

Abstract
Semen-derived enhancer of viral infection (SEVI), an amyloid fibril formed from a cationic peptide fragment of prostatic acidic phosphatase (PAP), dramatically enhances the infectivity of human immunodeficiency virus type 1 (HIV-1). Insoluble, sedimentable fibrils contribute to SEVI-mediated enhancement of virus infection. However, the SEVI-forming PAP(248–286) peptide is able to produce infection-enhancing structures much more quickly than it forms amyloid fibrils. This suggests that soluble supramolecular assemblies may enhance HIV-1 infection. To address this question, non-SEVI amyloid-like fibrils were derived from general amphipathic peptides of sequence Ac-Kn(XKXE)2-NH2. These cationic peptides efficiently self-assembled to form soluble, fibril-like structures that were, in some cases, able to enhance HIV-1 infection even more efficiently than SEVI. Experiments were also performed to determine whether agents that efficiently shield the charged surface of SEVI fibrils block SEVI-mediated infection-enhancement. To do this, we generated self-assembling anionic peptides of sequence Ac-En(XKXE)2-NH2. One of these peptides completely abrogated SEVI-mediated enhancement of HIV-1 infection, without altering HIV-1 infectivity in the absence of SEVI. Collectively, these data suggest that soluble SEVI assemblies may mediate infection-enhancement, and that anionic peptide supramolecular assemblies have the potential to act as anti-SEVI microbicides.
Introduction
Semen-derived enhancer of viral infection (SEVI) is a naturally occurring amyloidogenic enhancer of human immunodeficiency virus type 1 (HIV-1) infection that is derived from self-assembling cationic peptides found in semen, the most abundant of which is PAP(248–286) (1). Filtration of semen results in a substantial loss of infection-enhancing activity, suggesting that insoluble fibrils contribute to SEVI-mediated enhancement of virus infection (1). However, addition of freshly dissolved monomeric PAP(248–286) peptide to filtered semen restores its HIV-1 infection-enhancing activity within minutes—a time frame that is 1–2 orders-of-magnitude faster than the kinetics of fibril formation by the monomeric peptide, even under optimal in vitro conditions (1,2). Thus, it seems likely that soluble supramolecular assemblies (SSA) of PAP(248–286) may also contribute to enhancement of HIV-1 infection.
To address the question of whether (and how) soluble supramolecular peptide assemblies can enhance HIV-1 infection, non-SEVI amyloid-like fibrils were derived from general amphipathic peptides of sequence Ac-Kn(XKXE)2-NH2. Amphipathic peptides of this kind ([XKXE]y) efficiently form soluble fibril-like structures with a proposed bilayer architecture (3–5). Forces driving self-assembly of these peptides into β-sheet structures include a combination of hydrogen-bonding, hydrophobic interactions, aromatic interactions, and ionic complementarity (6–8); higher level assembly is believed to occur by the formation of a cofacial bilayer of β-sheets, which then laminate at the hydrophobic faces to form a hydrophobic core, exposing the hydrophilic faces of the β-sheets (Fig. 1) (3,5,9).
Figure 1.
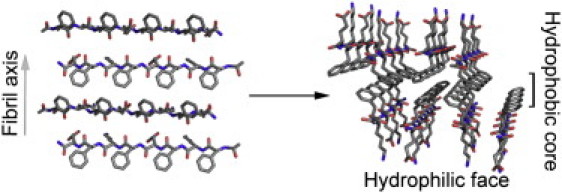
Proposed packing mode of the cofacial bilayer for (FKFE)2. (Left) (FKFE)2β-strands in an antiparallel registry forming a β-sheet; the figure is arranged to illustrate the direction of the fibril axis with the hydrophobic amino-acid side chains extending out of the page and the hydrophilic amino-acid side chains descending into the page. (Right) The cofacial assembly of two β-sheets to form the proposed bilayer architecture that comprises the basic fibrillar unit. This cofacial assembly is facilitated by burial of the hydrophobic side chains in a hydrophobic core, leaving the hydrophilic side chains exposed at the fibril surface. This arrangement accounts for the high water solubility of these peptide fibrils. (Colors: Red corresponds to oxygen, blue to nitrogen, and gray to carbon.)
The self-assembly of these amphipathic peptide domains can be modulated by using amino-acid residues with differing hydrophobicity, including the nonnatural amino acid, cyclohexylalanine (Cha), which has enhanced hydrophobicity relative to phenylalanine (Phe) (Table 1) (6). Increasing the hydrophobicity at X is also expected to thermodynamically stabilize the final fibril structure pushing the dynamic equilibrium between fibril and monomer further toward fibrils. By changing X, we can tune this equilibrium as a function of environment (temperature, pH, ionic strength). We substituted Cha for Phe in the motif Kn(XKXE)2, to derive synthetic peptides that were expected to exhibit an increased propensity for fibril formation due to more favorable desolvation of the hydrophobic face of the peptide. We also synthesized peptides in which Ala was substituted for X in the (XKXE)2 motif; these peptides were expected to be unable to undergo self-assembly due to their greatly reduced hydrophobicity (Table 1) (5).
Table 1.
Self-assembling synthetic peptides used in this study
| Peptide No. | Sequence | n | X | Π∗ | Mass (Da) | Charge (at pH 7)† | Charge/mass |
|---|---|---|---|---|---|---|---|
| Cationic Kn(XKXE)2 peptides | |||||||
| 1 | Ac-K2(FKFE)2-NH2 | 2 | Phe | 1.79 | 1419 | +2.00 | 1.41 × 10−3 |
| 2 | Ac-K4(FKFE)2-NH2 | 4 | Phe | 1.79 | 1675 | +4.00 | 2.39 × 10−3 |
| 3 | Ac-K2(ChaKChaE)2-NH2 | 2 | Cha | 2.72 | 1443 | +2.00 | 1.39 × 10−3 |
| 4 | Ac-K4(ChaKChaE)2-NH2 | 4 | Cha | 2.72 | 1658 | +4.00 | 2.41 × 10−3 |
| 5 | Ac-K2(AKAE)2-NH2 | 2 | Ala | 0.31 | 1114 | +2.00 | 1.80 × 10−3 |
| Anionic En(XKXE)2 peptides | |||||||
| 6 | Ac-E2(FKFE)2-NH2 | 2 | Phe | 1.79 | 1421 | −2.00 | 1.41 × 10−3 |
| 7 | Ac-E4(FKFE)2-NH2 | 4 | Phe | 1.79 | 1679 | −4.00 | 2.38 × 10−3 |
| 8 | Ac-E2(ChaKChaE)2-NH2 | 2 | Cha | 2.72 | 1445 | −2.00 | 1.38 × 10−3 |
| 9 | Ac-E4(ChaKChaE)2-NH2 | 4 | Cha | 2.72 | 1703 | −4.00 | 2.35 × 10−3 |
| SEVI-forming peptide | |||||||
| 10 | PAP(248–286) | NA | NA | NA | 4549 | +6.46 | 1.42 × 10−3 |
The bilayer fibrils formed by XKXE-based peptides remained soluble (i.e., did not spontaneously precipitate in solution, and were not readily sedimented by high-speed centrifugation), in contrast to SEVI fibrils. We then performed HIV-1 infection experiments using these peptides (and soluble supramolecular assemblies (SSA) derived thereof). The results showed that SSA formed by some of these cationic peptides enhanced HIV-1 infection of target cells even more efficiently than SEVI. In contrast, anionic peptides, neutral peptides and peptides that were unable to undergo self-assembly failed to enhance HIV-1 infection.
We also tested whether anionic SSA could shield the charged surface of SEVI fibrils and thereby block SEVI-mediated infection-enhancement. To do this, we generated self-assembling anionic peptides of sequence Ac-En(XKXE)2-NH2. One of these peptides completely abrogated SEVI-mediated enhancement of HIV-1 infection, without altering HIV-1 infectivity in the absence of SEVI. Collectively, these data suggest that soluble SEVI supramolecular assemblies may contribute to the infection-enhancing properties of seminal fluid, and that anionic peptide supramolecular assemblies have the potential to be developed into anti-SEVI microbicides.
Materials and Methods
Peptide synthesis
Fmoc amino acids and rink amide resin were purchased from AAPPTec (Louisville, KY) and Advanced ChemTech (Louisville, KY). All peptides were synthesized using standard Fmoc methods (HOBt and HBTU activation) on a microwave-equipped Liberty Peptide Synthesizer (CEM, Matthews, NC). Peptides were cleaved from the resin by treatment with trifluoroacetic acid (TFA), triisopropylsilane, and deionized water (95:2.5:2.5) for 2 h at room temperature. The solution was concentrated in vacuo to roughly 0.5 mL and then the peptide was precipitated with cold ether and centrifuged (repeated twice). The resulting peptide pellet was dissolved in dimethylsulfoxide. The crude samples were then purified by reverse-phase preparatory high-pressure liquid chromatography (HPLC) with purity analyzed by analytical HPLC. Product verification was performed by matrix-assisted laser desorption ionization time-of-flight mass spectroscopy. Matrix-assisted laser desorption ionization mass spectra for test peptides are provided in Fig. S12, Fig. S13, Fig. S14, Fig. S15, Fig. S16, Fig. S17, Fig. S18, Fig. S19, and Fig. S20 in the Supporting Material.
Purification
HPLC purification was performed on a LC-AD HPLC utilizing a variable wavelength absorbance detector (Shimadzu Scientific Instruments, Columbia, MD) with a reverse phase C18 column (model No. BEH300 10 μM, 19 × 250 mm; Waters, Milford, MA). A gradient of water (0.1% TFA) and acetonitrile (0.1% TFA) at 10 mL min−1 was used for separation while monitoring UV absorbance at 215 and 254 nm. The purity of collected fractions was verified by analytical HPLC using an RP-C18 column (BEH300 10 μM, 4.6 × 250 mm; Waters). The pure solutions were frozen and lyophilized. Analytical HPLC traces for test peptides are provided in Fig. S3, Fig. S4, Fig. S5, Fig. S6, Fig. S7, Fig. S8, Fig. S9, Fig. S10, and Fig. S11; analytical HPLC conditions are provided in Table S1 in the Supporting Material.
Self-assembly
Lyophilized peptide was dissolved in a mixture of acetonitrile and water (4:6) to prevent assembly, further diluted into dimethylsulfoxide, and concentrations were determined by HPLC correlation to standard concentration curves that were calibrated by amino-acid analysis of peptide stocks (amino-acid analysis done by AIBiotech, Richmond, VA). Peptides were aliquoted, frozen, and lyophilized. The lyophilized powder was then dissolved in deionized water followed by vortex treatment (1 min). When studying the ionic strength necessary to induce self-assembly, NaCl solutions were prepared (0 mM, 100 mM, 200 mM, etc.) and added directly to the lyophilized powder, followed by vortex treatment (1 min). Peptides were immediately analyzed by circular dichroism (CD) or Fourier transform infrared spectroscopy (FTIR) to assess secondary structure, which is an indicative measure for self-assembly of these peptides. SEVI amyloid fibrils were generated as described, using the PAP(248–286) peptide (10).
Circular dichroism spectroscopy
CD spectra were obtained on an AVIV 202 circular dichroism spectrometer (AVIV Biomedical, Lakewood, NJ). Wavelength scans were performed from 260 to 190 nm with a 1.0-nm step, 1.0-nm bandwidth, and a 3 s averaging collection time per step at 25°C in a 0.1-mm pathlength quartz cuvette (Hellma, Plainview, NY). The AVIV software (AVIV Biomedical) was used to perform background subtraction, conversion of raw data to molar ellipticity, and data smoothing with a least-squares fit. Additional CD spectra for test peptides in PBS are provided in Fig. S1 and Fig. S2.
Fourier transform infrared spectroscopy
FTIR was performed with a 8400 FT-IR spectrophotometer (Shimadzu). A quantity of 200 μL of peptide was placed in an IR cell with calcium fluoride plates (International Crystal Labs, Garfield, NY) and spectra were obtained from that solution. Scans were taken from 1400 to 1800 cm−1 at a resolution of 2.00 cm−1. IRSolution software (Shimadzu) was used to smooth the data, and perform a multipoint baseline correction.
Transmission electron microscopy
A quantity of 10 μL of the various peptide assemblies were placed onto a 200-mesh carbon-coated copper grid. K4(FKFE)2 and K4(ChaKChaE)2, however, were spotted onto a graphene-enhanced Lacey carbon film on 200-mesh nickel grids (Electron Microscopy Sciences, Hatfield, PA). Samples were allowed to stand on the grid for 1 min, and then aqueous solvent was removed by capillary action and the grids were subsequently stained with uranyl acetate for 2 min. The stain was removed by capillary action and the grid was allowed to dry for 5 min. The images were obtained on a model No. 7650 transmission electron microscope (Hitachi, Santa Clara, CA) at an accelerating voltage of 80 kV. The program ImageJ (National Institutes of Health, Bethesda, MD) was used to determine fibril diameter, reported as the average of 100 measurements on unique fibrils for each measurement.
HIV infectivity assays
These assays were performed using CEMx M7 cells, which contain a luciferase reporter gene under the transcriptional control of the HIV-1 LTR (10). Self-assembled cationic peptides were preincubated with HIV-1IIIB virus (21 ng/mL p24; ZeptoMetrix, Buffalo, NY) for 10 min at room temperature before being added to 5 × 104 CEMx M7 cells in a final volume of 150 μL (including tissue culture media). After incubation for 2 h at 37°C, cells were pelleted by centrifuging at 800 × g for 5 min, and the supernatant removed. This step was included to eliminate the possibility that the test peptides/aggregates might exert a late (post-entry) effect on virus infection. The cells were then washed 1× in phosphate-buffered saline, resuspended in culture medium, and incubated for 48 h at 37°C, after which luciferase activity was measured in cell lysates (10).
Inhibition of SEVI-mediated enhancement of HIV infection by self-assembled anionic peptides
SEVI fibrils were first incubated for 10 min with HIV-1IIIB virus at room temperature and then the self-assembled anionic peptides were added for an additional 10 min. Peptide-virus complexes were then added to CEMx M7 cells and HIV-1 infection was analyzed by measuring luciferase expression (see above).
Cytotoxicity assay
Viability was measured by resazurin cytotoxicity assay (alamarBlue assay; Invitrogen, Carlsbad, CA) (10).
Inhibition of cationic peptide-mediated enhancement of HIV-1 infection
CEMx M7 cells were pretreated for 30 min at room temperature with either Surfen or self-assembled Ac-K4-(FKFE)2-NH2. The cells were then washed once with RPMI-1640 and resuspended in culture medium. Cationic peptides and virus were combined with the cells as outlined above (HIV-1 infectivity assays). Surfen (bis-2-methyl-4-amino-quinolyl-6-carbamide) was obtained from the Open Chemical Repository in the Developmental Therapeutic Program at the National Cancer Institute (NSC 12155), National Institutes of Health.
Results
To test whether soluble, supramolecular peptide assemblies can enhance HIV-1 infection, cationic peptides were designed using the general sequence Ac-Kn(XKXE)2-NH2 (Table 1). First, CD spectroscopy and FTIR spectroscopy were used to evaluate the secondary structure of the synthetic cationic peptides in water containing increasing concentrations of NaCl. All peptides readily dissolved in water and presented clear aqueous solutions with no visibly insoluble material. CD spectra revealed that all of the peptides formed β-sheet assemblies, with the exception of the K2(AKAE)2 peptide (Fig. 2). The classical β-sheet CD signature is a minimum at 218 nm and a maximum between 190 and 200 nm. CD spectra for (XKXE)2 peptides display a minimum at 215–218 nm correlating to β-sheet, but also have an additional minimum at 205–210 nm which has been attributed to either π-π effects or twisting of the β-sheet (6,9,11,12). The CD spectrum of the K2(AKAE)2 peptide is consistent with an unordered structure orientation, with a strong minimum between 190 and 200 nm, indicating that this peptide fails to undergo self-assembly (11). Unlike (FKFE)2 (4–6), the K2(FKFE)2 and K4(FKFE)2 peptides did not immediately assemble upon dissolution in water due to the installation of positive charges at the N-terminus (Fig. 2, A and B), which provide an element of molecular frustration that inhibits self-assembly (13). However, the charge repulsion resulting from the additional lysine (Lys) residues that prevents β-sheet assembly could be overcome by increasing the ionic strength of the medium by inclusion of NaCl. In the presence of NaCl, the characteristic β-sheet signature indicates self-assembly was observed at 100 mM NaCl for the K2(FKFE)2 peptide and at 300 mM NaCl for the K4(FKFE)2 peptide (Fig. 2, A and C). The [NaCl] required to promote fibril formation was proportional to the degree of positive charge in the peptide sequence.
Figure 2.
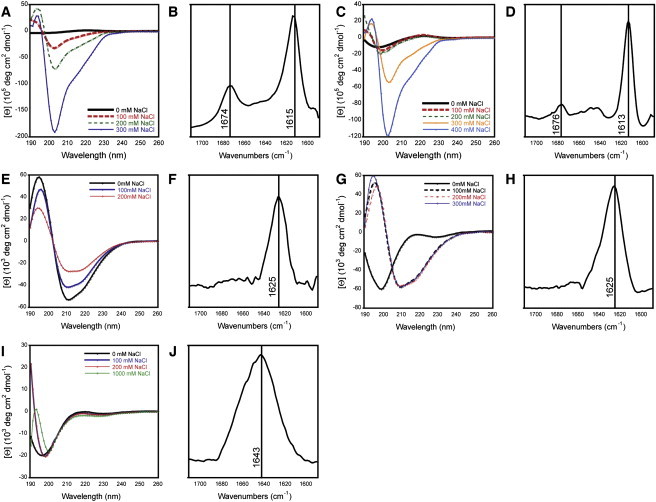
Spectral characterization of self-assembled structures formed by synthetic cationic peptides. (A, C, E, G, and I) CD spectra of cationic peptides in unbuffered water. Peptides were dissolved in unbuffered water at a peptide concentration of 0.9 mM, in the presence of increasing concentrations of sodium chloride. Spectra are labeled as follows: (A) K2(FKFE)2; (C) K4(FKFE)2; (E) K2(ChaKChaE)2; (G) K4(ChaKChaE)2; (I) K2(AKAE)2. (B, D, F, H, and J) FTIR spectra of cationic peptides in unbuffered water. The TFA counterions that are present with each peptide as a function of HPLC purification were exchanged by lyophilization from HCl: peptides were then lyophilized from D2O. Peptides were then dissolved in D2O (1.5 mM) containing the appropriate amount of NaCl necessary to induce the formation of β-sheet assemblies. IR spectra are labeled as follows: (B) K2(FKFE)2 [NaCl = 100 mM]; (D) K4(FKFE)2 [NaCl = 300 mM]; (F) K2(ChaKChaE)2 [NaCl = 0 mM]; (H) K4(ChaKChaE)2 [NaCl = 100 mM]; (J) K2(AKAE)2 [NaCl = 1 M].
K2(ChaKChaE)2 and K4(ChaKChaE)2 behaved similarly to their Phe-containing counterparts (Fig. 2, E and G). Because Cha has an increased hydrophobicity compared to Phe (Table 1) (6,14), less salt was required to induce assembly of these peptides. Indeed, K2(ChaKChaE)2 formed β-sheet assemblies in the complete absence of NaCl, and K4(ChaKChaE)2 formed β-sheet assembles at just 100 mM NaCl. The CD signatures of these Cha-containing peptides exhibited a more classical β-sheet trace, with a strong minimum at 218 nm and a maximum from 190–200 nm; an additional minimum at 209 nm, similar to that observed with (FKFE)2, was also resolvable (attributed to twisting of the β-sheet) (Fig. 2, E and G). The last cationic peptide examined was K2(AKAE)2. As expected, due to the relative hydrophilicity of Ala (Table 1), this peptide failed to form β-sheets, even at high NaCl concentrations (Fig. 2 I).
FTIR analysis of the cationic series of peptides was also performed to confirm β-sheet assembly. K2(FKFE)2 showed a distinct amide I stretch at 1615 cm−1 and K4(FKFE)2 showed a similar stretch at 1613 cm−1 (Fig. 2, B and D); these peaks are slightly lower than the expected amide I stretch for β-sheets from 1620 and 1640 cm−1; this is likely due to an antiparallel strand registry and subtle sheet twisting that may be induced by the Phe residues in the bilayer interface (6,8,15–18). These peptides also showed peaks between 1674 and 1676 cm−1 (Fig. 2, B and D), which may be due to hydrogen bonding (17) or vibrational coupling of the peptides (8,17). K2(ChaKChaE)2 and K4(ChaKChaE)2 both exhibited peaks at 1625 cm−1 which correlates to classical amide I stretch attributed to β-sheet architectures (Fig. 2, F and H).
In addition to the cationic peptides, four additional anionic peptides utilizing the general Ac-En(XKXE)2-NH2 sequence were synthesized and examined (Table 1). To determine the self-assembly conditions of these peptides, CD spectroscopy was performed in unbuffered water (pH 3–4 due to residual TFA), and water adjusted to pH 7 with NaOH. In acidic solutions, the peptides were not fully soluble as evidenced by slight cloudiness in the solutions; when the pH was adjusted to 7, the solutions clarified. The CD spectra for the E2(FKFE)2 and E4(FKFE)2 peptides showed evidence of β-sheet assembly at pH 3–4 (Fig. 3, A and C), although the assemblies were not completely soluble as indicated by slightly cloudy solutions. The CD spectrum for E4(FKFE)2 transitioned from β-sheet to unordered structure when solutions were adjusted to pH 7, indicating disassembly of the fibrils as a function of pH; β-sheet structure was restored by addition of high concentrations of NaCl which served to mask the negative charges of the glutamic acid (Glu) residues at pH 7 (Fig. 3 C).
Figure 3.
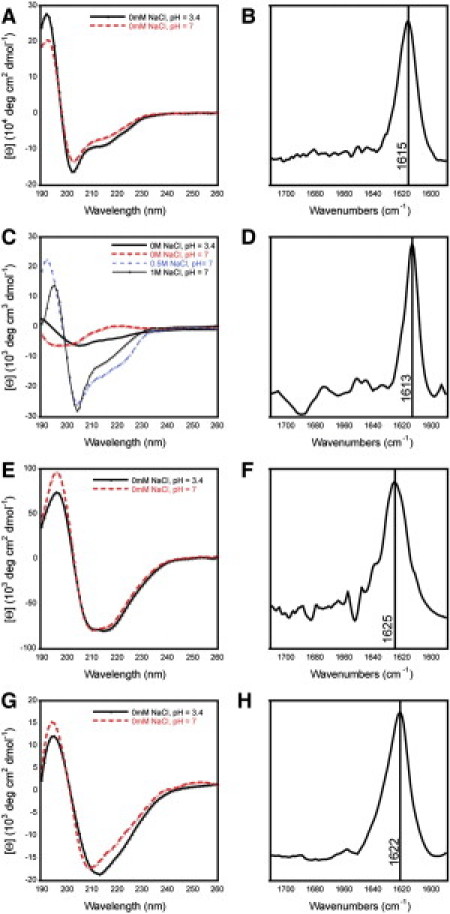
Spectral characterization of self-assembled structures formed by synthetic anionic peptides. (A, C, E, and G) CD spectra of anionic peptides in unbuffered water. Peptides were dissolved in unbuffered water (0.3 mM), and spectra were then obtained at acidic pH (pH was 3–4 as a function of residual TFA from HPLC purification); additional spectra were obtained by increasing the pH 7 by addition of NaOH. In the case of peptide E4(FKFE)2, an additional spectrum was obtained at neutral pH in the presence of NaCl, because this was required for formation of β-sheet assemblies. (A) E2(FKFE)2; (C) E4(FKFE)2; (E) E2(ChaKChaE)2; (G) E4(ChaKChaE)2. (B, D, F, and H) FTIR spectra of anionic peptides in unbuffered water. This analysis was performed as described in the legend to Fig. 2. (B) E2(FKFE)2 [NaCl = 0 mM]; (D) E4(FKFE)2 [NaCl = 1 M]; (F) E2(ChaKChaE)2 [NaCl = 0 mM]; (H) E4(ChaKChaE)2 [NaCl = 0 mM].
The Cha-containing anionic peptides exhibited similar properties to their Phe-containing counterparts. In unbuffered water at pH 3-4, both E2(ChaKChaE)2 and E4(ChaKChaE)2 exhibited a clear β-sheet structure (as reflected by the minimum at ∼218 nm, and a slightly larger minimum at 210 nm) and incomplete solubility (Fig. 3, E and G). When the pH was increased to 7, the peptides became more soluble, and retained a β-sheet conformation (Fig. 3, E and G). In contrast to the Phe-containing peptides, no NaCl was required to promote self-assembly of the more hydrophobic Cha peptides at pH 7.
FTIR studies yielded data that were consistent with the CD analysis, confirming the formation of β-sheet assemblies by the anionic peptides. Similar to CD analysis, the E4(FKFE)2 was unassembled at neutral pH without the addition of NaCl; however, at 1 M NaCl, β-sheet assembly was observed, indicated by the amide I stretch at 1613 cm−1 (Fig. 3 D). As with the cationic peptides, the Phe peptides had peaks at lower frequencies than their Cha counterparts (1615 and 1613 cm−1 for E2(FKFE)2 and E4(FKFE)2, respectively (Fig. 3, B and D), compared to 1625 and 1622 cm−1 for E2(ChaKChaE)2 and E4(ChaKChaE)2, respectively (Fig. 3, F and H)).
Spectral β-sheet signatures are usually an indication of fibril self-assembly in these systems, but transmission electron microscopy (TEM) imaging is necessary to confirm the presence of fibrils. We therefore performed TEM analysis on each peptide under conditions that resulted in β-sheet formation by spectral analysis. All the peptides, with the exception of K2(AKAE)2, showed positive confirmation of fibrillar assemblies in TEM images (Fig. 4). The observed fibrils were uniformly long (micrometers) and unbranched. The fibrils showed some morphological variability in diameter. K2(ChaKChaE)2 and K4(ChaKChaE)2 had the largest diameters of the assemblies studied, with fibril diameters of 5.9 ± 0.9 and 5.0 ± 0.8 nm, respectively. E4(FKFE)2 had a comparable fibril diameter to K2(ChaKChaE)2 with a diameter of 4.5 ± 0.6 nm. E2(FKFE)2, E2(ChaKChaE)2, E4(ChaKChaE)2, K2(FKFE)2, and K4(FKFE)2 all had similarly sized fibril diameters of 3.2 ± 0.6, 2.8 ± 0.5, 3.2 ± 0.4, 2.9 ± 0.5, and 3.4 ± 0.6 nm, respectively.
Figure 4.
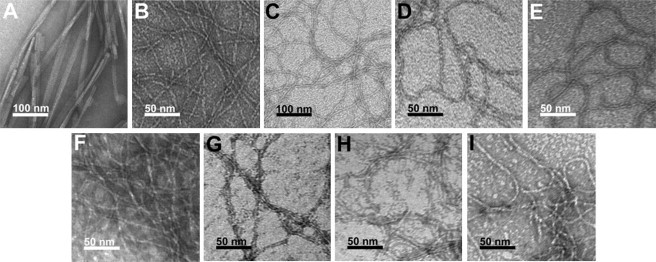
TEM images of cationic and anionic peptides. TEM images were obtained from solutions in which spectral evidence indicated the formation of β-sheet assemblies. (A) SEVI: [peptide] = 1.33 mM, [NaCl] = 150 mM (in phosphate-buffered saline); (B) K2(FKFE)2: [peptide] = 0.9 mM, [NaCl] = 100 mM, fibril diameter = 2.9 ± 0.5 nm; (C) K4(FKFE)2: [peptide] = 0.9 mM, [NaCl] = 200 mM, spotted onto a nickel grid, fibril diameter = 3.4 ± 0.6 nm. (D) K2(ChaKChaE)2: [peptide] = 0.3 mM, [NaCl] = 0 mM, fibril diameter = 5.9 ± 0.9 nm; (E) K4(ChaKChaE)2: [peptide] = 0.3 mM, [NaCl] = 100 mM, spotted onto a nickel grid, fibril diameter = 5.0 ± 0.8 nm; (F) E2(FKFE)2: [peptide] = 0.3 mM, [NaCl] = 0 mM, at pH 7, fibril diameter = 3.2 ± 0.6 nm; (G) E4(FKFE)2: [peptide] = 0.3 mM, [NaCl] = 1 M, at pH 7, fibril diameter = 4.5 ± 0.6 nm; (H) E2(ChaKChaE)2: [peptide] = 0.3 mM, [NaCl] = 0 mM, at pH 7, 2.8 ± 0.5 nm; (I) E4(ChaKChaE)2: [peptide] = 0.3 mM, [NaCl] = 0 mM, at pH 7, fibril diameter = 3.2 ± 0.4 nm.
The bilayer fibrils formed by XKXE-based peptides remained soluble, unlike SEVI fibrils. Fig. S21 shows that SEVI fibrils precipitated spontaneously, as indicated by the appearance of flocculent material suspended in these solutions. Conversely, XKXE-derived fibrils did not precipitate spontaneously but remained soluble (Fig. S21). TEM confirmed that the XKXE-based peptides formed amyloid fibrils, and that these fibrils remained soluble even after high-speed centrifugation (Fig. S22). In contrast, the SEVI fibrils efficiently sedimented upon high-speed centrifugation—confirming their insolubility (Fig. S22).
We next evaluated whether the SSA formed by our peptides were able to enhance HIV-1 infection of target cells. To do this, we infected CEMx M7 reporter cells in the presence of increasing concentrations of both cationic and anionic SSA. The cationic but nonfibrillar peptide [K2(AKAE)2] and the anionic amyloidlike fibrils [E4(FKFE)2, E2(ChaKChaE)2] had no effect on HIV-1 infection, while all of the cationic amyloidlike fibrils [K4(FKFE)2, K2(FKFE)2, K4(ChaKChaE)2, K2(ChaKChaE)2] significantly enhanced viral infection (Fig. 5). The cationic Phe-containing peptides were more efficient at enhancing HIV-1 infection than their Cha-containing counterparts, even though the former peptides were able to self-assemble more efficiently, especially under low salt conditions. The reason for this remains unclear, although it could be due to slightly decreased solubility of the Cha peptides at high ionic strength. This was sometimes observed as a slight cloudiness of the solutions, as noted above.
Figure 5.

Fibril structure and charge are critical for cationic peptide enhancement of HIV-1 infection. CEMx M7 cells were infected with HIV-1 in the presence of increasing concentrations of self-assembled peptides with 1), net positive charges (shown in red): SEVI, K4(FKFE)2, K2(FKFE)2, K4(ChaKChaE)2, K2(ChaKChaE)2; 2), net negative charges (shown in blue): E4(FKFE)2, E2(ChaKChaE)2; and 3), unassembled positively charged peptide (shown in orange): K2(AKAE)2. At 48 h post-infection, cells were harvested and luciferase measured as a readout of HIV-1 infection. Results represent mean values from three experimental replicates; error bars denote the standard deviation of these values. The results shown are representative of three independent experiments performed with a range of peptide concentrations all yielding similar results. Notes: 1), All peptide concentrations refer to final concentrations in cell culture (and not to concentrations during virion preincubation, which were higher); and 2), we have plotted the data on a semilog scale, to facilitate comparison of values obtained at low concentrations. Because the log of zero is not defined, we were obliged to use a discontinuous x axis to do this. The far left portion of the x axis is thus plotted with a linear (ordinary) axis to accommodate the value x = zero.
Several of the self-assembled cationic peptides tested were considerably more efficient than SEVI in enhancing HIV-1 infection at very low peptide concentrations. This effect was most pronounced for the K4(FKFE)2 and K4(ChaKChaE)2 peptides (Fig. 5), which have a considerably greater charge/mass ratio at pH 7 than the PAP(248–286) peptide from which SEVI is derived (Table 1). Not only was virus infection enhanced at much lower concentrations of the self-assembled K4(FKFE)2 peptide than SEVI, but the peak magnitude of infection enhancement by the self-assembled K4(FKFE)2 peptide was also greater. At high concentrations, however, infection-enhancement by the K4(FKFE)2 peptide was reduced (Fig. 5).
Similar results were obtained when infection experiments were repeated using a 10-fold lower concentration of virus (Fig. S23). Consistent with data that have been reported for SEVI, the infection enhancing effects of the cationic SSA were more pronounced at low virus concentrations. This is evident by the fact that the peak level of HIV infection in the presence of the cationic SSA was reduced by only approximately twofold when using this low virus concentration, even though the virus inoculum had been reduced by 10-fold, when compared to our standard experimental conditions.
One possible explanation for this concentration-dependent reduction in infection enhancement by K4(FKFE)2 might be cellular toxicity at high concentrations. To address this concern, we treated CEMx M7 cells with varying concentrations of our self-assembled peptides for 24 h, and then examined their viability using a resazurin-based cell viability assay (alamarBlue assay; Invitrogen). None of the fibrils demonstrated toxicity to the target cells in vitro at the concentrations used, when assayed over a 24 h incubation period (Fig. S24 A). Prolonged (72 h) incubation of cells with high concentrations of the K4(FKFE)2 and K4(ChaKCheE)2 peptides did, however, result in a modest decline in cell viability (Fig. S24 B).
We next examined whether SSA formed by the K4(FKFE)2 peptide might competitively inhibit the attachment of virus:SSA complexes to target cells. To do this, target cells were pretreated with increasing concentrations of self-assembled K4(FKFE)2 and then exposed to preformed complexes of HIV-1 virions plus either K4(FKFE)2 or SEVI. Pretreating the target cells with soluble, self-assembled K4(FKFE)2 resulted in a dose-dependent decrease in the efficiency of infection by both SEVI:virus and K4(FKFE)2:virus complexes (Fig. 6 A). These results suggest that free cationic SSA in solution are able to compete with fibril:virus complexes for attachment to target cells. It also suggests that SEVI and the self-assembled K4(FKFE)2 peptide may compete for attachment to the same (limiting) cell surface molecules (19).
Figure 6.
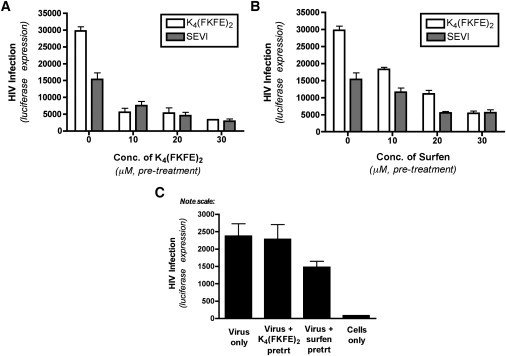
Cationic supramolecular assemblies enhance HIV-1 infection through a mechanism similar to SEVI. (A and B) CEMx M7 cells were pretreated with increasing concentrations of K4(FKFE)2 (A) or Surfen (B) for 30 min before being infected with HIV-1 in the presence of SEVI (10 μM) or K4(FKFE)2 (2 μM). At 48 h post-infection, cells were harvested and luciferase expression measured as a readout of HIV-1 infection. Results represent mean values from three experimental replicates; error bars denote the standard deviation of these values. The results shown are representative of three independent experiments performed with a range of pretreatment concentrations, all yielding similar results. (C) CEMx M7 cells were pretreated with a high concentration of K4(FKFE)2 (30 μM) or Surfen (30 μM) for 30 min being infected with HIV-1 alone, in the absence of any enhancing agent. At 48 h post-infection, cells were harvested and luciferase expression measured as a readout of HIV-1 infection. Results represent mean values from three experimental replicates; error bars denote the standard deviation of these values.
To examine this prediction further, we took advantage of previous results, showing that the small-molecule heparan sulfate antagonist, Surfen, can abrogate SEVI-mediated enhancement of HIV-1 infection (19). We pretreated our target cells with increasing concentrations of Surfen and subsequently exposed the cells to preformed complexes of HIV-1 virions plus either K4(FKFE)2 or SEVI. Surfen pretreatment resulted in a dose-dependent decrease in the efficiency of infection by both SEVI:virus and K4(FKFE)2:virus complexes (Fig. 6 B). This finding further supports the hypothesis that SEVI and the self-assembled K4(FKFE)2 peptide may compete for attachment to the same (limiting) cell surface molecules (19).
We also tested whether pretreatment of target cells with K4(FKFE)2 or Surfen had any affect on unenhanced HIV-1 infection. K4(FKFE)2 exerted no effect on unenhanced HIV-1 infection, and Surfen exerted only a slight inhibitory effect on unenhanced HIV-1 infection, even at a very high concentration (30 μM; Fig. 6 C).
Because cationic charge is essential for HIV-1 infection enhancement both by SEVI (20) and by our self-assembling, amphipathic peptides (Fig. 5), we reasoned that SSA formed by our anionic peptides might be capable of effectively shielding the charged surface of SEVI fibrils and thereby blocking SEVI-mediated infection-enhancement. To test this prediction, we infected target cells with preformed SEVI:virus complexes in the presence of increasing concentrations of our self-assembled anionic peptides. All of the anionic peptides demonstrated at least a modest ability to interfere with SEVI-mediated infection enhancement (Fig. 7 A), but the self-assembled E4(ChaKChaE)2 peptide was the most efficient at inhibiting SEVI-mediated enhancement of HIV-1 infection—completely abrogating SEVI's effects at high concentrations (20 μM). Importantly, the self-assembled E4(ChaKChaE)2 peptide was nontoxic to our CEMx M7 target cells (Fig. S24) and had no effect on the efficiency of HIV-1 infection in the absence of SEVI (Fig. 7 B). These data suggest the possibility that anionic peptide supramolecular assemblies have the potential to be developed into anti-SEVI microbicides.
Figure 7.
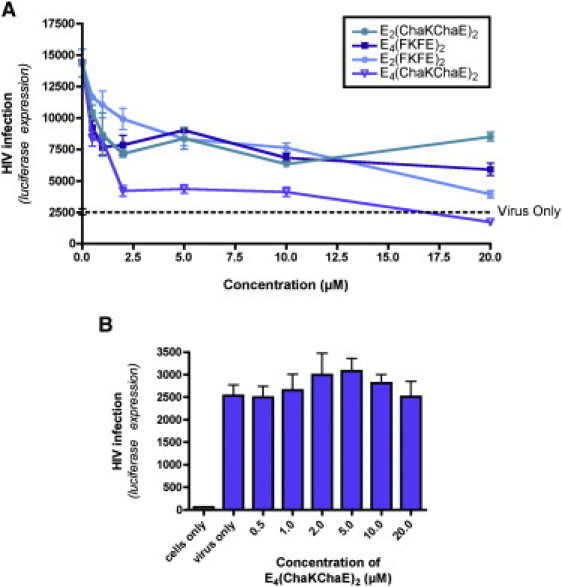
Anionic supramolecular assemblies have a limited capacity to inhibit SEVI-mediated enhancement of HIV-1 infection. (A) SEVI fibrils (5 μM) were allowed to form a complex with HIV-1 virions for 10 min at room temperature. The anionic peptide supramolecular assemblies were then added to the SEVI:virus solution and incubated for 10 min. This solution was then incubated on CEMx M7 cells. After 48 h, the cells were harvested and luciferase expression measured as a readout of HIV-1 infection. (B) HIV-1 infections were performed on CEMx M7 cells in the presence of increasing concentrations of E4(ChaKChaE)2 alone, in the absence of SEVI. Results in both panels represent mean values from three experimental replicates; error bars denote the standard deviation of these values. The results shown are representative of three independent experiments performed with a range of anionic peptide concentrations all yielding similar results.
Conclusions
SEVI is a naturally occurring insoluble amyloid fibril derived by self-assembly of a cationic peptide (PAP(248–286)), which has the ability to strongly enhance HIV-1 infection (1) and infection of other enveloped retroviruses (21,22). Consistent with this, filtration of semen results in a substantial reduction in its HIV-1 enhancing activity (1). However, other findings suggest that soluble supramolecular assemblies of the PAP(248–286) peptide may also contribute to HIV-1 infection enhancement (1,2,23). We therefore set out to examine the ability of SSA to enhance HIV-1 infection.
By generating non-SEVI, amyloidlike fibrils that do not sediment we were able to conclude that cationic SSA are capable of efficiently enhancing HIV-1 infection. This is consistent with the ability of other cationic polymers (such as polybrene) to enhance HIV-1 infection, by reducing the electrostatic repulsion between the virion and target cell surface (24–26)—thereby enhancing receptor-independent adsorption (27)—and accelerating the sedimentation velocity of virus particles by permitting their aggregation (26). For these cationic polymers, the net positive charge relative to the molecular weight of the polymer (26) directly influences the degree of retroviral infection enhancement, by determining the extent of polycationic bridging and the size of virus:polymer complex formed (thereby influencing virus sedimentation rate) (26,28). This may explain why the charge/mass ratio of our peptides seems to generally predict their effectiveness at enhancing HIV-1 infection.
We also determined that the presence of additional charge at the N-terminus of the peptides is able to frustrate the assembly process; however, we were able to overcome Coulombic interactions by increasing the ionic strength of the solution. Additionally, in the case of the fibrils formed from E4(FKFE)2, we found that the assembly process was dynamic and upon addition of NaOH without extra NaCl, we were able to perturb the assembly and the fibrils disassembled as a function of pH.
It remains unclear how the size and charge/mass ratio of cationic SSA or insoluble SEVI fibrils may influence the migration and sedimentation of attached HIV-1 particles in biological fluid such as human cervicovaginal mucus (CVM) (29,30). It is possible that insoluble fibrils may be better able to promote sedimentation of attached HIV-1 virions—thereby accelerating virus infection. However, it is also possible that very large virus:fibril complexes may become trapped by the mucin network found in CVM. The spacing between these mucin fibers creates a mesh of pores with a size range of 50–1800 nm (and an average size of 340 ± 70 nm) (30), which may be sufficient to allow free migration/sedimentation of complexes between HIV-1 virions and smaller SSA, but not of large complexes between HIV-1 virions and insoluble fibrils that are several microns in length. Future experiments will be necessary to compare the interaction of these different types of virus:fibril/SSA complexes with CVM.
Not only was the charge/mass ratio of our subunit peptides critical for enhancing HIV-1 infection, but so was their ability to form a self-assembled β-sheet structure—as evidenced by the inability of K2(AKAE)2 to enhance HIV infection despite its cationic charge (Fig. 4). The contribution of the β-sheet structure to HIV-1 infection enhancement is further supported by the ability of the amyloid-binding small molecule BTA-EG6, which intercalates into the β-sheet structure, to inhibit both SEVI- and semen-mediated enhancement of HIV-1 (10).
There are also interesting differences in the effect on infectivity enhancement by the Cha- and Phe-containing peptide sequences. The K4(FKFE)2 sequence more strongly promoted infection than the K4(ChaKChaE)2 peptide in the CEMx M7 cells. This may reflect the increased solubility of the Phe peptide relative to the Cha peptide as evidenced by slight cloudiness in solutions of the Cha peptide.
In our HIV-1 infectivity assays, preformed peptide structures were diluted in RPMI cell culture medium, which contains 103 mM NaCl. Direct assessment of the structure of the peptide assemblies in RPMI medium is technically difficult using IR or CD, due to the contents of the RPMI medium. However, we can infer from our functional (HIV-1 infection enhancement) data, that the previously self-assembled cationic peptides maintained their supramolecular assemblies in the RPMI medium.
In contrast, the unexpected inability of the self-assembled E4(FKFE)2 peptide to inhibit SEVI-mediated enhancement of HIV-1 infection suggests that this SSA may disassemble after being diluted in RPMI media. This would be consistent with the high salt requirement for initial self-assembly of this particular peptide (∼500 mM NaCl). On the other hand, the E4(ChaKChaE)2 peptide self-assembled efficiently in the absence of salt, and presumably was able to maintain its β-sheet content in the RPMI media—allowing it to efficiently inhibit SEVI-mediated enhancement of HIV-1 infection in vitro.
The ability of the self-assembled E4(ChaKChaE)2 peptide to abrogate SEVI-mediated enhancement of HIV-1 infection, without altering HIV-1 infectivity in the absence of SEVI, suggests that this and other anionic peptide supramolecular assemblies may have potential as anti-SEVI microbicides. A future direction will be to test whether anionic SSA can inhibit SEVI-mediated enhancement in the presence of semen. This is important because the microbicidal properties of other anionic polymers are greatly reduced in the presence of semen (31,32).
In summary, the results reported here show that amphipathic, cationic peptides of sequence Ac-Kn(XKXE)y-NH2 can efficiently self-assemble into soluble, fibril-like structures that, in some cases, were able to enhance HIV-1 infection even more efficiently than SEVI. This demonstrates that formation of insoluble, sedimentable fibrils is not a prerequisite for HIV-1 infection enhancement. Finally, our data also show that self-assembling anionic peptides can abrogate SEVI-mediated enhancement of HIV-1 infection, and thus have the potential to function as anti-SEVI microbicides.
Acknowledgments
We gratefully acknowledge the Open Chemical Repository in the Developmental Therapeutic Program at the National Cancer Institute for providing Surfen, and Dr. Nathaniel Landau (New York University School of Medicine) for providing CEMx M7 cells.
This work was supported by the following grants from the National Institutes of Health: No. R01AI084111 (to S.D., B.N., and D.E.) and No. T32AI049815 (to D.E.), as well as by a DuPont Young Professors Award (to B.N.) and a Creative and Novel Ideas in HIV Research (CNIHR) award (to B.N.). The mass spectroscopy facility in the Department of Chemistry was partially supported by a grant from the National Science Foundation (No. CHE-0840410).
Contributor Information
Stephen Dewhurst, Email: stephen_dewhurst@urmc.rochester.edu.
Bradley L. Nilsson, Email: nilsson@chem.rochester.edu.
Supporting Material
References
- 1.Münch J., Rücker E., Kirchhoff F. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell. 2007;131:1059–1071. doi: 10.1016/j.cell.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 2.Ye Z., French K.C., Makhatadze G.I. Mechanism of fibril formation by a 39-residue peptide (PAPf39) from human prostatic acidic phosphatase. Biochemistry. 2009;48:11582–11591. doi: 10.1021/bi901709j. [DOI] [PubMed] [Google Scholar]
- 3.Zhang S.G., Holmes T., Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. USA. 1993;90:3334–3338. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marini D.M., Hwang W., Kamm R.D. Left-handed helical ribbon intermediates in the self-assembly of a β-sheet peptide. Nano Lett. 2002;2:295–299. [Google Scholar]
- 5.Bowerman C.J., Ryan D.M., Nilsson B.L. The effect of increasing hydrophobicity on the self-assembly of amphipathic β-sheet peptides. Mol. Biosyst. 2009;5:1058–1069. doi: 10.1039/b904439f. [DOI] [PubMed] [Google Scholar]
- 6.Caplan M.R., Moore P.N., Lauffenburger D.A. Self-assembly of a β-sheet protein governed by relief of electrostatic repulsion relative to van der Waals attraction. Biomacromolecules. 2000;1:627–631. doi: 10.1021/bm005586w. [DOI] [PubMed] [Google Scholar]
- 7.Hamley I.W. Peptide fibrilization. Angew. Chem. Int. Ed. Engl. 2007;46:8128–8147. doi: 10.1002/anie.200700861. [DOI] [PubMed] [Google Scholar]
- 8.Wang K., Keasling J.D., Muller S.J. Effects of the sequence and size of non-polar residues on self-assembly of amphiphilic peptides. Int. J. Biol. Macromol. 2005;36:232–240. doi: 10.1016/j.ijbiomac.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Hwang W.M., Marini D.M., Zhang S.Q. Supramolecular structure of helical ribbons self-assembled from a β-sheet peptide. J. Chem. Phys. 2003;118:389–397. [Google Scholar]
- 10.Olsen J.S., Brown C., Dewhurst S. Amyloid binding small molecules efficiently block SEVI and semen mediated enhancement of HIV-1 infection. J. Biol. Chem. 2010 doi: 10.1074/jbc.M110.163659. DOI:jcb.M110.163659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manavalan P., Johnson W.C. Sensitivity of circular-dichroism to protein tertiary structure class. Nature. 1983;305:831–832. [Google Scholar]
- 12.Manning M.C., Illangasekare M., Woody R.W. Circular dichroism studies of distorted α-helices, twisted β-sheets, and β-turns. Biophys. Chem. 1988;31:77–86. doi: 10.1016/0301-4622(88)80011-5. [DOI] [PubMed] [Google Scholar]
- 13.Dong H., Paramonov S.E., Hartgerink J.D. Self-assembly of multidomain peptides: balancing molecular frustration controls conformation and nanostructure. J. Am. Chem. Soc. 2007;129:12468–12472. doi: 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- 14.Fauchère J.L., Charton M., Pliska V. Amino acid side chain parameters for correlation studies in biology and pharmacology. Int. J. Pept. Protein Res. 1988;32:269–278. doi: 10.1111/j.1399-3011.1988.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 15.Stuart B., Ando D.J. Published on behalf of ACOL (University of Greenwich) by John Wiley; Chichester and New York: 1997. Biological Applications of Infrared Spectroscopy. [Google Scholar]
- 16.Jackson M., Mantsch H.H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 1995;30:95–120. doi: 10.3109/10409239509085140. [DOI] [PubMed] [Google Scholar]
- 17.Kubelka J., Keiderling T.A. Differentiation of β-sheet-forming structures: ab initio-based simulations of IR absorption and vibrational CD for model peptide and protein β-sheets. J. Am. Chem. Soc. 2001;123:12048–12058. doi: 10.1021/ja0116627. [DOI] [PubMed] [Google Scholar]
- 18.Hiramatsu H., Kitagawa T. FT-IR approaches on amyloid fibril structure. Biochim. Biophys. Acta. 2005;1753:100–107. doi: 10.1016/j.bbapap.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Roan N.R., Sowinski S., Greene W.C. Aminoquinoline Surfen inhibits the action of SEVI (semen-derived enhancer of viral infection) J. Biol. Chem. 2010;285:1861–1869. doi: 10.1074/jbc.M109.066167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roan N.R., Münch J., Greene W.C. The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J. Virol. 2009;83:73–80. doi: 10.1128/JVI.01366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong S., Klein E.A., Silverman R.H. Fibrils of prostatic acid phosphatase fragments boost infections with XMRV (xenotropic murine leukemia virus-related virus), a human retrovirus associated with prostate cancer. J. Virol. 2009;83:6995–7003. doi: 10.1128/JVI.00268-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wurm M., Schambach A., Horn P.A. The influence of semen-derived enhancer of virus infection on the efficiency of retroviral gene transfer. J. Gene Med. 2010;12:137–146. doi: 10.1002/jgm.1429. [DOI] [PubMed] [Google Scholar]
- 23.Brender J.R., Hartman K., Ramamoorthy A. Helical conformation of the SEVI precursor peptide PAP248-286, a dramatic enhancer of HIV infectivity, promotes lipid aggregation and fusion. Biophys. J. 2009;97:2474–2483. doi: 10.1016/j.bpj.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coelen R.J., Jose D.G., May J.T. The effect of hexadimethrine bromide (polybrene) on the infection of the primate retroviruses SSV 1/SSAV 1 and BaEV. Arch. Virol. 1983;75:307–311. doi: 10.1007/BF01314897. [DOI] [PubMed] [Google Scholar]
- 25.Toyoshima K., Vogt P.K. Enhancement and inhibition of avian sarcoma viruses by polycations and polyanions. Virology. 1969;38:414–426. doi: 10.1016/0042-6822(69)90154-8. [DOI] [PubMed] [Google Scholar]
- 26.Davis H.E., Rosinski M., Yarmush M.L. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophys. J. 2004;86:1234–1242. doi: 10.1016/S0006-3495(04)74197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis H.E., Morgan J.R., Yarmush M.L. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys. Chem. 2002;97:159–172. doi: 10.1016/s0301-4622(02)00057-1. [DOI] [PubMed] [Google Scholar]
- 28.Landazuri N., Krishna D., Le Doux J.M. Retrovirus-polymer complexes: study of the factors affecting the dose response of transduction. Biotechnol. Prog. 2007;23:480–487. doi: 10.1021/bp060336y. [DOI] [PubMed] [Google Scholar]
- 29.Olmsted S.S., Padgett J.L., Cone R.A. Diffusion of macromolecules and virus-like particles in human cervical mucus. Biophys. J. 2001;81:1930–1937. doi: 10.1016/S0006-3495(01)75844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai S.K., Wang Y.Y., Hanes J. Nanoparticles reveal that human cervicovaginal mucus is riddled with pores larger than viruses. Proc. Natl. Acad. Sci. USA. 2010;107:598–603. doi: 10.1073/pnas.0911748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel S., Hazrati E., Keller M.J. Seminal plasma reduces the effectiveness of topical polyanionic microbicides. J. Infect. Dis. 2007;196:1394–1402. doi: 10.1086/522606. [DOI] [PubMed] [Google Scholar]
- 32.Keller M.J., Mesquita P.M.M., Herold B.C. Postcoital bioavailability and antiviral activity of 0.5% PRO 2000 gel: implications for future microbicide clinical trials. PLoS ONE. 2010;5:e8781. doi: 10.1371/journal.pone.0008781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MARVIN. 2010. MarvinSketch 5.3.6. ChemAxon. www.chemaxon.com. Accessed February 4, 2011.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.