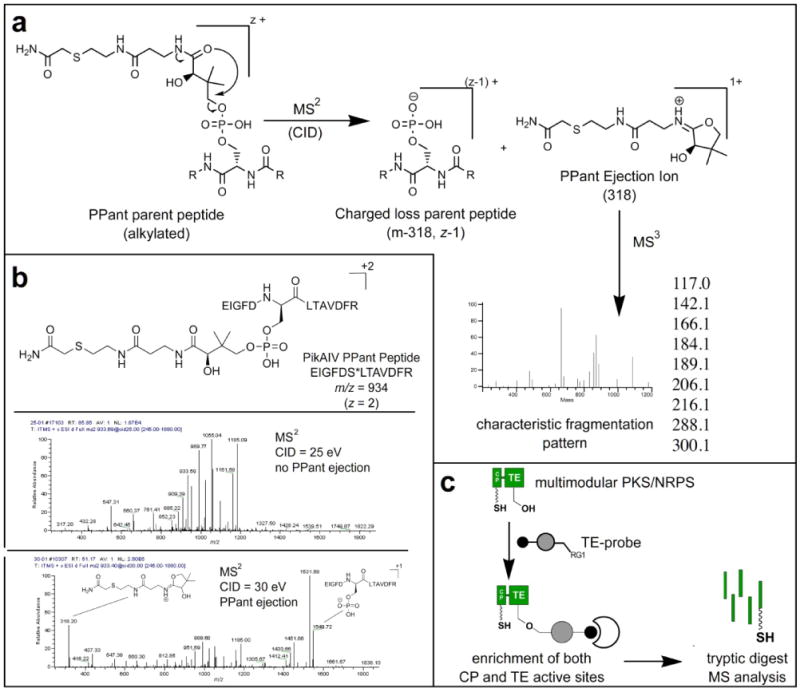
(a) PPant ejection during MS2 generates characteristic ejection ions and charged loss parent peptides (z-1). The PPant ejection ion (m/z 318) can be further fragmented in MS3 to generate a characteristic signature, allowing unambiguous detection of PPant peptides. (b) Impact of CID energy applied during MS2 on PPant ejection. (c) Mechanism of enrichment of PPant peptides by FP-biotin 1. RG1 = fluorophosphonate, which reacts covalently with the conserved serine residue of PKS/NRPS TE domains, to allow active site enrichment.
Recently Kelleher and coworkers reported the identification of CP active site peptides from fractionated proteomic samples of Bacilli using targeted multistage fragmentation (MSn) of peptides displaying characteristic PPant ejection masses.10 This study demonstrated sequence determination of CP active site peptides, facilitating primer design and discovery of a new NRPS gene cluster. However, despite the success of this approach, its reliance on the high mass accuracy of Fourier Transform mass spectrometry along with specialized MSn methods and manual de novo sequencing of the fragmented CP peptides requires levels of instrumentation and analyst expertise not accessible to many natural products laboratories and core facilities. Here we broaden the scope of methods for analysis of CP active site peptides from proteomic samples, developing experimental and computational solutions for identification of PPant peptides using low mass accuracy ion trap tandem mass spectrometry (Figure 1b). First we develop a multistage fragmentation strategy for detection of CP peptides from enriched proteomes based on their characteristic MS3 signature.11 Second, we demonstrate a data analysis pipeline that allows many of these putative PPant peptides to be identified directly from low resolution MS2 data by a modified database search. Finally, we apply insights from these studies to develop a computational supervised learning approach to directly detect PPant peptide spectra from only MS2 fragmentation data. This latter method obviates the necessity of multistage mass spectrometry methods in the proteomic and biochemical analysis of CP active sites and is validated by comparison with multistage fragmentation-based PPant detection. In this work, we make a distinction between detection and identification of PPant peptides in MS, where the former declares a spectrum representing a PPant peptide and the latter determines the amino acid sequence of the PPant peptide observed in a spectrum. By providing a detailed inquiry into the strengths and limitations of both experimental and computational methods for the identification of CP active sites from proteomic samples, this study represents a first step towards the standard integration of proteomic analysis of CP active sites into studies of polyketide and nonribosomal peptide biosynthesis.
2 Materials and Methods
2.1 Materials
Probe 1 was synthesized as previously described. Sfp, PikAIV, CouN5, Strop_4416, and YbbR were expressed and purified as previously described.11-13 Luria-Bertani (LB) media was purchased from Aldrich. PD10 desalting columns were purchased from GE Healthcare. Avidin-agarose was purchased from Aldrich. Capillary columns were prepared by drawing 100 μm inner diameter deactivated, fused silica tubing (Agilent) with a Model P-2000 laser puller (Sutter Instruments Co.) and packed at ∼ 600 psi with the appropriate chromatography resin (Aqua C18 reverse phase resin [Phenomex] or Partisphere strong cation exchange resin [Whatman]) suspended in methanol. Desalting columns were packed with 3 cm C18 resin, while biphasic MudPIT columns were packed with 10 cm C18 and 3 cm strong cation exchange (SCX) resin. LC-MS/MS analysis was performed using an LTQ ion trap mass spectrometer (ThermoFisher) coupled to an Agilent 1100 series HPLC.
2.2 Growth Conditions and Proteome Preparation
B. subtilis strains 168 was streaked on LB-agar and incubated overnight at 37 °C. A single colony of each strain was picked and used to inoculate individual 5 mL liquid LB starter cultures and rotated overnight at 37 °C. This starter culture (2 mL) was used to inoculate 1 L of autoclaved LB media and grown aerobically at 37 °C with vigorous agitation. Growth curves were plotted by analyzing optical density at 600 nm and cells were harvested in stationary growth phase (OD600 ∼1.3). After centrifugation (8000×g for 20 min at 4 °C) cell pellets were washed twice with lysis buffer (25 mM potassium phosphate, pH 7.0, 100 mM NaCl) and again centrifuged. After resuspension in lysis buffer (50-100 mL), cell lysis was performed by two passes through a French pressure cell, followed by treatment with DNase I for 30 minutes at 0 °C and clearing of cell debris by centrifugation (20,000×g for 30 min at 4 °C). Protein concentration was determined by BCA assay, resulting in isolation of unfractionated proteomes of ∼5-15 mg/mL. For MudPIT analyses 1 mg aliquots of proteomes were stored at -80 °C without glycerol and thawed immediately prior to enrichment, as the presence of glycerol was found to severely impede downstream analysis.
2.3 Proteome Labeling and Enrichment
Whole cell proteomes of B. subtilis 168 were adjusted to a final protein concentration of 1 mg/mL and labeled with activity-based probe 1 using a procedure identical to previous reports. Briefly, to a 1000 μL reaction mixture of B. subtilis 6051 proteome (1 mg/mL in 50 mM Tris-Cl, pH 8.0) was added fluorophosphonate-biotin (5 μM; 1 mM stock in DMSO). To ensure all B. subtilis CP active sites were present in holo-form, coenzyme A (25 μM; 1 mM stock in H2O), MgCl2 (10 mM; 0.5 M stock in H2O), and Sfp (8.8 μg) were also added. Samples were vortexed and incubated at room temperature for 2 hr, followed by addition of 1% Triton-X (to aid membrane protein solubilization) and rotation at 4 °C for 1 hour. Reactions were loaded onto a pre-equilibrated PD10 Desalting column (GE Healthcare) to remove excess biotin probe, collected, and denatured by addition of SDS to 0.5% and heating at 90 °C for 10 minutes. Samples were diluted to an SDS concentration of ∼ 0.2% and allowed to cool to room temperature before addition of 50 μL pre-washed avidin-agarose, whereupon samples were rotated at 4 °C for 1 hour to facilitate avidin binding of biotinylated proteins. Avidin-agarose bound samples were then washed sequentially with 1% SDS, 6 M urea, and 50 mM Tris-Cl pH 8.0 (two washes each), and resuspended in 200 μL 8M urea, 50 mM Tris-Cl pH 8.0. Samples were then prepared for on-bead digest by reduction with 10 mM tris(2-carboxyethyl)phosphine (TCEP) and alkylation with 12 mM iodoacetamide. Samples were diluted to 2 M urea with 50 mM Tris-Cl pH 8.0 (400 μL total volume), followed by addition of trypsin and 2 mM CaCl2. Digests were allowed to proceed overnight at 37°C overnight. After extraction, tryptic peptide samples were acidified to a final concentration of 5% formic acid and frozen at -80°C for MudPIT analysis.
2.4 Liquid Chromatography – Mass Spectrometry (MudPIT) Analysis of Enriched Proteomes
Enriched tryptic peptides were loaded onto a biphasic (strong cation exchange/reverse phase) capillary column and analyzed by 2D-LC separation in combination with tandem MS as previously described.14 Peptides were eluted in a five-step MudPIT experiment and data collected on a Thermo Scientific LTQ-MS set in a data-dependent acquisition mode with dynamic exclusion turned on (60 s). Each full MS survey scan was followed by 7 MS/MS scans. For the detection of PPant peptides MS2 scans containing an ion with an m/z of 318 were selected for an additional round of MS3. This MS3 scan event isolated the 318 ion specifically for fragmentation (isolation width 2 m/z). Spray voltage was set to 2.75 kV and the flow rate through the column was 0.25 μL/min. Peptides that were found to have a fragment ion at 318 were considered authentic PPant peptides only if 4 out of the 6 most significant pantetheinyl ions annotated by Meluzzi et al. (m/z 300, 288, 216, 184, 142, or 118)11 were observed among the top 15 most intense MS3 signals, allowing for a mass tolerance of ± 1 Da.
2.5 Peptide Identification by InsPecT and InsPecT:PPant
InsPecT is a fast peptide identification tool applying tag-based filtering, Bayesian network models of peptide MS2 fragmentation for scoring, and peptide match quality assessment.15 Unmodified and phosphorylated peptide identification was performed by InsPecT (CCMS LiveSearch server) with a 1% false discovery rate to show true enrichment of CP domain containing proteins. PPant peptide identification in MS2 scans was performed using an altered phosphorylation Bayesian network in InsPecT (InsPecT:PPant). Following Payne et al.16, a peptide is defined as a series of breaks with prefix mass characters m1, m2, …, m1. Given a spectrum S, the log-odds score of a break is denoted as score(mi, S).
A Bayesian network approach is applied to compute PrCID(I⃗ = [I0, I1, …]∣ mi, S), where I⃗ is the set of ion fragments supporting the break (see Fig. S3 for a pictorial example). These ion fragments are identified by their respective mass offsets from mi. In the case of InsPecT:PPant, these supporting ion fragments include PPant loss fragments and pantetheinyl (Pant) loss fragments, observed at -397 and -317 mass offsets. Due to insufficient PPant peptide examples, the previously trained phosphorylation Bayesian network was used as a PPant Bayesian network by exchanging phosphorylation related supporting fragments for PPant and Pant supporting fragments. The probabilities of these new supporting fragments were set to be the same as their equivalent phosphorylation support fragments (see Fig. S3).
All 11 discernible characteristic PPant ejection and peptide PPant labile loss ions were masked in MS2 spectra prior to scoring. InsPecT was run in non-tagging mode, searching against organism specific protein database for peptides within 2.5 Da of the inferred parent peptide mass M, M + 2 (isotopes), and M - H2O. The B. subtilis protein database consisted of all CDS regions of the B. subtilis strain 168 as annotated by Pasteur GenoList.17
InsPecT:PPant search reports a log-odds score for each peptide sequence that is matched to a CP active site spectrum. The log-odds score represents the probability identified peaks are generated by peptide fragmentation as opposed to random background. Each peptide match is also assigned a delta score, corresponding to the difference between the log-odds scores of a peptide match and the next best peptide match. True peptide hits were assessed by a delta score 1% p-value cutoff along and a UniProtKB validation 5% p-value cutoff (see Section 3.3). To determine the delta score based p-value cutoff for InsPecT:PPant search results, we accumulated the delta scores of the top 50 matches for each MS3 validated spectrum and generated an empirical distribution. In this distribution, 1% of the matches fell above 3.84; therefore, peptide matches with delta scores above 3.84 passed delta score 1% p-value cutoff. Additionally, we define a UniProtKB validation p-value as the probability that a CP domain related peptide match is in the ith or better position in the log-odds score ranked list of peptide matches for a given spectrum (geometric distribution).
The probability of mapping to a CP domain related peptide, p, is the fraction of peptides derived from CP domain containing protein (proteins with a “pp-binding site” annotation in UniProtKB) out of all peptides matched against the given spectrum. The two p-value cutoffs were applied to InsPecT:PPant search results of MS3 PPant peptide detected spectra and remaining best peptide matches for spectra were additionally verified for matching plausible CP active sites (exact “pp-binding site” location predicted by UniProtKB). This method of PPant peptide validation was repeated for InsPecT:PPant search results of SVM detected PPant peptide spectra, where the 1% p-value delta score cutoff was 3.35.
2.6 Generation of Authentic PPant MS/MS Datasets from Recombinant CP Domains
To obtain holo-CP domains (PikAIV, Strop_4416, CouN5, Ybbr), 60 μg recombinant purified CP proteins were incubated with CoA and Sfp as described previously.11 In order to simulate proteomically enriched CP peptides, PikAIV was subjected to reductive alkylation using iodoacetamide and subjected to tryptic digest as previously described.14 Strop_4416, CouN5, and Ybbr were analyzed in the unalkylated form, which produces a 261 pantetheine MS2 ion rather than a 318 alkylated pantetheine ion. These samples were subjected to partial tryptic digest using Trypsin Singles (Sigma). After 5 minutes, digest reactions were quenched with 1:1 (v/v) 10% formic acid-water and placed on ice. Samples were subjected to C18 ZipTip (Millipore) treatment and eluted in 15 μL 85:2:13 acetonitrile-acetic acid-water. Immediately before injection into the mass spectrometer, 15 μL 50:49:1 methanol-water-formic acid was added to the eluted sample and mixed thoroughly. The prepared sample was infused by nano-electrospray ionization and analyzed on a Thermo Scientific LTQ-MS running Tune Plus software version 1.0. The tune file was calibrated to cytochrome c. Broadband MS, MS2, and MS3 scans were acquired for each ACP domain and averaged with QualBrowser software version 1.4 SR1 (Thermo). Parameters for a characteristic experiment (Strop_4416 CP domain) are as follows: MS2 (normalized collision energy: 15; width: 5; data type: profile; precursor m/z: 857.08; average: 342 scans) and MS3 (precursor m/z: 261; normalized energy: 15; width: 5; data type: profile; average: 98 scans). In all cases CP phosphopantetheinylation was manually verified via MS3 fragmentation at precursor m/z 318 or 261 as described above.
2.7 Characteristic PPant Ion Fragments Assignment in MS2
Analysis of PPant peptide spectra from the purified recombinant CP domains PikAIV, CouN5, YbbR, and Strop_4416 led to identification of 11 ion types corresponding to PPant ejection and a fully intact peptide backbone, characterized as: 3 ejected PPant ions, 4 intact backbone ions with PPant +1 charged ejection, and 4 intact backbone peptide ions with PPant neutral ejection (see Section 3.1 for PikAIV example). The neutral ejections are prevalent in higher charged (≥ +3) PPant peptides (data not shown). Exact m/z values for each ion type are calculated from the parent PPant peptide's monoisotopic parent mass and charge (Table S2). A species is identified as a particular PPant characteristic ion type if the species' observed m/z falls within 800 parts-per-million (ppm) error of the ion type expected m/z. Because LTQ mass spectrometers only report the parent peptide's precursor m/z, charge and monoisotopic parent mass are inferred by testing combinations of +2 and +3 charge and up to 3 isotopic states to find the combination that maximizes the explained intensity in observed PPant ion types. Figure 4 shows a representative spectrum before (Figure 4a) and after (Figure 4b) removal of the dominating PPant fragmentation ions.
Figure 4.
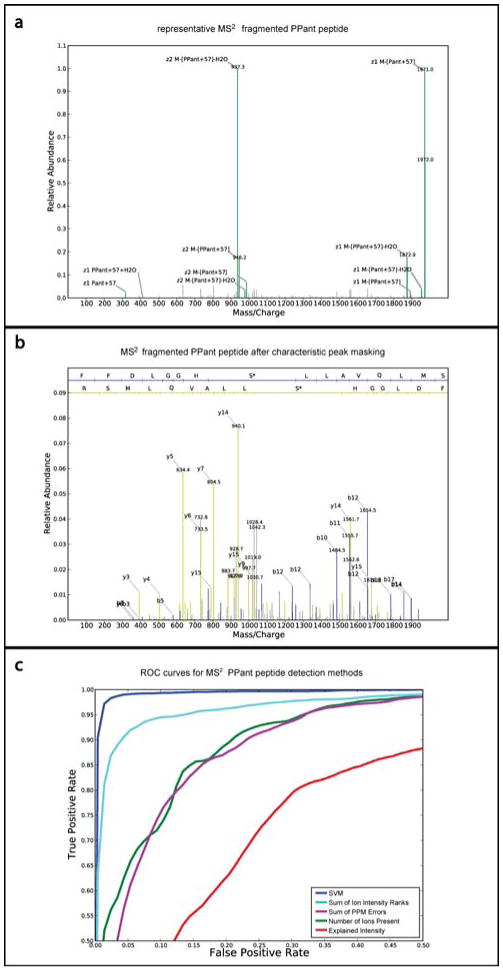
Ejection species dominate the MS2 spectrum of a PPant peptide. (a) MS2 spectrum (scan 13673, run fp3-04) of +2 charged peptide “FFDLGGHS*LLAVQLMSR” from DhbF protein (asterisk denotes site of PPant modification). The parent peptide (M), neutral and charged ejection parent peptides, and 318 m/z alkylated pantetheine (Pant+57) account for a majority of the spectrum's ion intensity (Table S2). The relative abundance is scaled such that the max intensity peak is 1.0. (b) Same spectrum after removal of characteristic PPant species. The b and y ions associated with each peptide bond cleavage are more visible permitting improved peptide identification. Relative abundance is depicted on the same scale as in “a”. (c) Receiver Operating Characteristic (ROC) curves of true positive and false positive rates of SVM approach for detecting PPant spectra from MS2 data. SVM detection is compared to detection of PPant peptides based on Sum of Intensity Rank of Ions, Sum of PPM Error, Number of Ions Present, and Explained Intensity of annotated PPant ions. The curves show average test performance from 30 rounds of 5-fold cross validation for each method.
2.8 Detecting PPant Spectra in MS2
PPant peptide detection can be performed using only information found in MS2 spectra, due to PPant fragmentation resulting in ion species with no breaks in the peptide backbone. The expected 11 PPant characteristic ions (features) are assigned to peaks observed in the spectrum with an 800 ppm tolerance. Isotopes for each of these ion species are subsequently identified if present in the spectrum. The maximum intensity and best intensity rank in the isotopic profile of a feature is used as the feature representative.
We applied a machine learning approach, SVM 18, 19, to solve our two class computational problem, does a MS2 spectrum represent a PPant peptide or non-PPant ion species. To do this we curated a training dataset (Table S4) consisting of 463 CP active site MS2 spectra (derived from manual analysis of recombinant PikAIV, CouN5, YbbR, and Strop_4416, as well as B. subtilis proteomic peptides bearing authentic PPant MS3 signatures), and 5127 non-CP active site spectra (derived from expert analysis, failure to meet minimal PPant MS2 signature criteria, and no MS3 fragmentation). From these spectra we extracted the 11 characteristic CP active site ions (annotated in Section 2.7), and compared PPant and non-PPant peptides for different combinations of intensity metrics and ppm error of observed PPant ejection ions, charged loss parent peptide ions, and neutral loss parent peptide ions. The most distinguishing feature set was found to consist of intensity rank and ppm error of all expected characteristic ions (see Supporting Information for optimal feature and parameter selection).
The trained SVM reports a score suggesting the extent to which a feature set spectrum representation belongs to the PPant class or non-PPant class. To estimate an accurate false positive rate and true positive rate for our supervised learning approach, we performed 30 rounds of 5-fold cross validation on our training dataset of PPant peptide spectra (defined here as TP) and non-CP active site spectra (TN) using the best SVM classifier (selected by the choice of kernels and parameters showing best performance, details in Supporting Information). This ensures that the SVM model generated is not specific to only our training dataset, but has unbiased predictive power when applied to a general dataset. In each round of cross validation, the training data examples were randomly separated into five separate bins, where learning was performed using data from four bins and then the trained classifier was tested on the last bin. Five experiments are completed such that each bin is used as a test bin. SVM classifier accuracy is measured in terms of true positive rate (TP/(TP+FN)) and false positive rate (FP/(TN+FP)), where the number of true positives, false positives, true negatives and false negatives are established by choosing an SVM score cutoff.
3 Results and Discussion
3.1 Investigating the Effect of CID Energy on PPant Ejection and Fragmentation of Tryptic CP Peptides
While a number of studies have examined the PPant ejection of intact CP proteins using “top-down” methods on high resolution FT-instruments, comparatively little work has systematically examined the behavior of PPant peptides using the nano-electrospray ionization (ESI) and collisionally induced dissociation (CID) tandem mass spectrometry conditions commonly applied in “bottom-up” shotgun proteomics analyses. Therefore, before attempting proteomic analysis of CP active site peptides, the impact of CID energy used during tandem mass spectrometry was examined using a tryptic digest of heterologously expressed PikAIV, a module of the type I PKS responsible for biosynthesis of pikromycin by the organism Streptomyces venezualae.12 The range of CID energies tested mimics conditions typically used in shotgun proteomics analyses and are similar to those reported by Stein and coworkers during their pioneering studies of the gramicidin NRPS by ESI-MS.20 Figure 2b displays characteristic MS2 spectra of an ion at m/z of 934 corresponding to the in silico predicted CP peptide of PikAIV in the +2 charge state after the PPant modification has been reductively alkylated by iodoacetamide. Characteristic PPant ejection ions can be grouped into three main classes: 1) the PPant ejection ions 2) parent peptides which have undergone charged ejection of PPant, and 3) parent peptides which have undergone neutral ejection of PPant.7, 9 While all three species are not observed for every PPant peptide, specific combinations of species are highly indicative of PPant. For example, at 30-35 eV the first two species dominate the MS2 spectra of the PikAIV PPant peptide (Figure 2b), while correspondingly weak fragmentation of the peptide backbone is observed. Charged ejection peptides corresponding to the singly charged (z -1) phosphorylated parent mass which has undergone loss of pantetheine (m/z 1549), the same species following dehydration (m/z 1531), and the cognate pantetheine ejection ion (m/z 318) can be clearly visualized. Lower intensity peaks can also be seen for the singly charged dehydroalanine containing peptide (m/z 1451) and corresponding alkylated PPant fragment, which had been lost (m/z 416). This mass difference of 98 m/z from dephosphorylation provides an initial diagnostic tool in manual analysis of MS2 spectra with the purpose of identifying possible PPant peptides of interest. Similar results were observed upon MS/MS analysis of tryptic digests of the CP domains CouN5 (Streptomyces rishiriensis), YbbR (Bacillus subtilis), and Strop_4416 (Salinispora tropica) (data not shown). Along with facilitating the optimization of CID energies for PPant ejection during proteomic MudPIT analyses, these studies provided insight into charge state, ion intensity, and pathways of PPant peptide fragmentation and served as a training dataset of our supervised learning approach for detecting PPant peptides based on their characteristic MS2 signatures (described in Section 3.4).
3.2 Proteomic Enrichment of CP Active Sites by Activity-Based Protein Profiling (ABPP)
Following optimization of CID energy for PKS/NRPS proteomic experiments, we examined the utility of serine-hydrolase directed activity-based protein profiling (ABPP) probes for enrichment of PKS and NRPS CP domains.21 We first applied this approach to B. subtilis st. 168, a model natural product producer which we have previously demonstrated produces high levels of PKS and NRPS enzymes. Because this strain is known to be deficient in the gene encoding for PPant posttranslational modification (sfp),22 prior to enrichment we incubated cell lysate with recombinant Sfp and CoA to ensure that all CP domains were PPant modified for the experiment, mimicking the conditions found in most natural product producing organisms. The ABPP probe used for enrichment was fluorophosphonate (FP)-biotin conjugate, a class-wide inhibitor of serine hydrolases which has known reactivity with PKS and NRPS thioesterase (TE) domains.6, 21, 23 Because of the multidomain nature of type I PKS and NRPS enzymes, TE-directed probes can also enrich CP domains located on the same polypeptide chain of the megasynthase (Figure 2c). Enrichment of serine hydrolases from B. subtilis whole-cell lysate was carried out as previously described, using FP-biotin and avidin-agarose beads. Enriched samples were subjected to on-bead tryptic digestion to produce peptides for MudPIT LC-MS/MS analysis.14 Experiments were designed to acquire data-dependent MS2 spectra on the 7 most intense parent ions, while an additional MS3 step was triggered upon observation of the pantetheine ejection ion at 318 m/z.
As seen previously, application of FP-biotin to the B. subtilis st. 168 proteome results in enrichment and identification of a number of modular biosynthetic enzymes when analyzed by the database search tool InsPecT (Table S1). Among proteins identified are the terminal modules of the NRPS enzymes responsible for biosynthesis of bacillibactin (DhbF), surfactin (SrfAC) and plipastatin (PpsC and PpsE). DhbF contains two CP active sites (unmodified m/z 1891 and 2004), while SrfAC, PpsC, and PpsE contain one CP active site each (unmodified m/z 1855, 1727, and 2474 respectively). Consistent with our earlier reports, none of the four CP active site peptides were identified in either apo- or 4′-PPant modified (+397 m/z after alkylation) form by InsPecT analysis.6 InsPecT did identify a single CP active site peptide, from DhbF, in its phosphorylated (+80 m/z) state. Similar results have been previously observed in phosphoproteomic analysis of B. subtilis24, and likely represent artifactual posttranslational modifications resulting from PPant ejection during ionization prior to MS1.
To verify the ability of ABPP to enrich PKS/NRPS CP domains and unambiguously detect PPant peptides we analyzed MS3 scans from enriched (probe-treated, P) and non-enriched (no-probe, NP) B. subtilis proteomes for signature pantetheine fragmentation patterns (Figure 3).11 Non-enriched samples refer to MudPIT analysis of background peptides enriched due to non-specific interaction with avidin beads or endogenous biotinylation when no probe 1 is added. MS3 scans were called a positive hit for PPant if at least 4 of the 15 most intense peaks in the MS3 spectrum corresponded to the PPant signature peaks 300, 288, 216,184, 142, or 118 m/z within a tolerance of 1 m/z. Figure 3 shows that approximately equal numbers (300-400) of MS3 spectra are collected in both enriched and non-enriched samples, indicating the occurrence of ions with an m/z of 318. This is not unexpected, as a number of b3-tripeptide fragment ions have the same molecular formula as the alkylated pantetheine fragment at 318 (Figure S2). However, when MS3 data are analyzed using the above signature match criteria, spectra collected from FP-biotin enriched samples clearly have a much higher proportion of true PPant ejection events (Figure 3). This indicates ABPP probes such as 1 can be used to specifically enrich CP activeites from proteomic samples, and the presence of PPant can be determined from enriched samples by MS3 assay using low-resolution ion trap instrumentation. Kelleher and coworkers have previously shown that further targeted fragmentation of the backbone peptide can be used to determine the peptide sequence of the CP active site itself10, and our studies suggest such an approach should also be compatible with the ABPP enrichment strategy shown here.
Figure 3.
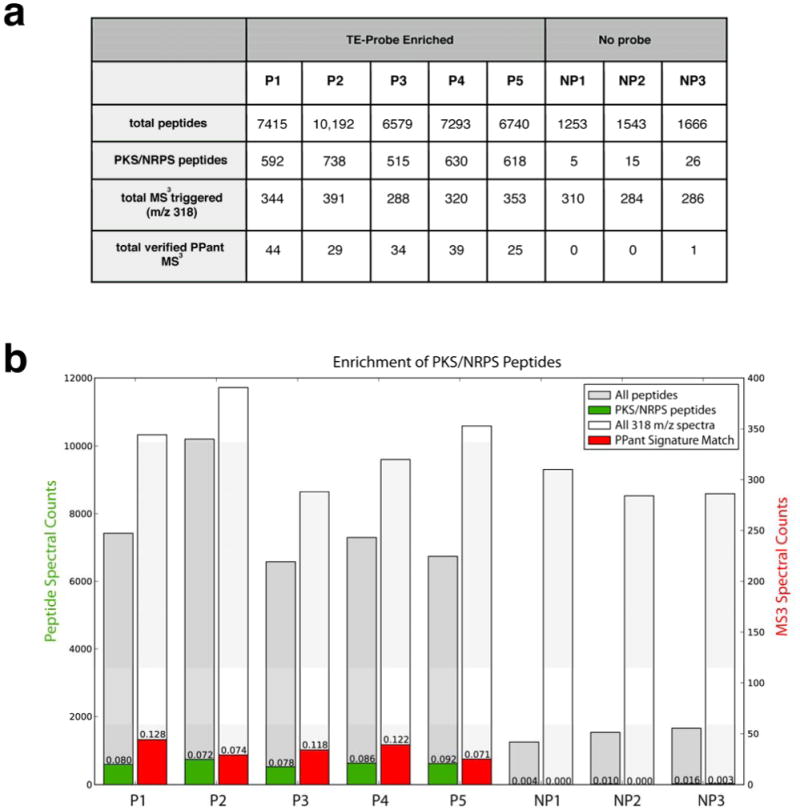
PPant peptide enrichment statistics. (a) Table denoting total number of peptides identified by database search of individual replicates of enriched (probe-treated, P) and non-enriched (no probe, NP) B. subtilis proteome, as analyzed by MudPIT. PKS/NRPS peptides refer to peptides whose protein products are encoded by the srf, dhb, pps, or pks gene clusters in B. subtilis. PPant signature match refers to how many 318 m/z MS3 spectrum from each MudPIT dataset contain 4 out of the 6 most significant pantetheinyl ions as annotated by Meluzzi et al. (b) Same data presented in graphical form.
3.3 Identification of CP Active Sites by InsPecT:PPant with UniProtKB Validation
Having validated our methods for enrichment and detection of PPant peptides, we next sought to develop a computational toolkit for their identification. In addition to poor fragmentation of the PPant peptide backbone, a major source of difficulty in assigning PPant peptide sequence from MS2 data stems from the preference of the species to break apart the PPant modification itself during fragmentation. Therefore, masking of these dominating species, as seen in Figure 4b, aided in the assignment of amino acid sequence to PPant peptide spectra.
Although many advances have been made in the development of computational methods to match experimentally-derived MS2 spectra to those generated in silico from database entries, the identification of both native and posttranslationally modified peptides remains a challenging task. InsPecT identifies the atypical but biologically important phosphopeptides utilizing Bayesian networks to model the unique neutral loss fragmentation expected from peptide bond cleavages of the phosphopeptide.15 Application of a similar approach to generate accurate PPant peptide cut Bayesian networks is stymied by the limited availability of authentic PPant peptide MS2 spectra. To circumvent this obstacle, we modified the trained phosphopeptide networks to score PPant modified peptide sequences against CP active site spectra, a reasonable approach since both posttranslational modifications share the same mechanism of phospho-ester attachment to modify serine residues.
A 1% delta score p-value filtering of InsPecT:PPant results on B. subtilis MS2 spectra showing positive MS3 PPant signatures led to the identification of 78 spectra. Of these, 43 passed our 5% CP domain related peptide p-value cutoff (see Fig. 5a). By comparison, only 5 out of the 94 spectra failing our delta score based p-value passed the CP domain related peptide criteria, demonstrating the power of our approach. The 43 spectra contained 4 unique CP active sites. In each case, the CP active site identified was identical to the site predicted in UniProtKB (Table 1).25 By contrast, none of the 5/94 spectra matched a UniProtKB prediction.
Figure 5.
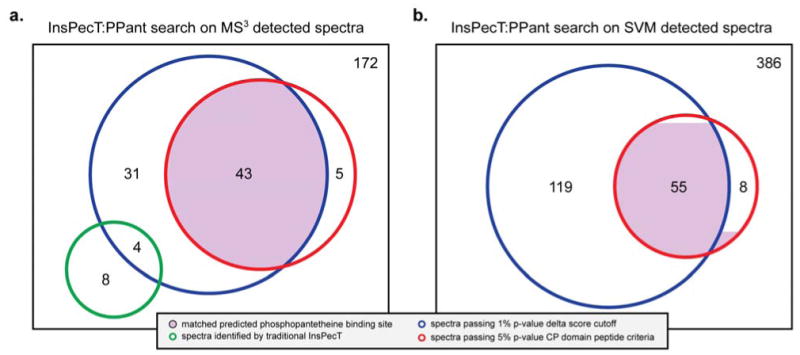
Breakdown of InsPecT:PPant search results for MS3-based detected MS2 spectra and SVM detected MS2 spectra. The rectangle represents the number of spectra detected as PPant peptides.
Table 1. Summary of CP Active Site Peptides Identified by Targeted Fragmentation and SVM.
| CP Active Site | Peptide Mass [M+H]+ (Da) | Protein | Number identified spectra from MS3 detection | Number identified spectra from SVM detection |
|---|---|---|---|---|
| FFDLGGHSppant+57LLAVQLMSR | 2288.0973 | BG11243∣DhbF: involved in 2,3-dihydroxybenzoate biosynthesis | 23 | 29 |
| FFDLGGHSppant+57LLAVQLM | 2044.9641 | |||
| FFDLGGHSppant+57LLAVQL | 1913.9237 | |||
| IGIEDSFFELGGDSppant+57IK | 2123.9612 | BG10972∣PpsC: plipastatin synthetase | 2 | 3 |
| KQIGIHDDFFALGGHSppant+57LK | 2380.1525 | BG10170∣SrfAC: surfactin synthetase / competence | 13 | 15 |
| QIGIHDDFFALGGHSppant+57LK | 2252.0575 | |||
| QVLGVNTISIDDDFFAIGGHSppant+57LR | 2871.3752 | BG11962∣PpsE: plipastatin synthetase | 5 | 6 |
| TISIDDDFFAIGGHSppant+57LR | 2261.0314 | |||
| Fraction of Identified Spectra | 43/172 | 53/386 | ||
The 43 spectra represent the highest confidence annotations and also have the highest delta scores (see Fig. S4). For example, in B. subtilis the UniProtKB database shows 18 NRPS and PKS enzymes containing a total of 43 annotated PPant binding sites in which exact PPant modified serine residues have been predicted by protein sequence homology or MS phosphoproteomic studies.13, 24 An additional 35 candidate spectra passed the delta score cut-off potentially corresponding to novel active sites uncharacterized in B. Subtilis. Of these, 20 map to proteins with unknown function. The remaining 15 spectra map to known proteins which include FabD and other proteins not previously known to have a PPant binding site.
Interestingly, 12 of the 172 spectra selected for MS3 analysis had corresponding MS2 spectra that were annotated with high significance by InsPecT (non-PPant mode) as peptides with no PPant modification (Table S1.1). InsPecT:PPant annotated 4 of these spectra as PPant peptide sequences with delta scores passing the 1% p-value criteria (but not containing a known CP domain). From manual inspection of the MS2 spectra, the InsPecT annotated peptides explain numerous high intensity peaks, except for the characteristic PPant peaks such as those represented in Figure 2. These spectra possibly represent composite/mixture spectra generated by concurrent fragmentation of biologically distinct and unrelated precursor ions due to the following reasons: a) one fragment (m/z 318) in the MS2 spectra was MS3 verified as pantetheine, b) InsPecT identified the MS2 spectrum to be a single peptide with a high score, and c) removing InsPecT identified peaks, the remaining ion peaks appear to be highly similar to the MS2 fragmentation signature expected from a PPant peptide (see Fig. S5).
Typically, MS2 datasets from ion-traps report only a 10-20 percent identification rate.28 By contrast, we confirmed at least 43 of 172 spectra detected by MS3 fragmentation. This suggests a strong enrichment for Ppant spectra. The robustness of the InsPecT:PPant is further evidenced by its identification of several non-tryptic DhbF peptides (Table 1), which are misidentified or go completely unmatched using alternative search algorithms, such as InsPecT and OMMSA27 with comparable PPant modification parameters (data not shown). In addition to its utility in identifying PPant peptides detected by MS3 fragmentation, this strategy proved equally compatible in the identification of CP active sites detected by our machine learning approach (vide infra).
3.4 Development of a Machine Learning Approach for Discovery of PPant Peptides Directly from MS2 Data
While MS3 provides a useful diagnostic tool for analyzing the presence of CP active site peptides, we were also interested in developing a more general method that could be applied to detect PPant containing peptides without the need for targeted fragmentation methods. Such an approach would allow PPant peptide modifications to be mined from existing microbial proteomic datasets, enriching their information content and potentially providing valuable insights into natural products biosynthesis. As an initial step towards this goal we sought to define the MS2 spectral features, which distinguish PPant and non-PPant peptides. Defining a PPant feature set for spectra allowed us to utilize a support vector machine (SVM) approach to learn the distinguishing patterns between PPant and non-PPant spectra and thereby detect whether an arbitrary MS2 spectrum represent a PPant peptide. 18, 19
As no independent PPant detection tool is available for comparison, to evaluate performance of the SVM classifier we compared it to simpler methods of sum of ion intensity ranks, sum of ppm errors, number of ions present, and explained intensity (see Supporting Information for alternate scoring method details) by executing the same five fold cross validation for each method and plotting receiver operating characteristic (ROC) curves (Figure 4c). The ROC curve represents the averaged true positive rate vs. false positive rate across all 150 tests while varying the score cutoffs. At a 1.5% false positive rate, our SVM PPant classifier has a true positive rate of 97.5%, a significant improvement over 82% for the next best intensity ranked scoring method.
Finally, to show the benefit of using the machine learning approach, we compared Inspect:PPant search results of SVM detected PPant spectra with those from MS3 validated PPant spectra, applying the same 1% delta score and 5% CP domain p-value filtering criteria for peptide identification. Classifying spectra as PPant peptides based on a 97% true positive rate and 1% false positive rate, the SVM approach identified an additional 10 spectra mapping to the same B. subtilis CP active sites (Table 1). These represent PPant spectra undetectable by other methods, as analysis showed all 10 to represent instances in which the mass spectrometer failed to select the pantetheinyl ion for MS3 targeted fragmentation. Of the 55 spectra identified using the PPant peptide validation criteria, only 2 matches were not previously predicted as PPant binding sites by UniProtKB (Figure 5b). Conversely, a single DhbF PPant peptide identification did not meet the delta score criteria due to low spectrum quality. While modest, the identification of additional spectra exhibits the power of our specialized SVM approach as well as the limitations of even stringent MS3 based PPant authentification. Additionally, the SVM method requires no additional experimental costs or expertise, as it only depends on MS2 data such as that typically acquired in large-scale proteomic studies.
4 Conclusion
We have presented three core components in an improved practical strategy for identifying PPant active sites commonly found within enzymes involved in PKS and NRPS biosynthesis. Our strategy involved activity-based enrichment and targeted PPant fragmentation, followed by application of a novel computational approach for MS2 PPant active site identification. Finally, we demonstrate proof-of-principle development of a computational MS2 detection method to replace the MS3-based PPant detection. Applying this strategy allowed experimental confirmation of 4 PPant active sites from Bacillus subtilis st. 168 for the first time utilizing a shotgun proteomics approach and low-resolution ion trap instrumentation commonly available in mass spectrometry core facilities.
While the methods here present significant advances in proteomic analysis of PPant peptides, the complex nature of PPant loss together with the limited peptide backbone fragmentation of PPant peptides means these species will remain a challenge to efficiently identify by computational techniques. B. subtilis contains several PKS/NRPS enzymes containing enrichable TE domains; however, enrichment of natural product biosynthetic enzymes from alternative organisms will likely benefit from application of probes targeting more commonly occurring active sites, such as acyltransferase (AT) or ketosynthase (KS) domains. Notably in this regard, Mann and coworkers reported identification of several PPant active sites from B. subtilis proteomes after titanium oxide-based enrichment, suggesting this may be a viable method for PPant enrichment as well.24 In addition, while we have focused on developing a computational approach compatible with spectra generated utilizing standard CID fragmentation, the use of milder fragmentation techniques such as electron transfer dissociation (ETD) may prove useful in reducing the complexity of netural and charged loss PPant peptide species observed under CID. Importantly, an analogous approach has been applied in phosphoproteomics to allow sequence assignment of phosphorylated peptides without neutral loss.28 Similarly, there has been considerable work done to increase the robustness of PPant identification based on iterative rounds of MSn and parent mass analysis, as demonstrated in the recently reported PrISM workflow.10 The continued refinement and mainstream integration of such techniques and instrumentation promise to expand the scope of the PPant detection methods demonstrated here.
To our knowledge, this study represents the first application of an MS2 based machine learning approach to filter for specific posttranslationally modified spectra. The additional peptide identifications discovered through our SVM PPant peptide detection approach demonstrates that SVM can be as effective as targeted fragmentation methods, while requiring less specialized experimentation. In addition to potential applicability of this method for mining existing microbial proteomic datasets for PPant peptides, in the future SVM-based approaches may also prove useful in facilitation of MSn-based PKS/NRPS proteomic discovery, providing confirmation of PPant modification and facilitating de novo sequencing approaches.29 Notably, many computational methods have been developed for database independent (de novo) unmodified and phosphorylated peptide identification,30-32 species that would be observed in targeted fragmentation of MS2 fragmented PPant peptides. Another persistent challenge in the future for this field will be the further development of experimental and computational techniques to allow direct observation of biosynthetic intermediates directly from natural product producer proteomes. Preliminary studies are currently underway to test the applicability of our SVM and InsPect:PPant to detect and identify PPant modified peptides hosting known small intermediate substrates in vitro. In this regard, it is our hope that the new methods for analysis of this cryptic posttranslational modification reported here represent a first step towards the study of natural product biosynthesis in proteomic settings.
Supplementary Material
Acknowledgments
Funding was provided by the University of California, San Diego, Department of Chemistry and Biochemistry, NSF CAREER Award MCB-0347681, NIH GM075797, NIH P41RR024851, NIHGM086283, and NIH RO1-HG004962-01. JYY is supported by a Ruth L. Kirschstein National Research Service Award, NIH 1 T32 EB009380-01.
Footnotes
Supporting Information Supplementary Figures, datasets, source code, and computational details. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Michael D. Burkart, Email: mburkart@ucsd.edu.
Vineet Bafna, Email: vbafna@cs.ucsd.edu.
References
- 1.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106(8):3468–96. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.Mercer AC, Burkart MD. The ubiquitous carrier protein--a window to metabolite biosynthesis. Nat Prod Rep. 2007;24(4):750–73. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]
- 3.Walsh CT, Fischbach MA. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 132(8):2469–93. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen T, Ishida K, Jenke-Kodama H, Dittmann E, Gurgui C, Hochmuth T, Taudien S, Platzer M, Hertweck C, Piel J. Exploiting the mosaic structure of trans-acyltransferase polyketide synthases for natural product discovery and pathway dissection. Nat Biotechnol. 2008;26(2):225–33. doi: 10.1038/nbt1379. [DOI] [PubMed] [Google Scholar]
- 5.Challis GL. Genome mining for novel natural product discovery. J Med Chem. 2008;51(9):2618–28. doi: 10.1021/jm700948z. [DOI] [PubMed] [Google Scholar]
- 6.Meier JL, Niessen S, Hoover HS, Foley TL, Cravatt BF, Burkart MD. An orthogonal active site identification system (OASIS) for proteomic profiling of natural product biosynthesis. ACS Chem Biol. 2009;4(11):948–57. doi: 10.1021/cb9002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45(42):12756–66. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffert JD, Knepper MA. Taking aim at shotgun phosphoproteomics. Anal Biochem. 2008;375(1):1–10. doi: 10.1016/j.ab.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorrestein PC, Kelleher NL. Dissecting non-ribosomal and polyketide biosynthetic machineries using electrospray ionization Fourier-Transform mass spectrometry. Nat Prod Rep. 2006;23(6):893–918. doi: 10.1039/b511400b. [DOI] [PubMed] [Google Scholar]
- 10.Bumpus SB, Evans BS, Thomas PM, Ntai I, Kelleher NL. A proteomics approach to discovering natural products and their biosynthetic pathways. Nat Biotechnol. 2009;27(10):951–6. doi: 10.1038/nbt.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg Med Chem Lett. 2008;18(10):3107–11. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S, Xue Y, Sherman DH, Reynolds KA. Mechanisms of molecular recognition in the pikromycin polyketide synthase. Chem Biol. 2000;7(12):907–18. doi: 10.1016/s1074-5521(00)00039-9. [DOI] [PubMed] [Google Scholar]
- 13.Yin J, Straight PD, Hrvatin S, Dorrestein PC, Bumpus SB, Jao C, Kelleher NL, Kolter R, Walsh CT. Genome-wide high-throughput mining of natural-product biosynthetic gene clusters by phage display. Chem Biol. 2007;14(3):303–12. doi: 10.1016/j.chembiol.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, 3rd, Jeffrey SS, Cravatt BF. A streamlined platform for high-content functional proteomics of primary human specimens. Nat Methods. 2005;2(9):691–7. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 15.Tanner S, Shu H, Frank A, Wang LC, Zandi E, Mumby M, Pevzner PA, Bafna V. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77(14):4626–39. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 16.Payne SH, Yau M, Smolka MB, Tanner S, Zhou H, Bafna V. Phosphorylation-specific MS-MS scoring for rapid and accurate phosphoproteome analysis. J Proteome Res. 2008;8(7):3373–81. doi: 10.1021/pr800129m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbe V, Cruveiller S, Kunst F, Lenoble P, Meurice G, Sekowska A, Vallenet D, Wang T, Moszer I, Medigue C, Danchin A. From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology. 2009;155(Pt 6):1758–75. doi: 10.1099/mic.0.027839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vapnik VN. The Nature of Statistical Learning Theory. Springer; New York: 1995. [Google Scholar]
- 19.Joachims T. Estimating the Generalization Performance of a SVM Efficiently. Morgan Kaufmann Publishers Inc.; San Francisco: 2000. pp. 431–438. [Google Scholar]
- 20.Stein T, Vater J, Kruft V, Wittmann-Liebold B, Franke P, Panico M, Mc Dowell R, Morris HR. Detection of 4′-phosphopantetheine at the thioester binding site for L-valine of gramicidinS synthetase 2. FEBS Lett. 1994;340(1-2):39–44. doi: 10.1016/0014-5793(94)80169-x. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96(26):14694–9. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakano MM, Corbell N, Besson J, Zuber P. Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol Gen Genet. 1992;232(2):313–21. doi: 10.1007/BF00280011. [DOI] [PubMed] [Google Scholar]
- 23.Meier JL, Mercer AC, Burkart MD. Fluorescent profiling of modular biosynthetic enzymes by complementary metabolic and activity based probes. J Am Chem Soc. 2008;130(16):5443–5. doi: 10.1021/ja711263w. [DOI] [PubMed] [Google Scholar]
- 24.Macek B, Mijakovic I, Olsen JV, Gnad F, Kumar C, Jensen PR, Mann M. The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol Cell Proteomics. 2007;6(4):697–707. doi: 10.1074/mcp.M600464-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38(Database issue):D142–8. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mujezinovic N, Schneider G, Wildpaner M, Mechtler K, Eisenhaber F. Reducing the haystack to find the needle: improved protein identification after fast elimination of non-interpretable peptide MS/MS spectra and noise reduction. BMC Genomics. 11 1:S13. doi: 10.1186/1471-2164-11-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res. 2004;3(5):958–64. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 28.Swaney DL, Wenger CD, Thomson JA, Coon JJ. Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2009;106(4):995–1000. doi: 10.1073/pnas.0811964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bandeira N, Olsen JV, Mann JV, Mann M, Pevzner PA. Multi-spectra peptide sequencing and its applications to multistage mass spectrometry. Bioinformatics. 2008;24(13):i416–23. doi: 10.1093/bioinformatics/btn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dancik V, Addona TA, Clauser KR, Vath JE, Pevzner PA. De novo peptide sequencing via tandem mass spectrometry. J Comput Biol. 1999;6(3-4):327–42. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- 31.Frank AM, Savitski MM, Nielsen ML, Zubarev RA, Pevzner PA. De novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res. 2007;6(1):114–23. doi: 10.1021/pr060271u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson RS, Taylor JA. Searching sequence databases via de novo peptide sequencing by tandem mass spectrometry. Mol Biotechnol. 2002;22(3):301–15. doi: 10.1385/MB:22:3:301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.