

Abstract
Lanthanide-binding-tags (LBTs) are valuable tools for investigation of protein structure, function, and dynamics by NMR spectroscopy, X-ray crystallography and luminescence studies. We have inserted LBTs into three different loop positions (denoted L, R, and S) of the model protein interleukin-1β and varied the length of the spacer between the LBT and the protein (denoted 1-3). Luminescence studies demonstrate that all nine constructs bind Tb3+ tightly in the low nanomolar range. No significant change in the fusion protein occurs from insertion of the LBT, as shown by two X-ray crystallographic structures of the IL1β-S1 and IL1β-L3 constructs and for the remaining constructs by comparing 1H-15N-HSQC NMR spectra with wild-type IL1β. Additionally, binding of LBT-loop IL1β proteins to their native binding partner in vitro remains unaltered. X-ray crystallographic phasing was successful using only the signal from the bound lanthanide. Large residual dipolar couplings (RDCs) could be determined by NMR spectroscopy for all LBT-loop-constructs and revealed that the LBT-2 series were rigidly incorporated into the interleukin-1β structure. The paramagnetic NMR spectra of loop-LBT mutant IL1β-R2 were assigned and the Δχ tensor components were calculated based on RDCs and pseudocontact shifts (PCSs). A structural model of the IL1β-R2 construct was calculated using the paramagnetic restraints. The current data provide support that encodable LBTs serve as versatile biophysical tags when inserted into loop regions of proteins of known structure or predicted via homology modelling.
Keywords: Lanthanide-binding-tag, LBT, loop-LBT, peptide fusion tag, encodable lanthanide-binding tag, paramagnetic tag, X-ray crystallography, NMR spectroscopy, luminescence, pseudocontact shift, residual dipolar coupling
Introduction
Peptide-based tags find widespread application in molecular, cellular, and structural biology.1,2 The recently introduced lanthanide-binding tags (LBTs) represent a versatile class of such tags given their potential utility in luminescence-based measurements3, NMR spectroscopy4 and X-ray-crystallography5. The spectral characteristics of LBTs arise from the photophysical properties of the selected trivalent lanthanide ions. In particular, Tb3+ and Eu3+ ions6 are luminescent upon sensitization by organic fluorophores and exhibit distinct and long-lived7 emission profiles allowing cellular localization and binding interaction studies of LBT-tagged proteins.8 The photophysical properties make lanthanide ions useful probes for imaging and resonance energy transfer experiments.9,10 The indole side chain of tryptophan serves as a sensitizer to induce luminescence of the bound Tb3+ in LBTs.11 In X-ray crystallography, lanthanides are used as heavy atoms and provide high phasing power due to their strong anomalous scattering that can be used for single- or multi-wavelength anomalous phasing.12-15 In NMR-spectroscopy, the paramagnetic properties of lanthanide ions can be exploited to weakly align biomolecules along the magnetic field leading to structural and dynamic restraints such as residual dipolar couplings (RDCs), pseudo-contact shifts (PCSs), paramagnetic relaxation enhancement (PRE) or Curie cross-correlated relaxation (CCR).16-24 These parameters have been shown to be sufficient to determine the overall fold of a protein even in the absence of NOE information. Compared to short-range distance restraints such as NOEs (< 5 Ǻ) and scalar couplings, RDCs provide long-range orientation information.25 Additionally, PCSs can be used to determine structures of protein-protein and protein-ligand complexes.26-29 Paramagnetic centers therefore augment the repertoire of methods which include liquid crystals,30 polyacrylamide gels31 and phage32 to induce partial alignment. The use of paramagnetic centers overcomes the problem of external alignment interactions of the target protein with the media. In addition, the determination of relative domain motion in multidomain proteins or RNA strictly requires internal alignment to provide an independent frame of reference.33,34
The first biomolecular applications of paramagnetic alignment in NMR spectroscopy were introduced utilizing naturally-occurring metal ion binding sites substituted with paramagnetic lanthanide ions35 or a heme-cofactor as the paramagnetic center bound to myoglobin.36 In particular, similarities between the ionic radii of the divalent metal ions Ca2+, Mg2+, and Zn2+ and the trivalent lanthanide ions led to applications in which either one or two of these metal ions were replaced with paramagnetic lanthanide ions.37,38 This approach was then extended to diamagnetic proteins lacking native metal-binding sites by fusion with entire paramagnetic protein domains such as zinc finger proteins,39 EF hand motifs,40 or calmodulin-binding peptides16,41 loaded with paramagnetic lanthanide ions for alignment. However, use of such domains results in a considerable increase in molecular weight, which may cause a subsequent loss of signal intensity and also compromise the function of the protein. Furthermore, the high mobility of the tags relative to the protein scaffold reduces the extent of alignment and therefore may result in a low number of measureable structural restraints.16
Other strategies to introduce lanthanides exploited small organic metal-binding chelators based on DTPA,42 EDTA43-45 or DOTA46 attached to the protein via cysteine-modification chemistry. Although such chemical tags have been shown to induce alignment, they result in highly overlapping spectra due to peak doubling resulting from the diastereomeric nature of the tag.19,47 Such chelators have also been stably attached at two points via cysteine disulfide bridges (CLaNP-5).48,49 Additionally, the smallest known lanthanide binding tag, DPA, chelates the lanthanide ion using proximal carboxyl groups of the protein.50 These tags need to be positioned carefully taking into account a suitable distance between the thiol group and nearest carboxyl group. Additionally, free cysteine thiols need to be available in the protein of interest or need to be engineered by site-directed mutagenesis.
Exploiting the strategy of paramagnetic protein fusion domains for alignment, a family of closely related single-LBTs (sLBTs)18 were designed by optimizing naturally occurring calcium-binding loops to avidly bind Ln ions.51,52 These tags are short peptide sequences comprising up to 23 amino acids, which enable incorporation via standard molecular biology strategies. Design and engineering studies have resulted in tags that bind lanthanide ions tightly with low nanomolar KDs and which are selective for lanthanides over other common metal ions.53-56 Attached to either the N- or C-terminus, sLBTs were successfully introduced for NMR structure determination of proteins.18,57 Based on previous investigations, the alignment induced by paramagnetic lanthanide ions was found to depend on the mobility of the tag relative to the protein frame.25 Rigidification and site-specific tagging could also be achieved by linking a sLBT to a single cysteine within the protein via a disulfide bridge.58,59 Two-point anchoring has been shown to further reduce the mobility of the tag compared to single-point anchoring.60 However, this approach necessitates appropriate placement of a cysteine residue close to the N-terminus. The strategy using an encodable multi-functional peptide-based tag at the protein terminus was further improved by the design of a double LBT (dLBT) with two lanthanide binding motifs concatenated in a single 32-residue peptide.5 The X-ray studies and NMR-spectroscopic analysis of the subnanosecond dynamics of the tag61 demonstrated that the increased tag size results in a less mobile tag that is sufficiently ordered with respect to the fusion protein ubiquitin. An increase in luminescence output was also shown for the dLBT-tagged ubiquitin fusion protein.61
In this report, we systematically investigate the possibility of further rigidifying LBTs with respect to the protein by incorporating a sLBT into defined loop regions. This design approach is based on our previous structure determination of Ln-bound LBTs. From these structural studies, we reasoned that integration of the LBT unit into proteins should be feasible with minimal disruption of the loop structures of the target protein. Therefore, the loop position and the length of the linker between the protein and the tag were systematically investigated using interleukin-1β (IL1β), a protein comprising three loops and a β-sheet core, as a model system. In the case of IL1β, the incorporation of the lanthanide binding tag into any of three different protein loops did not impact the overall fold of the protein, the in vitro affinity for native binding partner, or the binding affinity of Ln3+ to the LBT. The present report demonstrates a new and potentially general application of encoded lanthanide-binding tags.
Results and Discussion
Design of IL1β-LBT
In order to utilize lanthanide-binding coexpression tags in NMR spectroscopy and in phasing for X-ray crystallography in macromolecular structure determination, the lanthanide tag must be well ordered with respect to the protein. A survey of structures deposited in the protein data bank reveals that in over 50% of the structures submitted to the PDB, the N-terminal and/or C-terminal residues are disordered. These statistical studies suggest that without significant interaction between the LBT and fusion protein or other means of decreasing domain-domain dynamics, the LBT might be ordered, but adopt several different orientations relative to the core of the protein of interest, therefore limiting its utility.
One method to reduce the conformational dynamics is to restrain both termini of the LBT to decrease interdomain motion, which could be achieved by making the LBT integral to the proteins sequence. Although such placement requires some knowledge of either the secondary or tertiary structure of the protein, secondary structure prediction and homology modelling programs now allow for considerable accuracy in the prediction of β-turns from amino-acid sequences. Therefore, it may be possible to predict appropriate placement of LBTs in structures based upon analyses of protein structures or directly from the protein sequence.62,63 As a model system, the LBT was placed into three different loops (denoted L, R, and S) of IL1β differing in spacer length (denoted LBT-1-3) between the LBT and IL1β resulting in nine different loop-LBT constructs (Table 1). The choice of the LBT sequence GYIDTNNDGWIEGDELY was based upon inspection of the crystal structure of ubiquitin with the dLBT tag.5 Specifically, the termini of one terbium-binding loop were chosen so that they could insert into a pre-existing protein loop allowing overlap of the two short β-strands of the dLBT with the native β-turn of the protein. It was envisioned that the metal-binding residues would remain appropriately positioned to bind the lanthanide without disrupting the protein fold. Based on published crystal and NMR structures of IL1β, three loops were identified as well suited for LBT insertion (Figure 1).64,65 Previously, the S loop has been replaced in a similar, but not identical position, (residues 50-53 versus those replaced herein 52-55) with the result that the protein was well folded, as assessed by NMR structure determination and the loop insertion (alpha 1-antitrypsin inhibitor) retained biological activity.66 In this case, the construct did not have the same number of residues removed on either side of the loop and yet still folded robustly. This provides evidence that precise pre-knowledge of loop structure is not an absolute requirement for the success of insert design.
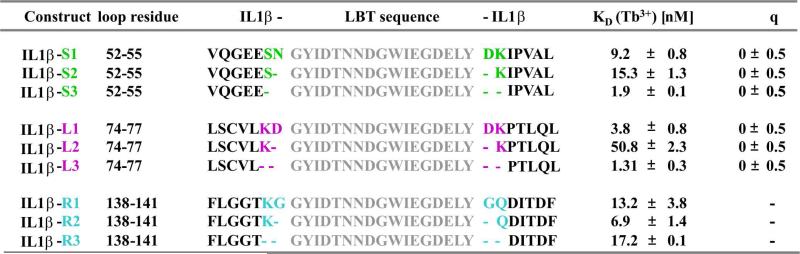
Table 1.
Summary of IL1β-LBT constructs, LBT sequences, KD and q values.
|
Figure 1.

NMR structure of interleukin-1β (IL1β).65 sLBTs have been incorporated into three different loops shown here in purple for the L-loop (residues 74 to 77), in green for the S-loop (residues 52 to 55) and in cyan for the R-loop (residues 138 to 141).
Three constructs of each S-, L- and R-series were generated such that in the LBT-1 series (IL1β-S1/L1/R1), the LBT was inserted between the middle loop residues. In the LBT-2 series (IL1β-S2/L2/R2), the flanking residues were removed and the LBT was inserted in their place. In the LBT-3 series (IL1β-S3/L3/R3), all four protein loop residues were removed and replaced by LBT. Dissociation constants (KD) were determined by luminescence titration of LBTs by Tb3+, in 100 mM NaCl and 10 mM HEPES buffer at pH 7.0. All values are the average of at least three titrations. The number of bound water molecules, q, was determined by luminescence decay experiments (see Materials and Methods).
An additional goal for incorporation of the tag was to retain binding affinity for the native IL1β receptor, s-IL-1R1. For the S-series proteins, it is known from the literature that modification of this loop does not impair receptor binding, and binding is not related to the size of the inserted loop.67 The second loop (L-series) targeted for LBT insertion is a β-turn. A previous co-crystal structure of IL1β with the IL1 receptor showed that this second loop is not involved in receptor recognition, and from computational studies it was predicted that it is also not involved in binding of an accessory protein.64,68 Since residues 138-141 were also shown not to be involved in binding to the receptor, this loop was chosen for the third (R) series.64,68,69 It is also known that derivatization of a IL1β-K138C mutant with iodoactamidofluorescein does not alter receptor recognition, and that fusion with a large (275 kDa) protein did not significantly affect receptor binding.69 Thus, inserting the relatively small LBT at this position may result in a construct retaining its ability to bind the receptor.
Preparation of IL1β-LBT
For luminescence measurements, NMR spectroscopy and receptor binding assays, full-length IL1β-S1 – S3, IL1β-R1 – R3 and IL1β-L1 – L3 were obtained via heterologous expression in Escherichia coli using a glutathione S-transferase (GST) fusion strategy for purification. For luminescence measurements and NMR spectroscopy, the expressed GST-IL1β-LBT proteins were cleaved with TEV protease at the inserted TEV site between the GST and IL1β and purified by size-exclusion chromatography to yield the desired LBT-tagged IL1β products.
For all NMR spectroscopic applications, 15N-labeled proteins were expressed in P-5052 minimal-auto-induction media.70 Expression of the R- and S-series resulted in very good yields of the soluble fusion protein (40-100 mg/L). In contrast, under the same conditions, proteins of the L-series expressed as inclusion bodies in 15N-minimal media. Therefore, the temperature was lowered to 16 °C and proteins of the L-series expressed in acceptable yields (ca. 20 mg/L; see Supplementary Information Figure S1). Constructs of the S-series to be used for crystallization were expressed without a GST-tag and refolded from inclusion bodies (see Materials and Methods).
Receptor-binding assays
The IL1s including IL1α and IL1β are proinflammatory cytokines, which participate in the regulation of numerous immunological and inflammatory processes.71 To control biological activity of IL-1, initiation of signal transduction occurs upon binding of agonist ligands (IL1α or IL1β) to a specific membrane receptor, the transmembrane glycoprotein of the immunoglobulin superfamily IL-1R1.72
Receptor-binding studies were performed using a pull-down assay, in which GST-tagged IL1β-LBT bound to glutathione sepharose beads served as bait for the soluble receptor s-IL-1R1. As confirmed by SDS-PAGE and Western blot analysis (see Supplementary Information, Figure 2 and 3) the incorporation of the LBT does not impair the receptor binding capability of the engineered loop-LBT IL1β mutants. Representative data for the IL1β-S-series are shown in Figure 2 (lanes 4, 7 and 10).
Figure 2.

Receptor-binding assay results, with representative 12% SDS-PAGE gel (left) and anti-sIL-1R1 Western blot (right) shown for GST-IL1β-S1, -S2 and -S3. All experiments were performed in 10 mM HEPES, 100 mM NaCl, pH 7.0 with 0.1% BSA. 1 mg of each protein was preloaded with 1 equivalent of Tb3+.
Photophysical characterization
We determined the luminescence properties and the number of Tb3+-bound water molecules assessed of the LBT-tagged constructs. Luminescence titration studies revealed that the LBTs in all three insert sites bind Tb3+ tightly, with binding constants in the low nanomolar range (Figure 3 and Table 1). The binding affinity of Tb3+ to the loop-LBTs is similar to those found for the first binding event of Tb3+ binding to the dLBT.18,61 While IL1β-L2 shows a slightly higher dissociation constant, we note that two positively charged lysine residues flank the LBT incorporation site in this construct. It is possible that the positively charged side chains surrounding the LBT slightly impede binding of Tb3+. From these results, it can be concluded that covalently linking the LBT into protein loops does not compromise binding affinity for Tb3+.
Figure 3.

Tb3+ titration data and fit, shown for IL1β-R1. Data points represent the average of three independent measurements.
LBTs were originally developed to exclude water from the lanthanide coordination sphere, since coordinated water molecules cause excited Tb3+ to undergo rapid, non-radiative energy transfer to the vibrational states of the water O-H bonds leading to a decrease in luminescence intensity and lifetime.73 The number of bound water molecules was thus evaluated for six of the nine IL1β-LBT proteins (see Table 1 and Supplementary Information Figure S4) using standard methods.74 It was confirmed that there are no water molecules bound to the metal center in any of the loop-LBT proteins as shown by near-zero q values (Table 1). These findings are consistent with data reported previously for sLBTs and dLBTs.61
Characterization of loop-LBT by NMR spectroscopy
In solution, dipolar couplings are averaged to zero due to molecular tumbling. Lanthanides with an anisotropic magnetic susceptibility tensor Δχ induce partial alignment within the magnetic field leading to residual dipolar couplings (RDCs)25. The RDC between two heteronuclear nuclei A and B, DAB, is described by
| (1) |
where θ and ϕ describe the orientation of the internuclear vector in the principal axes system of the alignment tensor, Aa and Ar are the axial and rhombic components of the alignment tensor, rAB is the internuclear distance, S is the generalized order parameter, γA and γB are the gyromagnetic ratios of nuclei A and B, ħ is Planck`s constant divided by 2π and μ0 is the magnetic permeability of vacuum.25,59
Furthermore, pseudocontact shifts (PCSs, δΔ)75 induced by the anisotropic magnetic susceptibility of paramagnetic centers correspond to a change in the observed chemical shifts between paramagnetic and diamagnetic samples that depend on distance and position relative to the paramagnetic center and are described by
| (2) |
where Δχax and Δχrh are the axial and rhombic components of the anisotropic magnetic susceptibility tensor describing the magnetic moment of the paramagnetic center and r, θ, and ϕ are the spherical coordinates of the nucleus in the frame of the Δχ tensor.76
The alignment tensor is proportional to the Δχ tensor:
| (3) |
where B0 is the external magnetic field, kB is the Boltzmann constant, T is the temperature in Kelvin, and Δχa,r are the axial and rhombic components of the anisotropic magnetic susceptibility tensor. The observed RDC depend on the magnetic field strength B02, and the Δχax and Δχrh for the different lanthanides.59,77
Paramagnetic properties differ between lanthanides.75 Therefore, the extent of alignment and observed RDC and PCS can be fine-tuned by the choice of paramagnetic ion. The peptide-based LBTs bind Tb3+, Tm3+, Er3+, Lu3+, La3+,Dy3+, Yb3+, Ho3+ and NMR studies were performed for the described lanthanide ions.18,57-61 The possibility of measuring RDCs for LBTs as fusion tags was first shown for sLBTs and dLBTs attached to the N-terminus of ubiquitin using Tm3+ as the paramagnetic ion. Compared to the sLBT, an additional β-sheet was formed in the structure of the dLBT peptide chain which resulted in decreased tag flexibility and improved alignment. For the sLBT, RDC values between -2 to 6 Hz could be measured on a spectrometer with a 1H frequency of 800 MHz. The size of the RDC was increased for the dLBT by a factor of three with RDCs in the range of -18 Hz to 12 Hz due to the decreased mobility of the tag relative to the protein as inferred from analysis of order parameters from 15N relaxation data. For the sLBT complexed with Tb3+, RDCs were observed between -7.6 and 5.5 Hz.
Previous studies have shown that fusion to the highly flexible N- and C- termini limits the possible attachment sites and does not confer maximum rigidity of the tag. Attaching the sLBT sequence via a disulfide linkage to a cysteine residue yielded RDC values from -12 to 21 Hz for Tm3+ at 800 MHz. Recently, increased rigidification could also be achieved by linking the sLBT via two anchoring points providing RDC values in the range of -12 to +11 Hz for Tb3+ at 600 MHz.60 The present study was conducted to improve the rigid attachment of the sLBT. NMR analyses were performed for all nine IL1β-LBT constructs in the IL1β-R, -S and L series. We titrated 15N-labeled protein with a 1.1-fold excess of paramagnetic Tb3+ and Tm3+ ions or Lu3+ ions as a diamagnetic reference. Paramagnetic shifts and signal broadening could be observed in the 1H-15N-HSQC spectra of the paramagnetic protein complexes demonstrating the utility of loop-LBTs for the measurement of paramagnetic effects.
Magnitude of observed RDCs
RDC values for the amide 1H-15N backbone amide spin pairs (1DHN) were obtained by subtracting the scalar coupling (1JHN) in the presence of diamagnetic Lu3+ from the sum of scalar and dipolar coupling in the presence of paramagnetic Tb3+, both measured in 1H-15NIPAP-HSQCs. Large RDC values could be observed for the IL1β-S-series in the range of -14 to 18 Hz for Tb3+, for the IL1β-R-series of about -12 to 12 Hz and for IL1β-Lseries of -16 Hz to 18 Hz at 600 MHz. Additionally, RDCs were measured for the loop mutant IL1β-R2 using Tm3+ as a second paramagnetic lanthanide ion. RDCs with values from -12 to 10 Hz were observed (see Supplementary Information, Figure S6). Compared to RDC values of sLBT, the loop-LBTs provide a more than threefold improvement demonstrating that the LBT is rigidly attached within the protein frame. Furthermore, the RDC values depend on the linker length, where the LBT-2 series was found to be optimal (Figure 4).
Figure 4.

(a) RDC histogram for IL1β-S1, -S2 and -S3. (b) RDC histogram for IL1β-R1, -R2 and -R3. (c) RDC histogram for IL1β-L1, -L2 and –L3. (d) . Estimated values of and R are obtained by nonlinear least-squares optimization of equations ; , and . The values of Dzz, Dyy and Dxx were measured from the histograms of the RDCs for IL1β-LBT by taking the extreme high and low values such that . Dxx corresponds to the most populated value in the histogram of the RDCs.78,79
In order to make use of RDCs for structure refinement, the magnitude of the axial component of the molecular alignment tensor and the rhombicity must be known. A generalized expression for the RDC can be described by
| (4) |
Where Da is the magnitude of the residual dipolar coupling tensor with and R the rhombicity.78,80 The histogram of the ensemble of RDCs approximates a powder pattern from which and R are readily extracted in the absence of any prior structural information for all IL1β-LBT loop constructs.78,80,81 The histograms for the IL1β-S, -R and -L series suggest asymmetric shapes of the molecules with calculated values for the rhombicity greater than zero (R > 0) (Figure 4).
Diamagnetic assignment of the loop-LBT construct IL1β-R2
To assign the 1H-15N-HSQC cross-peaks of the diamagnetic spectra of IL1β-R2, a 3D-1H-15N-NOESY-HSQC was recorded and assigned utilizing the known assignments of wt-IL1β.82 The similarity of the 1H-15N-HSQC spectra of wt-IL1β and the LBT insertion mutants shows the minimal structural perturbation induced by the LBTs onto the protein structure (see Supplementary Information Figure S5). Additional peaks for the LBT sequence could be detected by comparing the diamagnetic spectra with a reference sample missing the lanthanide ion. In this sample, 89% of the backbone amide signals of diamagnetic spectra could be assigned. Figure 5 shows the paramagnetic and diamagnetic 1H-15N HSQC spectra of IL1β-R2. The effects of the paramagnetic lanthanide ion are shown in representative one-dimensional traces for residues N119, I104, and M44 because residues in close proximity to the lanthanide ion experience larger paramagnetic effects than those more distant from the paramagnetic center.
Figure 5.

1H-15N-HSQC spectrum showing the assigned backbone amide resonances of the sLBT-tagged interleukin-1β-R2 in the presence of 1.1 equivalents of diamagnetic Lu3+ (blue) and paramagnetic Tb3+ (pink) and Tm3+ (green). PCS vectors are indicated as black lines between corresponding diamagnetic and paramagnetic peaks. 1D spectra from the diamagnetic (blue) and paramagnetic (pink, Tb3+) experiments are shown for representative resonances indicating paramagnetic shift and signal broadening. Additional peaks resulting from the LBT sequence are annotated in orange.
Paramagnetic assignment of the loop-LBT construct IL1β-R2
The 1H-15N HSQC cross peaks of the paramagnetic spectra were assigned using the diamagnetic assignment. The resonances in the 1H-15N-HSQC spectra for Tb3+, Tm3+ and the diamagnetic sample with Lu3+ are displaced on diagonal lines due to the similarity of the PCS values of the bonded 1H and 15N spins by what most of the paramagnetic shifts could be assigned (Figure 5).83 Signals that remained unassigned were assigned using a bootstrapping procedure. The initial experimental PCS values were used to predict a first position of the lanthanide ion based on the wild type crystal structure (PDB entry 9ILB) using the program Numbat84. Prediction of the unassigned PCSs onto the wild type crystal structure was achieved using the program Echidna,83 where the initial lanthanide position, the diamagnetic assignment, and the peak table of the paramagnetic 1H-15N-HSQC including some of the reliable and unambiguously assigned paramagnetic peaks were used as input data.84 The iterative procedure resulted in the assignment of 102 out of 140 of the backbone amide cross-peaks peaks for the spectrum with Tb3+ and 99 cross-peaks for Tm3+. PCS values of up to 1 and 0.6 ppm could be detected for Tb3+ and Tm3+, respectively (see Supplementary information, Table 2).
A cross-validation of the experimental RDCs showed a correlation of 0.93 with the three-dimensional crystal structure of native IL1β (PDB entry 9ILB) using the program PALES.79 A structure minimization was performed using the crystal structure of native IL1β and the previously optimized alignment tensor parameters for Tb3+ showing only small changes (see Supplementary Information Figure S6). Cross-validation of the final experimental RDCs and PCS values showed an excellent fit with the refined crystal structure of wt-IL1β (Figure 6a). The Δχ tensor values were then calculated based on the PCS values and the refined wild-type crystal structure. These values are, however, smaller than those reported in the literature, the reason for which remains unclear.85 The data were proven to be robust by performing a Monte-Carlo error analysis where 30% of the experimental PCS were randomly deleted. The Monte-Carlo error analysis and the principal tensor axis orientations are shown in Sanson-Flamsteed-projections. Additionally, using the assigned PCS values, perfect metal ion positions located adjacent to the R-loop were calculated with an optimal distance of 10Å with respect to the loop residues. Alignment tensors for IL1β-R2 with Tb3+ were calculated based on the RDC values and the refined crystal structure of wt-IL1β using PALES.79 To compare the RDC and PCS datasets the alignment tensor was converted to the Δχ tensor using equation 2. The result shows a tensor scaled by 20% - 30% in comparison with . The RDCs seem to be more sensitive to the mobility of the lanthanide ion than the PCS values (Figure 6), such observations have been previously discussed.86
Figure 6.

(a) Scatter plot showing the correlation between observed 1DHN dipolar shifts [Hz] for loop-LBT mutant IL1β-R2 and those back-calculated using the program PALES79 based on the refined crystal structure of Il1β (PDB entry 9ILB). (b) Cross-validation of the final experimental PCSs using Numbat84. Orientations of the Δχ tensor axis components of Tb3+ in complex with IL1β-R2 are visualized in Sanson-Flamsteed projections with the z-, y- and x- axis in blue, green and red respectively. 500 sets of plots represent the result of the Monte Carlo error analysis in which 30% of the data was randomly deleted. (c) The position of the lanthanide ion with respect to the protein is indicated as a black sphere for Tb3+ and as a orange sphere for Tm3+. (d) The calculated axial and rhombic components of the anisotropic magnetic susceptibility (Δχ) tensor and the alignment tensor based on the refined crystal structure of IL1β (PDB entry 9ILB).
Model of the loop-LBT construct IL1β-R2
In an effort to invert the iterative assignment procedure, we modeled the structure of the loop from experimental RDCs, Diffusion Anisotropy and PCSs using CNS.87 The X-ray crystal structure of IL1β (PDB entry 9ILB) was used as starting structure. Distance information calculated for the lanthanide position from PCS values using Numbat84 and the experimental RDCs were used as restraints. Calculation of the loop-LBT-IL1β-R2 model structure was performed keeping the stable secondary structure core parts of the molecule by fixing the heavy atom coordinate positions and by the addition of hydrogen bond restraints. Based on the X-ray crystal structure of IL1β-S1 (vide infra), heavy atom distance restraints were added for the LBT and the lanthanide ion to keep the structure of the tag intact. The linker region of the tag was fully unrestrained. Figure 7 shows the final calculated model of the loop-LBT-construct IL1β-R2. Following the calculation, a cross-validation of the model was performed using the paramagnetic restraints in Numbat.84 The back-calculated PCS for the backbone amide protons are in excellent agreement with the model. In addition, the overlay of the lanthanide position resulting from the model and from the Numbat back-calculated position is in excellent agreement and gives further support for the calculated model of IL1β-R2 and to the applicability of loop-LBTs for protein structure determination.
Figure 7.

(a) Model of the loop-LBT IL1β-R2 with the metal position indicated as a black sphere. (b) From PALES79 and Numbat84 estimated alignment tensor and χ tensor values. (c) Scatter plot showing the correlation between observed 1DHN dipolar shifts [Hz] and those back-calculated using the program PALES79 based on the model of IL1β-R2. (d) Cross-validation of the final experimental PCS values using Numbat84. Orientations of the χ tensor axis components of Tb3+ in complex with IL1β-R2 are visualized in Sanson-Flamsteed projections with the z-, y- and x- axis in blue, green and red respectively. 500 sets of plots represent the result of the Monte Carlo error analysis in which 30% of the data was randomly deleted.
Spin-lattice 15N-R1 and spin-spin 15N-R2 relaxation rates as well as the heteronuclear {1H}-15N nuclear Overhauser effect were measured for most backbone amide groups of loop-LBT IL1β-R2 and used to calculate the general order parameter S2, characterizing the amplitude of internal amide bond vector motion on time scales faster than the overall correlation time τc, by a Lipari-Szabo analysis88,89 with the program TENSOR290 using an anisotropic diffusion tensor (Figure 8).
Figure 8.

Generalized order parameters S2 for the 1H-15N amide bond vector shown for loop-LBT IL1β-R2. Analysis was performed using the programme TENSOR2. An overall τc of 11.49 ns and a axially symmetric diffusion tensor was determined with an asymmetry of 1.37.
Mean order parameters for loop-LBT IL1β-R2 show lower values for the loop-LBT residues than those of the remaining protein indicating some mobility of the loop residues (G1-Y17) compared to the β-sheet core structure of IL1β. However, these values are of the same order as those for the dLBT sequence.61 The increased dynamics in the loop-LBT residues might account for the measured Δχ tensor values resulting in averaged tensor values.
Characterization of IL1β-S1 by X-ray crystallography
Comparison to native IL1β and known LBT structures
The structure of IL1β-S1 complexed with Tb3+ (Figure 9) determined by X-ray crystallography comprises an IL1β with the LBT domain inserted at the end of a pair of anti-parallel β-sheets. The structure of IL1β is essentially unchanged. Notably, initial phasing was accomplished utilizing only the anomalous scattering from the bound Tb3+. Comparison of the structure of IL1β-S1 to that of the known 1.5 Å IL1β structure (PDB accession code 2NVH) shows an RMSD of 0.918 Å for 152 of 153 residues. The largest deviations are found in residues 32-34, 105-109 (both surface turns) and in 49-53. The 49-53 stretch leads up to the insertion with a Cα displacement of 1.6 to 3.3 Å. Notably, after the insertion point and the first atom of residue 54, the displacement is only 0.2-0.6 Å. Thus, no significant change in the fusion protein occurs from insertion of the LBT, as shown by the X-ray crystallographic structure. Only some minor, local deviations are apparent at the insertion site itself. The generality of the finding that the LBT does not affect the scaffold protein structure is also supported by the X-ray crystallographic structure determination of IL1β-L3 to 1.7 Å resolution (Supplementary Information Table S4). The LBT loop in this structure showed disorder (7 of the 17 LBT residues could not be modelled and the lanthanide could not be built into the electron density). However, the overall protein fold does not differ from that of wild-type IL1β with RMSD = 0.87Å. The observed disorder in the LBT electron density in IL1β-L3 is also consistent with the conclusion from NMR that the mobility of the LBT is dependent on the insertion topology.
Figure 9.

(a) Crystal structure of IL1β-S1 colored from N-terminus (red) to C-terminus (blue). (b) Overlay of the IL1β-S1 model (green) on the reference 2NVH (red) (c) IL1β-S1 structure colored by temperature factor (blue, low to red, high). The LBT (inserted in the S-loop) is marked by the Tb3+ depicted as a sphere.
The LBT inserted in the IL1β-S1 construct was designed based on the structure of the double LBT domain from the dLBT-ubiquitin structure. The RMSD of the 20 matching atoms of the IL1-S1-LBT and the dLBT is 0.682 Å. Therefore, the structure of the LBT is essentially unchanged in the environment of the insertion site in the protein (Figure 10).
Figure 10.

Overlay of the LBT portion of IL1β-S1 (red) with the dLBT portion of the ubiquitin structure (green) showing how the dLBT forms a pair of anti-parallel beta strands which fit into the beta sheet of IL1β.
B-factor analysis
In a typical globular protein, the temperature factors of atoms in the model are lower in the core of the protein and increase radially from the center, reflecting the mobility inherent in the protein. Such is the case with the reference IL1β (PDB entry 2NVH). However, in the IL1β-S1 model, the relatively low B-factors in the core are shared by those residues in the LBT. The atoms of the LBT domain have temperature factors of 11-17 Å2 compared to the overall average B-factor of 31 Å2 for the entire structure. The lower values are for atoms chelating the Tb+3 and the tryptophan which makes a crystal contact. This is consistent with the LBT domain acting like a “core”. The stability of the LBT domain allows for the scattering from Tb3+ to be used for phasing. Additional stability is afforded by crystal packing interactions between IL1β molecules.
Crystal Packing
With any X-ray crystal structure, one must be cognizant of the role that crystal packing may have on the target molecule. In this structure, as in the dLBT-ubquitin model, the tryptophan of the LBT fits into a groove of a hydrophobic pocket created by the symmetry related molecule, thereby stabilizing and immobilizing the LBT. The pocket is formed on the face of the symmetry related 105-109 region, which also showed a 1.0-3.9 Å displacement relative to the reference model 2NVH. Although the relative mobility of the LBT is restricted in this design by the insertion into a β-turn, a potential additional source of stability is the formation of this crystal contact. However, the results from NMR presented herein reflect the solution mobility of the LBT domain in these fusion proteins and rule out crystal contacts as the primary source of inter-domain stability.
Conclusions
Overall, this study provides a comprehensive analysis of the effect of inserting LBTs into protein loops, using the model protein IL1β. Analysis of the loop position shows that a canonical β-turn or loop motif provides a suitable insertion site. Luminescence studies demonstrate that the binding affinity is not correlated to loop mobility (determined by NMR) and is fairly insensitive to flanking sequence or to length of spacer between LBT and protein. However, effects correlated with motion as assessed by NMR show that the LBT-2 series (IL1β-S2, -R2 and –L2) is superior in terms of conferring a low relative mobility of the LBT with respect to the fusion partner.
Through a systematic analysis, we show that LBTs are versatile tools for the exploitation of the photophysical, paramagnetic, and diffraction properties of lanthanides in NMR and crystallographic structure determination and luminescence applications. LBTs can be attached to proteins via the N-terminus and/or C-terminus, attached to 1 or 2 cysteine side chains or, as shown here, incorporated into loop regions of proteins, relying on well-established molecular biology protocols. For IL1β, incorporation of the LBT neither impairs the overall fold of the protein nor the binding affinity of Ln3+ to the LBT. As such, the insertion of and LBT can be used for phasing via X-ray crystallography. Furthermore, the loop insertion does not impair binding of IL1β to the soluble domain of the cognate receptor. Incorporation of LBTs into loop regions of proteins provides a broadly applicable strategy for protein research. Such an improved toolkit is of importance to future applications in phase determination in X-ray crystallography and for the structure determination of large protein complexes by NMR where the LBT can be fused into one NMR-silent binding partner without increased mobility in a similar manner.
Materials and Methods
Preparation of IL1β-LBT
Starting from a plasmid encoding human IL1β obtained from ATCC (S595) the cloning of IL1β−S2 , IL1β-L2 and -R2 was accomplished via a two-step PCR procedure. First, the two halves of IL1β were separately amplified by PCR, with primers that included overhangs encoding the desired LBT. (GYIDTNNDGWIEGDELY). The two PCR fragments contained complementary sequences at their termini that could be annealed to each other. Extension via PCR then yielded a product encoding full-length IL1β-S2, -L2 or -R2 respectively, with the LBT at the desired position. This insert was then inserted into the vector pGEX-4T-2. A TEV (Tobacco Etch Virus) protease cleavage site (ENLYFQM) was included to facilitate removal of the N-terminal GST (glutathione-S-transferase) fusion protein. The final construct expresses GST-ENLYFQM-IL1β(LBT). To generate IL1β-S1 and IL1β-S3, site-directed mutagenesis was used to insert or remove codons corresponding to the appropriate amino acids (see Table 1). IL1β-R1 and –R3 were similarly generated from IL1β-R2, and IL1β-L1 and –L3 were similarly generated from IL1β-L2.
Initially, proteins were expressed using IPTG induction with excellent protein yields of ~ 25-50 mg/L, identified by SDS-PAGE, and purified using glutathione sepharose. The temperature for ITPG-induced protein expression was lowered to 16°C since expression at higher temperatures led to truncation products. Improved yields could be obtained using the Studier auto-induction method for protein expression.70 The proteins were expressed in BL21-CodonPlus(DE3)-RIL using ZYM-5052 complex auto-inducing media with excellent yields of about 40–200 mg/L. Protein expression was conducted overnight at 37°C, resulting in no significant truncation products. Following purification on glutathione sepharose, the GST-IL1β-LBT proteins were cleaved with TEV protease and purified by size-exclusion chromatography to yield the desired LBT-tagged IL1β products. For protein expression of the L-series the temperature was lowered to 16°C to avoid expression in inclusion bodies.
For crystallization experiments, constructs of the S-series were cloned starting from IL1-AT(4) which was kindly provided by Prof. T. Pochapsky, Brandeis University. The gene encoding IL1β was modified to remove the chymotrypsin recognition site, insert the LBT domain, and transfer the gene to a pET3a vector with a T7 promoter for IPTG inducible expression. Standard molecular biology tools were used via a QuickChange protocol to insert the LBT using an “inchworm technique”, adding approximately a third of the LBT with each round of modification. Plasmids were sequenced at the Tufts University Core Facility.
Luminescence Titrations
Titrations were recorded on a Fluoromax-3 spectrometer (Jobin Yvon Horiba) in a 1 cm path length quartz cuvette. Tryptophan-sensitized Tb3+ emission spectra were collected by exciting the sample at 280 nm and recording emission at 544 nm. A 315 nm long-pass filter was used to avoid interference from harmonic doubling. Slit widths of 5 nm were used with one second integration times. Luminescence spectra were recorded at room temperature and were corrected for intensity using the manufacturer-supplied correction factors. For all titrations the buffer was 10 nM HEPES and 100 mM NaCl. Titrations were performed in 3 mL of buffer by adding aliquots of the appropriate lanthanide to 50 nM solutions of the appropriate protein to obtain a titration curve. The protein concentration of the stock solutions used in photophysical experiments was determined by UV absorption at 280 nm in a 6 M guanidinium hydrochloride solution using the known extinction coefficient of Trp. After recording a background data point, five 1 μL aliquots of 40 μM Tb3+ were added, followed by five aliquots of 100 μM Tb3+ and 3 aliquots of 200 μM Tb3+. After each addition, the solution was mixed and a data point taken. All data points represent the average values from three independent titrations. The data was fit using SPECFIT/32, using a 1:1 binding model, which determines log β values where β is the binding constant. Errors reported for the KD measurements represent the standard deviation of the results from three independent titrations.
Determination of Tb3+-Bound Water Molecules
Luminescence lifetimes were measured for all proteins in buffered solutions in a Fluoromax-3 (Jobin Yvon Horiba) spectrometer, equipped with a Spex 1934D3 phosphorimeter. The intensity at 544 nm (Tb3+) was monitored at 60 μsec increments for 12 ms after an initial delay of 50μsec, following a lamp pulse at 280 nm from a Xenon flash lamp. Reported data is the average of 3 runs. Using Kaleidagraph, the curves were fit to a monoexponential [I(t) = I(0)*e(−t/τ)], where I(t) is the luminescence intensity at time t after the excitation pulse, I(t) is the initial intensity at t = 0 , and τ is the lifetime. The number of bound water molecules q can be determined by measuring the rate constant of luminescence decay τ-1 in pure H2O and pure D2O. The value of τ-1 for D2O is determined by measuring the lifetime in varying concentrations of D2O and H2O. The value of q can than be calculated using the equation where A is the sensitivity of lanthanide to vibronic quenching, τ is the lifetime in the specified solvent and 0.06 ms is the correction factor for outer-sphere water molecules.74
NMR Experiments
NMR experiments were performed in buffer containing 10 mM HEPES, 100 mM NaCl, 5 mM β-ME, 10 μM DSS an 9/1 (v/v) H2O/D2O. Samples were prepared by careful titration of a protein solution below 0.1 mM with 10 times 0.11 equivalents of lanthanide either Tb3+ or Lu3+. The final sample with 1.1 equivalents of lanthanide was concentrated to 0.5 mM using Amicon Centriprep/Centricon centrifugal concentrator devices. All spectra were recorded at 293K on a Bruker Avance 600 MHz spectrometer equipped with a 5 mm TXI CryoProbe H-C/N-D with single-axis. 1H-15N-HSQC were recorded using 2304 × 512 data points in t2 and t1, respectively, spectral widths of 14 × 40 ppm in ω2 and ω1 and 16 scans per t1 increment with a Z-gradient.
RDCs were obtained by subtraction of the scalar 1J (HN,N) coupling of the superposition of scalar and dipolar coupling, both measured in 1H-15N-IPAP-HSQCs. They were recorded using 2304 × 1024 data points in t2 and t1, respectively, spectral widths of 14 × 40 ppm in ω2 and ω1 and 32 scans per t1 increment with a Z-gradient at 293K. Scalar couplings were measured using diamagnetic lutetium (Lu3+), whereas paramagnetic terbium (Tb3+) was used for RDC measurements. Deconvolution of the picked peaks was performed in Topspin2.1 using dcon2d.
The 3D-1H-15N-NOESY-HSQC experiment was recorded on a Bruker AV600 NMR spectrometer. The mixing time was 150 ms and the ω3, ω2 and ω1 sweep widths were 8417.5, 1762.9 and 7498.5 Hz (corresponding to 29 ppm in the 15N dimension), respectively.
The bootstrapping procedure was performed using Echidna83 to assign paramagnetic peaks and the back-calculation of the experimental PCS as well as the estimated lanthanide position, Δχa,r tensors components and distances of the backbone amide protons to the metal center were determined using the program Numbat84.
Back-calculated RDC values and alignment tensors were determined using the program PALES79, assuming an order parameter S = 1.
15N longitudinal relaxation rates (T1) were obtained using relaxation delays of 100, 200, 400, 600, 800, 1200, 1600, 2000, and 2800 ms. For measurement of 15N transversal relaxation rates (T2), delays of 0, 17.6, 35.2, 52.8, 70.4, 105.6, 140.8, 176.0, and 281.6 ms were used. The spectra were recorded at 298 K using 2048 × 160 data points in t2 and t1, respectively, spectral widths of 13.3 × 28 ppm in ω2 and ω1, and 24 scans per t1 increment on Bruker DRX600 spectrometers equipped with a 5 mm TXI RT 1H{13C/15N} with Z-gradient. The {1H}-15N HetNOE was measured interleaved, using 2048 × (2 × 128) data points, spectral widths of 13.3 × 21.7 ppm, and 64 scans per t1 increment on Bruker DRX600 spectrometers with a 5 mm TXI CryoProbe 1H{13C/15N} with Z-gradient.
Crystallization and Data Collection of IL1β-S1
For crystallization, proteins were loaded with TbCl3 following a previously established protocol5. Briefly, protein was diluted to 1 mg/mL in storage buffer, sodium acetate was added to 10 mM and terbium chloride (in 1 mM HCl) was added in 10 equal aliquots to a final molar ratio of Tb3+: protein of 1.1:1. Protein was concentrated to 30 mg/ml in preparation for crystallization. Initial screening used the Hampton index screen. IL1β-S1 crystallized readily from 100mM sodium acetate trihydrate pH 4.5, 3.0 M NaCl and the conditions did not require further optimization. Crystals were not obtained for IL1β -S2 and IL1β -S3.
Data were collected at beamline X12C at the National Synchrotron Light Source. IL1β-S1 crystals were cryoprotected by soaking the crystals in 15% glucose in mother liquid and then transferred to 30% glucose solution plus mother liquor. Crystals were flash frozen in the gaseous cryogenic N2 stream. Data were collected at a wavelength of 0.95 Å and processed with DENZO/SCALEPACK91. Crystals diffracted to 2.1 Å and belong to space group P6(3)22. Data collection statistics are presented in Table 2.
Table 2.
Data collection, Structure Determination and Refinement Statistics of IL1β-S1.
| Data Collection of IL1β-S1 | |
|---|---|
| Space group and unit cell | P6322; a = b = 120.6 A, c = 74.9Å |
| Wavelength (Å) | 0.95000 |
| Resolution limits (Å) (highest resolution shell) no. of reflections | 50-2.10 (2.18-2.10) |
| Measured | 294259 |
| Unique | 19092 |
| Completeness (%) | |
| All data (highest resolution shell) | 99.0 |
| Rsyma (on I) (highest resolution shell) | 0.075 (0.274) |
| [I/σ(I)] | |
| all data (highest resolution shell) | 21.2 (3.9) |
| | |
| Structure Determination | |
| <m>SOLVE | 0.25 |
| <m>RESOLVE | 0.64 |
| | |
| Refinement | |
| Resolution (Å) | 50-2.1 |
| R factor | 0.192 |
| R free | 0.224 |
| Reflections in test set | 3225 |
| non-hydrogen atoms | 1486 |
| RMS deviations | |
| Bond lengths (Å) | 0.008 |
| Angles (°) | 1.16 |
| Average B factor (Å2) (all atoms) | 31.0 |
Rsym = Σ|Iobs – <I>|/ΣIobs
X-ray crystal structure solution
The space group assignment was confirmed by systematic absences and presence of a 20 sigma peak on the Harker section of the anomalous Patterson map calculated to 2.1 Å. At the wavelength of data collection f’ ~ -0.45e- and f” ~= 6.8e-; phases were determined by the program Phenix92 (FOM 0.49) followed by phase improvement and automatic building (final FOM 0.70) resulting in 83.6% of the backbone being built in an automated fashion including most of the LBT. Protein rebuilding was in COOT93 and refinement was carried out in Phenix92 using phase recombination with the starting phases determined from Tb. The final model contains the entire IL1β molecule including LBT domain with Tb3+, 2 acetate molecules and 131 waters. Only the first two residues of the construct and the C-terminal residue could not be observed in the electron density map.
Supplementary Material
Acknowledgment
H.S. is member of the DFG-funded Cluster of Excellence: Macromolecular Complexes. This work was supported by NSF MCB 0744415 to KNA and BI. A.M.R. acknowledges the NIH for a Ruth L. Kirschstein National Research Service Award. The work was further supported by EU-funded SPINE2 project. Data for this study were measured at Beamline X12C of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the National Institutes of Health. NMR data were obtained at the Center for Biomolecular Magnetic Resonance (BMRZ) supported by the state of Hesse.
Footnotes
Coordinates of the IL1β-S1 and IL1β-L3 structure have been deposited in the Protein Data Bank as entry 3LTQ and 3POK, respectively.
References
- 1.Pazos E, Vazquez O, Mascarenas JL, Vazquez ME. Chem. Soc. Rev. 2009;38:3348–3359. doi: 10.1039/b908546g. [DOI] [PubMed] [Google Scholar]
- 2.Marks KM, Nolan GP. Nat. Methods. 2006;3:591–596. doi: 10.1038/nmeth906. [DOI] [PubMed] [Google Scholar]
- 3.Allen KN, Imperiali B. Curr. Opin. Chem. Biol. 2010;14:247–254. doi: 10.1016/j.cbpa.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Su X-C, Otting GJ. Biomol. NMR. 2010;46 doi: 10.1007/s10858-009-9331-1. [DOI] [PubMed] [Google Scholar]
- 5.Silvaggi NR, Martin LJ, Schwalbe H, Imperiali B, Allen KN. J. Am. Chem. Soc. 2007;129:7114. doi: 10.1021/ja070481n. [DOI] [PubMed] [Google Scholar]
- 6.Bünzli J-CG. Acc. Chem. Res. 2006;37 doi: 10.1021/ar0400894. [DOI] [PubMed] [Google Scholar]
- 7.Richardson FS. Chem. Rev. 2002;82:541–552. [Google Scholar]
- 8.Bünzli J-CG, Piguet C. Chem. Soc. Rev. 2005;34:1048–1077. doi: 10.1039/b406082m. [DOI] [PubMed] [Google Scholar]
- 9.Selvin PR. Annu. Rev. Biophys. Biomol. Struct. 2002;31:275–302. doi: 10.1146/annurev.biophys.31.101101.140927. [DOI] [PubMed] [Google Scholar]
- 10.Sandtner W, Bezanilla F, Correa AM. Biophys. J. 2007;93:L45–L47. doi: 10.1529/biophysj.107.119073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sculimbrene BR, Imperiali B. J. Am. Chem. Soc. 2006;128:7346. doi: 10.1021/ja061188a. [DOI] [PubMed] [Google Scholar]
- 12.Weis WI, Kahn R, Fourme R, Drickamer K, Hendrickson WA. Science. 1991;254:1608–1615. doi: 10.1126/science.1721241. [DOI] [PubMed] [Google Scholar]
- 13.Molina R, Stelter M, Kahn R, Hermoso JA. Acta Crystallogr., Sect. D. 2009;65:823–831. doi: 10.1107/S0907444909017958. [DOI] [PubMed] [Google Scholar]
- 14.Ku S-Y, Smith GD, Howell PL. Acta Crystallogr., Sect. D. 2007;63:493–499. doi: 10.1107/S0907444907006592. [DOI] [PubMed] [Google Scholar]
- 15.Girard E, Stelter M, Vicat J, Kahn R. Acta Crystallogr., Sect. D. 2003;59:1914–1922. doi: 10.1107/s0907444903020511. [DOI] [PubMed] [Google Scholar]
- 16.Feeney J, Birdsall B, Bradbury AF, Biekofsky RR, Bayley PM. J. Biomol. NMR. 2001;21:41–48. doi: 10.1023/a:1011924017938. [DOI] [PubMed] [Google Scholar]
- 17.Gaponenko V, Altieri AS, Li J, Byrd RA. J. Biomol. NMR. 2002;24:143–148. doi: 10.1023/a:1020948529076. [DOI] [PubMed] [Google Scholar]
- 18.Wöhnert J, Franz KJ, Nitz M, Imperiali B, Schwalbe H. J. Am. Chem. Soc. 2003;125:13338. doi: 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
- 19.Ikegami T, Verdier L, Sakhaii P, Grimme S, Pescatore B, Saxena K, Fiebig KM, Griesinger C. J. Biomol. NMR. 2004;29:339–349. doi: 10.1023/B:JNMR.0000032611.72827.de. [DOI] [PubMed] [Google Scholar]
- 20.Boisbouvier J, Gans P, Blackledge M, Brutscher B, Marion D. J. Am. Chem. Soc. 1999;121:7700–7701. [Google Scholar]
- 21.Pintacuda G, Hohenthanner K, Otting G, Müller N. J. Biomol. NMR. 2003;27:115–132. doi: 10.1023/a:1024926126239. [DOI] [PubMed] [Google Scholar]
- 22.Iwahara J, Schwieters CD, Clore GM. J. Am. Chem. Soc. 2004;126:5879–5896. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- 23.Donaldson LW, Skrynnikov NR, Choy W-Y, Muhandiram DR, Sarkar B, Forman-Kay JD, Kay LE. J. Am. Chem. Soc. 2001;123:9843–9847. doi: 10.1021/ja011241p. [DOI] [PubMed] [Google Scholar]
- 24.Bertini I, Cavallaro G, Cosenza M, Kümmerle R, Luchinat C, Piccioli M, Poggi L. J. Biomol. NMR. 2002;23:115–125. doi: 10.1023/a:1016341507527. [DOI] [PubMed] [Google Scholar]
- 25.Blackledge M. Prog. NMR Spectrosc. 2005;46:23–61. [Google Scholar]
- 26.Pintacuda G, Park AY, Keniry MA, Dixon NE, Otting G. J. Am. Chem. Soc. 2006;128:3696–3702. doi: 10.1021/ja057008z. [DOI] [PubMed] [Google Scholar]
- 27.John M, Pintacuda G, Park AY, Dixon NE, Otting G. J. Am. Chem. Soc. 2006;128:12910–12916. doi: 10.1021/ja063584z. [DOI] [PubMed] [Google Scholar]
- 28.Pintacuda G, John M, Su XC, Otting G. Acc. Chem. Res. 2007;40:206. doi: 10.1021/ar050087z. [DOI] [PubMed] [Google Scholar]
- 29.Saio T, Yokochi M, Kumeta H, Inagaki F. J. Biomol. NMR. 2010;46:271–280. doi: 10.1007/s10858-010-9401-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tjandra N, Bax A. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 31.Tycko R, Blanco FJ, Ishii Y. J. Am. Chem. Soc. 2000;122:9340–9341. [Google Scholar]
- 32.Clore GM, Starich MR, Gronenborn AM. J. Am. Chem. Soc. 1998;120:10571–10572. [Google Scholar]
- 33.Bertini I, Gupta YK, Luchinat C, Parigi G, Peana M, Sgheri L, Yuan JJ. Am. Chem. Soc. 2007;129:12786–12794. doi: 10.1021/ja0726613. [DOI] [PubMed] [Google Scholar]
- 34.Dethoff EA, Hansen AL, Zhang Q, Al-Hashimi HM. J. Magn. Reson. 2009;202:117–121. doi: 10.1016/j.jmr.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee L, Sykes BD. Biochemistry. 1981;20:1156–1162. doi: 10.1021/bi00508a017. [DOI] [PubMed] [Google Scholar]
- 36.Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. Proc. Natl. Acad. Sci. U.S.A. 1995;92:9279–9283. doi: 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allegrozzi M, Bertini I, Janik MBL, Lee Y-M, Liu G, Luchinat C. J. Am. Chem. Soc. 2000;122:4154–4161. [Google Scholar]
- 38.Bertini I, Gelis I, Katsaros N, Luchinat C, Provenzani A. Biochemistry. 2003;42:8011–8021. doi: 10.1021/bi034494z. [DOI] [PubMed] [Google Scholar]
- 39.Gaponenko V, Dvoretsky A, Walsby C, Hoffman BM, Rosevear PR. Biochemistry. 2000;39:15217–15224. doi: 10.1021/bi001381w. [DOI] [PubMed] [Google Scholar]
- 40.Ma C, Opella SJ. J. Magn. Reson. 2000;146:381–384. doi: 10.1006/jmre.2000.2172. [DOI] [PubMed] [Google Scholar]
- 41.Tuechelmann A, Schwalbe H, Griesinger C. 3rd European Conference Oxford (1998); Oxford. 1998. [Google Scholar]
- 42.Franklin SJ, Raymond KN. Inorganic Chemistry. 2002;33:5794–5804. [Google Scholar]
- 43.Gaponenko V, Sarma SP, Altieri AS, Horita DA, Li J, Byrd RA. J. Biomol. NMR. 2004;28:205–212. doi: 10.1023/B:JNMR.0000013706.09264.36. [DOI] [PubMed] [Google Scholar]
- 44.Haberz P, Rodriguez-Castaneda F, Junker J, Becker S, Leonov A, Griesinger C. Org. Lett. 2006;8:1275–1278. doi: 10.1021/ol053049o. [DOI] [PubMed] [Google Scholar]
- 45.Pintacuda G, Moshref A, Leonchiks A, Sharipo A, Otting G. J. Biomol. NMR. 2004;29:351–361. doi: 10.1023/B:JNMR.0000032610.17058.fe. [DOI] [PubMed] [Google Scholar]
- 46.Häussinger D, Huang J.-r., Grzesiek S. J. Am. Chem. Soc. 2009;131:14761–14767. doi: 10.1021/ja903233w. [DOI] [PubMed] [Google Scholar]
- 47.Prudêncio M, Rohovec J, Peters JA, Tocheva E, Boulanger MJ, Murphy MEP, Hupkes H-J, Kosters W, Impagliazzo A, Ubbink M. Chem Eur J. 2004;10:3252–3260. doi: 10.1002/chem.200306019. [DOI] [PubMed] [Google Scholar]
- 48.Keizers PHJ, Saragliadis A, Hiruma Y, Overhand M, Ubbink M. J. Am. Chem. Soc. 2008;130:14802–14812. doi: 10.1021/ja8054832. [DOI] [PubMed] [Google Scholar]
- 49.Keizers PHJ, Desreux JF, Overhand M, Ubbink M. J. Am. Chem. Soc. 2007;129:9292–9293. doi: 10.1021/ja0725201. [DOI] [PubMed] [Google Scholar]
- 50.Su X-C, Man B, Beeren S, Liang H, Simonsen S, Schmitz C, Huber T, Messerle BA, Otting G. J. Am. Chem. Soc. 2008;130:10486–10487. doi: 10.1021/ja803741f. [DOI] [PubMed] [Google Scholar]
- 51.Brittain HG, Richardson FS, Martin RB. J. Am. Chem. Soc. 1976;98:8255–8260. doi: 10.1021/ja00441a060. [DOI] [PubMed] [Google Scholar]
- 52.MacManus JP, Hogue CW, Marsden BJ, Sikorska M, Szabo AG. J Biol. Chem. 1990;265:10358–10366. [PubMed] [Google Scholar]
- 53.Franz KJ, Nitz M, Imperiali B. ChemBioChem. 2003;4:265–271. doi: 10.1002/cbic.200390046. [DOI] [PubMed] [Google Scholar]
- 54.Nitz M, Franz KJ, Maglathlin RL, Imperiali B. ChemBioChem. 2003;4:272–276. doi: 10.1002/cbic.200390047. [DOI] [PubMed] [Google Scholar]
- 55.Nitz M, Sherawat M, Franz KJ, Peisach E, Allen KN, Imperiali B. Angew. Chem., Int. Ed. 2004;43:3682–3685. doi: 10.1002/anie.200460028. [DOI] [PubMed] [Google Scholar]
- 56.Martin LJ, Sculimbrene BR, Nitz M, Imperiali B. QSAR Comb. Sci. 2005;24:1149. [Google Scholar]
- 57.Zhuang T, Lee H-S, Imperiali B, Prestegard JH. Protein Sci. 2008;17:1220–1231. doi: 10.1110/ps.034561.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su X-C, Huber T, Dixon NE, Otting G. ChemBioChem. 2006;7:1599–1604. doi: 10.1002/cbic.200600142. [DOI] [PubMed] [Google Scholar]
- 59.Su X-C, McAndrew K, Huber T, Otting G. J. Am. Chem. Soc. 2008;130:1681–1687. doi: 10.1021/ja076564l. [DOI] [PubMed] [Google Scholar]
- 60.Saio T, Ogura K, Yokochi M, Kobashigawa Y, Inagaki F. J. Biomol. NMR. 2009;44:157–166. doi: 10.1007/s10858-009-9325-z. [DOI] [PubMed] [Google Scholar]
- 61.Martin LJ, Hähnke MJ, Nitz M, Wohnert J, Silvaggi NR, Allen KN, Schwalbe H, Imperiali B. J. Am. Chem. Soc. 2007;129:7106–7113. doi: 10.1021/ja070480v. [DOI] [PubMed] [Google Scholar]
- 62.Zheng C, Kurgan L. BMC Bioinformatics. 2008;9:430. doi: 10.1186/1471-2105-9-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwede T, et al. Structure. 2009;17:151–159. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vigers GPA, Anderson LJ, Caffes P, Brandhuber BJ. Nature. 1997;386:190–194. doi: 10.1038/386190a0. [DOI] [PubMed] [Google Scholar]
- 65.Clore GM, Wingfield PT, Gronenborn AM. Biochemistry. 2002;30:2315–2323. doi: 10.1021/bi00223a005. [DOI] [PubMed] [Google Scholar]
- 66.Arico-Muendel CC, Patera A, Pochapsky TC, Kuti M, Wolfson AJ. Protein Engineering. 1999;12:189–202. doi: 10.1093/protein/12.3.189. [DOI] [PubMed] [Google Scholar]
- 67.Wolfson AJ, Kanaoka M, Lau F, Ringe D, Young P, Lee J, Blumenthal J. Biochemistry. 1993;32:5327–5331. doi: 10.1021/bi00071a007. [DOI] [PubMed] [Google Scholar]
- 68.Casadio R, Frigimelica E, Bossù P, Neumann D, Martin MU, Tagliabue A, Boraschi D. FEBS Letters. 2001;499:65–68. doi: 10.1016/s0014-5793(01)02515-7. [DOI] [PubMed] [Google Scholar]
- 69.Wingfield P, Graber P, Alan RS, Alan R, Gronenborn AM, Clore MG, MacDonald RH. Eur J Biochem. 1989;179:565–571. doi: 10.1111/j.1432-1033.1989.tb14584.x. [DOI] [PubMed] [Google Scholar]
- 70.Studier FW. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 71.Dinarello CA. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 72.Martin MU, Falk W. Eur. Cytokine Netw. 1997;8:5–17. [PubMed] [Google Scholar]
- 73.Horrocks WD, Sudnick DR. J. Am. Chem. Soc. 2002;101:334–340. [Google Scholar]
- 74.Beeby A, Clarkson IM, Dickins RS, Faulkner S, Parker D, Royle L, de Sousa AS, Williams JAG, Woods M. J. Chem. Soc., Perkin Trans. 2. 1999:493–503. [Google Scholar]
- 75.Bleaney B. J. Magn. Reson. 1972;8:91–100. [Google Scholar]
- 76.Bertini I, Luchinat C, Parigi G. Prog. NMR Spectrosc. 2002;40:249–273. [Google Scholar]
- 77.Otting G. J. Biomol. NMR. 2008;42:1–9. doi: 10.1007/s10858-008-9256-0. [DOI] [PubMed] [Google Scholar]
- 78.Clore GM, Gronenborn AM, Bax A. J. Magn. Reson. 1998;133:216–221. doi: 10.1006/jmre.1998.1419. [DOI] [PubMed] [Google Scholar]
- 79.Zweckstetter M. Nat. Protoc. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 80.Rule GS, Hitchens TK. Fundamentals of Protein NMR Spectroscopy. Springer; Dordrecht: 2006. [Google Scholar]
- 81.Bryce DL, Bax A. J. Biomol. NMR. 2004;28:273–287. doi: 10.1023/B:JNMR.0000013701.16162.0c. [DOI] [PubMed] [Google Scholar]
- 82.Driscoll PC, Clore GM, Marion D, Wingfield PT, Gronenborn AM. Biochemistry. 1990;29:3542–3556. doi: 10.1021/bi00466a018. [DOI] [PubMed] [Google Scholar]
- 83.Schmitz C, John M, Park AY, Dixon N, Otting G, Pintacuda G, Huber T. J. Biomol. NMR. 2006;35:79–87. doi: 10.1007/s10858-006-9002-4. [DOI] [PubMed] [Google Scholar]
- 84.Schmitz C, Stanton-Cook M, Su X-C, Otting G, Huber T. J. Biomol. NMR. 2008;41:179–189. doi: 10.1007/s10858-008-9249-z. [DOI] [PubMed] [Google Scholar]
- 85.Bertini I, Janik MBL, Lee Y-M, Luchinat C, Rosato A. J. Am. Chem. Soc. 2001;123:4181–4188. doi: 10.1021/ja0028626. [DOI] [PubMed] [Google Scholar]
- 86.Bertini I, Del Bianco C, Gelis I, Katsaros N, Luchinat C, Parigi G, Peana M, Provenzani A, Zoroddu MA. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6841–6846. doi: 10.1073/pnas.0308641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr., Sect. D, Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 88.Lipari G, Szabo A. Journal of the American Chemical Society. 1982;104:4546–4559. [Google Scholar]
- 89.Lipari G, Szabo A. Journal of the American Chemical Society. 1982;104:4559–4570. [Google Scholar]
- 90.Dosset P, Hus JC, Blackledge M, Marion D. J. Biomol. NMR. 2000;16:23. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 91.Otwinowski Z, Minor W, Carter CWJ. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 92.Adams PD, et al. Acta Crystallograph., Sect. D. 2009;66:213–221. [Google Scholar]
- 93.Emsley P, Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.