

Abstract
It is well known that nucleic acids play an essential role in living organisms because they store and transmit genetic information and use that information to direct the synthesis of proteins. However, less is known about the ability of nucleic acids to bind specific ligands and the application of oligonucleotides as molecular probes or biosensors. Oligonucleotide probes are single-stranded nucleic acid fragments that can be tailored to have high specificity and affinity for different targets including nucleic acids, proteins, small molecules, and ions. One can divide oligonucleotide-based probes into two main categories: hybridization probes that are based on the formation of complementary base-pairs, and aptamer probes that exploit selective recognition of nonnucleic acid analytes and may be compared with immunosensors. Design and construction of hybridization and aptamer probes are similar. Typically, oligonucleotide (DNA, RNA) with predefined base sequence and length is modified by covalent attachment of reporter groups (one or more fluorophores in fluorescence-based probes). The fluorescent labels act as transducers that transform biorecognition (hybridization, ligand binding) into a fluorescence signal. Fluorescent labels have several advantages, for example high sensitivity and multiple transduction approaches (fluorescence quenching or enhancement, fluorescence anisotropy, fluorescence lifetime, fluorescence resonance energy transfer (FRET), and excimer-monomer light switching). These multiple signaling options combined with the design flexibility of the recognition element (DNA, RNA, PNA, LNA) and various labeling strategies contribute to development of numerous selective and sensitive bioassays. This review covers fundamentals of the design and engineering of oligonucleotide probes, describes typical construction approaches, and discusses examples of probes used both in hybridization studies and in aptamer-based assays.
Figure.
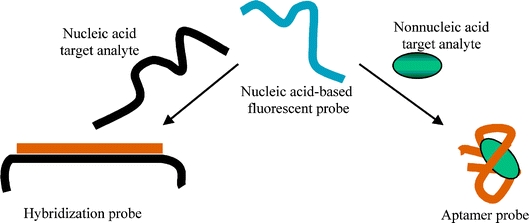
Hybridization with a nucleic acid target or affinity interactions with a nonnucleic acid target generate changes in the fluorescence characteristics of a nucleic acid-based fluorescent probe
Keywords: Bioanalytical methods; Bioassays; Biosensors; Chemical sensors; Fluorescence/luminescence; Nucleic acids (DNA, RNA)
Introduction
Fluorescent biosensors enable highly sensitive and selective detection of many target bioanalytes [1]. With the recent advent of DNA probe technology, a variety of fluorescent biosensors that exploit nucleic acids as bioreceptors have been engineered. The development of sensitive and selective sensors based on nucleic acid fragments has become a very active research field in recent years, because of their numerous applications including detection and visualization of DNA and RNA in genomic analyses, monitoring amplification progress in quantitative PCR (QPCR) assays, and detection of nonnucleic acid analytes (proteins, small molecules, metal ions) with aptamer-based sensors [2, 3].
In general, these probes are synthetic DNA or RNA molecules (with a sequence designed for a specific target molecule) containing a reporter group that can be monitored using fluorescence spectroscopy. The chemistry of nucleic acids is well-known, and their assembly into probe oligonucleotides can be achieved easily, by use of commercially available synthesizers. A simple probe of this kind labels its target by binding, because of specific probe–target interactions (hybridization, molecular recognition). Alteration of the fluorescence characteristics of reporter group(s) is exploited as an analytical signal.
The objective of this review is to introduce oligonucleotide-based fluorescent probes as versatile biorecognition tools to detect or monitor various biological important targets. Subsequent sections provide an overview of the design approaches, applications, and characterization of fluorescent oligonucleotide sensors.
Principles of biosensing
Oligonucleotide-based fluorescent probes can be regarded as typical examples of biosensor with the general layout shown in Fig. 1, consisting of a biologically relevant molecular recognition element integrated to a signal-transduction element.
Fig. 1.

Schematic layout of a fluorescent biosensor
Nucleic acid fragments are used as recognition element but, depending on the recognition mechanism (target–probe interactions), one can distinguish two categories of oligonucleotide biosensors. One group of probes is known as hybridization probes and the recognition interactions depend on complementary matching between target and probe molecules. Hybridization probes are widely used in many fields that require detection or monitoring of nucleic acids including gene microarrays, DNA chips, detection of gene translocation, monitoring intracellular mRNA and amplification progress in real-time PCR [4–12]. Sensors belonging to the second group are known as aptamer probes; these can bind nonnucleic acid analytes acting as three-dimensional bioreceptors. Applications of aptamer sensors include detection and monitoring of proteins, small bioactive molecules, or even metal cations [3, 13, 14]. Transduction of the analyte–bioreceptor binding into an analytical signal is achieved by attachment of one or more fluorescent groups to the oligonucleotide chain. Successful design of the fluorescent probe is apparent from advantageous changes in the fluorescence properties of labels (fluorescence quantum yield, lifetime, or other fluorescence characteristic) resulting from binding of the target analyte.
Recognition element
The natural nucleic acids (DNA, RNA) are generally used as recognition elements but some of their synthetic analogues, for example the peptide nucleic acids (PNA) and the locked nucleic acids (LNA) have recently been proposed [15–20]. PNAs are DNA mimics in which a peptide-like repeat of the (2-aminoethyl)glycine unit replaces the sugar–phosphate backbone [15, 16]. PNAs recognize and bind to their complementary nucleic acid sequences with higher thermal stability and specificity than the corresponding oligodeoxyribonucleotides; this was exploited to develop Light-up probes for microchip applications and detection of PCR products [17]. Locked nucleic acids, defined as oligonucleotides containing at least one LNA monomer, are a new class of bicyclic high-affinity DNA analogues [18]. The LNA monomer contains a natural phosphodiester linkage, but its sugar is conformationally locked by an O2′ to C4′ methylene linkage. LNAs are capable of recognizing complementary DNA and RNA with increased mismatch discrimination compared with natural nucleic acids that enable full control of the melting point of the hybridization reaction [18]. With these unique properties, LNAs have found widespread applications in biotechnology (drugs, primers) and in bioanalysis as effective detection probes. They are particularly well suited to SNP (single nucleotide polymorphism) genotyping microarrays, QPCR (quantitative polymerase chain reaction), and FISH (fluorescence in-situ hybridization) assays [19, 20].
Although the nucleic acid sequences for hybridization probes are related to the genomic DNA or mRNA, the aptamer sequences may vary freely and depend on the target analytes. Aptamers that are able to bind desired target molecules selectively are generated from a random library of 1015–1018 candidates by in-vitro selection known as the SELEX (systematic evolution of ligands by exponential enrichment) technique [21, 22].
Fluorescence transduction
With progress in automated DNA synthesis and phosphoroamidite chemistry, it has become possible to attach fluorophores covalently to an oligonucleotide of interest. Different fluorescent dyes have been used to engineer oligonucleotide probes, for example fluorescein, TAMRA, Cy dyes, Texas red, HEX, JOE, Oregon green, rhodamine 6 G, coumarin, pyrene, and others. Additionally, in fluorescence quenching approaches, quencher molecules have also been covalently attached to the fluorescent oligonucleotides. Dimethylaminophenylazobenzoic acid (DABCYL) has been widely used as a universal quencher for many fluorophores [23]. The fluorophores and quenchers may be easily chosen for the desired probe application. However, for successful rational design of oligonucleotide probes some other considerations must be taken into account, for example the photostability of the fluorophore, the pH-dependence of quantum yield, the nature and length of a linker, and the position of the fluorophore attachment (end vs. internal modification) [24–26]. Dye–nucleotide interactions were reported to be responsible for the sensitivity of fluorescent conjugates to environmental conditions and it was proposed that photoinduced electron transfer between dye and guanosine is crucially important in the process [25–29].
The fluorophores used for oligonucleotide labeling can be divided into two classes:
dyes that change their fluorescence properties on binding nucleic acids, used mainly in single-labeled probes, and
fluorophores with intrinsically strong fluorescence, for example fluorescein and rhodamine derivatives, which through structural design are brought into contact with each other or with a quencher molecule.
The mechanism of signal transduction for single-labeled probes may rely on several different processes including the above mentioned electron transfer quenching or dequenching with the participation of nucleobases, fluorescence enhancement as result of energy transfer from nucleobases to the acceptor [30], or steric restrictions imposed on the fluorophore that operate in Light-up probes [17]. Fluorescence intensity-based transduction, however, suffers from the sensitivity of the analytical signal to matrix effects and assay conditions. To overcome these problems, more advanced fluorescence techniques have been used, for example fluorescence polarization or anisotropy [31, 32] and fluorescence lifetime [33, 34].
Transduction processes responsible for the performance of probes containing two dyes may rely on fluorescence quenching (static or dynamic mechanisms), fluorescence resonance energy transfer (FRET), and monomer–excimer emission switching with pyrene fluorophores. Energy transfer, quenching, and excimer formation processes are distance-dependent, thus structural rearrangement of the probe upon binding to the target that changes the distance between the attached fluorophores should result in an alteration of the fluorescence characteristics of the system.
Fluorescence resonance energy transfer
Fluorescence resonance energy transfer (FRET) is a spectroscopic method that provides distance information about macromolecules in solution and is particularly suited to analysis of structural changes in proteins and nucleic acids [35]. In a typical FRET experiment, a biopolymer is labeled with two different fluorophores, a donor and an acceptor, covalently attached at different locations. Interactions between the electronic excited states of these dye molecules lead to the transfer of excitation energy in a nonradiative process from the donor molecule to the acceptor molecule. The occurrence of FRET depends upon such conditions as the distance between the donor and acceptor fluorophores (typically 10–80 Å), the spectral overlap between the donor emission and acceptor absorption spectra, and the orientation of transition moments of both fluorophores. [35, 36]. The efficiency (E) of FRET can be measured by looking at the decrease in the fluorescence (or lifetime) of the donor or at the increase in the fluorescence of the acceptor. Förster showed that FRET efficiency depended on the inverse sixth power of the distance (R) between the two fluorophores (Eq. 1) and this is the basis of the use of the technique to provide structural information [36].
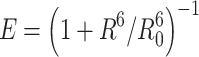 |
1 |
where R 0 is the characteristic Förster radius for a given donor–acceptor pair, which is given by:
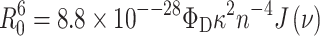 |
2 |
where Φ D is the fluorescent quantum yield of the donor in the absence of the acceptor, κ 2 is a term that depends on the relative orientation of the donor and acceptor transition moments, n is the refractive index of the medium, and J(ν) is the spectral overlap between the donor emission and acceptor absorption spectra. From Eq. 1 it is clear that for R = R 0, the efficiency of FRET is 50%.
Detection and the quantification with FRET can be accomplished in a number of ways. If the acceptor molecule is fluorescent, the ratio of the quenched fluorescence of the donor to the sensitized fluorescence of the acceptor can be used to detect FRET. This method has the advantage that the signal ratio is independent of the absolute concentration of donor and acceptor, in contrast with the approach with a quencher as the acceptor, where knowledge of absolute concentrations is required. FRET techniques have been used in a number of studies focused on oligonucleotide probes. Two design strategies are commonly used, the approach in which both energy transfer partners are attached to the same oligonucleotide at the 3′ and 5′ termini (e.g., molecular beacons (MB), hydrolysis probes) and the two-oligonucleotide strategy (binary probes, BP), used mainly in hybridization assays, in which two short oligonucleotides are single-labeled with the donor and acceptor fluorophores. The BP and MB probes will be described in detail in subsequent sections.
Excimer–monomer switching
It is well known that an excited pyrene molecule can form an excited-state weak association complex with another ground-state pyrene, forming an excimer [36, 37]. This excimer has a broad red-shifted emission band near 480 nm, compared with the structured emission of the pyrene monomer at 400 nm. A properly designed oligonucleotide probe will switch between excimer and monomer emissions depending on the presence of analyte target (free and bound states of the probe). Labeling of the oligonucleotide probe with pyrene can be carried out in a variety of ways, including simple end-labeling via a linker [12, 38–40], a 2′ sugar modification [41], attachment of pyrene to the nucleobase [42, 43], incorporation of bis-pyrene label [44–46], and by arraying multiple pyrene residues along the DNA backbone [47].
Another important photophysical property of pyrene is its relatively long fluorescence lifetime approaching 100 ns in aqueous solution [37]. Moreover, the excimer emission is also long-lived [48]; this can be exploited in a time-resolved emission spectra (TRES) approach. Short-lived autofluorescence background of biological samples can be eliminated using TRES that greatly improves signal to background (S/B) ratio [39].
Hybridization probes
Detection of DNA hybridization is widely used in molecular biology, genetics, and medical diagnostics. The most frequently studied systems are related to the polymerase chain reaction (PCR), microarray technology, and fluorescence in-situ hybridization (FISH). There are many formats for hybridization assays. All approaches require at least one analytical oligonucleotide (probe) with complementary sequence to the oligonucleotide analyte, also referred to as the oligonucleotide target. Many hybridization formats are heterogeneous with a probe or an analyte attached to a solid support. This facilitates the separation of hybridized and unhybridized oligonucleotides. These assays do not need the fluorescence characteristics of the label to be altered by the hybridization between probe and target, because excess reagent is usually washed out. Unfortunately, the requirement that the unhybridized probe has to be removed by washing, precludes the use of this format for on line monitoring of hybridization. The solid-state-supported approach is widely used in DNA chips, microarrays, and DNA sensor technology and will be not considered in this review.
The homogeneous, or solution-phase, assays have the advantage that insoluble supports are absent (elimination of the problem with nonspecific adsorption) but rely on the strategy that requires alteration of the fluorescence properties of the label(s) on hybridization. Several design strategies have been used to detect hybridization with homogeneous assays. Some possible approaches are shown schematically in Fig. 2. The most important strategies are the binary probe (Fig. 2a, b) and molecular beacon (Fig. 2c, d) approaches.
Fig. 2.

Schematic representation of nucleic acid hybridization probes: quenching-type binary probe (a), FRET-type binary probe (b), quenching-type molecular beacon (c), and FRET-type molecular beacon (d)
Binary probes
An early homogeneous assay was reported by Heller and Morrison [49]. They used two probes complementary to the target oligonucleotide. The probes were single-labeled with fluorophores at the 3′ and 5′ ends. After hybridization to complementary oligonucleotide at the adjacent locations, the labels were close enough to enable strong fluorescence quenching (Fig. 2a). In another approach a competitive hybridization between a labeled DNA duplex probe and target DNA was exploited [50]. Two single-labeled probes on 3′ and 5′ termini were also used, but the probes were complementary to one another. Efficient static quenching of the hybridized probe duplex was observed when one strand was labeled with a fluorescence quencher. Competitive hybridization with the target DNA produced a fluorescence signal as a result of a dequenching process (quencher labeled strand dissociation).
Recent applications of binary probes rely mainly on the FRET approach [2, 5, 6, 51]. Typically, one oligonucleotide is labeled with a donor fluorophore and the other is labeled with an acceptor molecule (dyes are selected to form a FRET pair). In the absence of target DNA, the donor and acceptor strands are randomly distributed in solution, and, because their concentration is very low, the intermolecular FRET signal is not observed. On addition of target DNA, a hybrid is formed in which the acceptor and donor strands hybridize in the adjacent sites on the target (Fig. 2b). Close proximity of the donor and acceptor fluorophores results in generation of a FRET signal. In an ideal BP assay, selective excitation of the donor fluorophore enables observation of only donor fluorescence in the absence of target and strongly sensitized emission of the acceptor with quenched donor fluorescence when the target DNA is present. In practice, the acceptor emission is also present in the absence of target, because of direct excitation, which together with the limited FRET efficiency causes lowering of the signal/background ratio [5, 52]. The challenge of improving the S/B ratio has been addressed by constructing probes that rely on the three-fluorophore approach reported by Marti et al. [53]. Their probe system contains the FAM–TAMRA–Cy5 FRET triad. One probe is labeled with FAM and TAMRA separated from one another by four nucleotides; the second probe is labeled with Cy5. In the absence of target, excitation of FAM gives only TAMRA emission (efficient FRET); on hybridization of probes to the target, TAMRA and Cy5 are brought into proximity and FRET operates between TAMRA and Cy5, which results in the appearance of Cy5 fluorescence and a decrease in TAMRA emission. This relayed FRET results in good spectral separation (Stokes shift) between the primary donor (FAM) and the final acceptor (Cy5). Another way to improve the S/B ratio of binary probes is by application of pyrene labels that are able to produce excimer emission [12, 54, 55]. When the two probes are free in solution, the pyrene groups are separated from each other and pyrene monomer fluorescence is observed without a contribution from excimer emission. When the probes are hybridized to the target, the pyrene groups are close to one another, favoring the formation of the excimer. S/B for the probe in buffer is excellent, but in the presence of the protein extract the “autofluorescence” of the biological background results in disturbance, leading to a poor S/B ratio. A time-resolved emission spectra (TRES) approach, used instead of steady-state measurements, eliminated the background autofluorescence as the result of longer lifetime of pyrene emission [12].
Among other attempts to improve the sensitivity and selectivity of hybridization monitoring, one can mention the use of the pyrene–perylene FRET pair coupled to a 2′-amino-LNA oligonucleotide probe with clear distance-dependent FRET efficiency [56], or application of micellar medium (cetylpyridinium bromide) that enhanced FRET between FAM and TAMRA-labeled probes in a sandwich-type assay [57].
An advantage of binary probes is that they do not yield false positive or nonspecific signals similar to those observed for molecular beacons. There are, however, some disadvantages of binary probes which have stimulated intense research yielding novel and creative approaches. First, the entropy of a binary probe system decreases more when bound to the target (three independent species becoming one hybrid), which reduces the equilibrium stability of the hybrid [58]. Furthermore, the hybridization kinetics of binary probes is slower than that of MB because BP hybridization depends on the binding of two different components to the target. To challenge these limitations, two separate binary probes have been covalently linked with a poly(ethylene glycol) chain; this significantly improved the hybridization kinetics and the thermodynamic stability of the hybrid [59].
Molecular beacons
The most common DNA and RNA hybridization probes are molecular beacons (MB). The first MB, described by Tyagi et al. [4], was based on fluorescence quenching and complementary pairing principles. Figure 2c shows the general structure of the MB and its working principle. The classic MB is a fluorescently-labeled oligonucleotide comprising three parts:
a loop of 15–30 nucleotides, which can recognize a target molecule;
a stem of 5–8 bases, which dissociates during the hybridization of the MB with a target molecule; and
a fluorescent group connected to the 5′ end and a quenching group linked to the 3′ end of the MB.
Unhybridized MB has the structure of a hairpin with fluorophore and quencher probes placed close to each other. As a result, the fluorescence quenching or FRET occur between the fluorophore and the quencher. However, in the presence of the target, the probe region of the MB hybridizes to it, promoting the opening of the hairpin conformation and separating the fluorophore and the quencher from each other. The fluorophore fluorescence is not quenched after hybridization because of the separation of the fluorophore and quencher. This results in the appearance of fluorescence emission from the fluorophore only after the probe has selectively hybridized to the target. The substitution of quencher molecule with another fluorophore enables construction of FRET-based molecular beacons (Fig. 2d). FRET MBs have several advantages, for example the possibility of ratiometric analysis to improve the S/B ratio, and use of two-channel detection that enables monitoring of free and bound probe in cells [60]. Briefly, in the hairpin conformation, the two fluorophores (the donor and acceptor molecules) are close to each other; this leads to FRET between them, whereas on hybridization with the target, the donor and acceptor fluorophores become separated in space leading to strong, unquenched emission of the donor (FRET is not observed).
A variety of sensitive assays have been based on the structural changes of MBs in the presence of complementary nucleic acids. Because of their simplicity and high specificity, MBs have been used to detect SNPs and gene mutations, to quantify the concentration of a target molecule during real-time or quantitative PCR (Q-PCR), or to carry out intracellular imaging of nucleic acids [5–12, 61–74]. These applications involve both quencher-based and FRET MBs. A mixed quenching/FRET approach has been also proposed to improve the signal/background ratio for detection of mRNA in living cells [61, 62]. Using a pair of classical molecular beacons, one with a donor and the other with an acceptor fluorophore, that hybridize to adjacent regions on the same mRNA target, FRET-based monitoring of mRNA was achieved with an advantageous S/B ratio when compared with the single MB approach. Detection of the FRET signal significantly reduced false positives, leading to sensitive imaging of mRNAs in live cells [61].
Despite many obvious advantages and applications there are some disadvantages and limitations that are challenged by intense research. Most of the problems arise either from the complexity of living environments or the intrinsic properties of MBs. For instance, MBs can be easily degraded by intracellular enzymes or non-specifically opened by single-strand-binding proteins (SSBs) causing false-positive or false-negative signals. Application of non-standard nucleic acids (2′OMe-modified RNA, PNA, and LNA) was helpful in solving this problem [63–65]. Another advantage of replacing the natural nucleic acids with their analogs includes higher affinity, increased specificity, faster hybridization kinetics, and fluorescence background suppression, because of reduction of dynamic opening of stem structure [66]. For improvement of the MB signaling properties several different approaches have been reported. One strategy is to apply brighter fluorophores, for example, quantum dots or organic polymers [67, 68]. Another approach used to increase S/B ratio relies on application of more effective metal quenchers (e.g., Au nanoparticles) or multiple quenchers [69, 70]. For example, use of MB with fluorescein and multiple DABCYL quencher resulted in 14, 81, and 320-fold signal enhancement as the number of quenchers was increased from one to three, respectively [70]. For FRET-based MBs, it is, in practice, difficult to eliminate a direct excitation of the acceptor at the donor excitation wavelength or to avoid an overlap between emission spectra of the donor and the acceptor. Tailoring new FRET fluorophores [75–77] and modifications of the “classical” architecture of MBs [78–81] have enabled enhancement of detection capabilities with better S/B ratio and lower LOD values. Comprehensive papers have been published recently that discuss and compare FRET conditions for numerous donor–acceptor pairs of organic fluorophores [75] and quantum dots [66, 76]. Quantum dots (QDs) are especially well suited to FRET applications. The broad excitation spectra and large absorption cross section of QDs, combined with the narrow excitation spectra of acceptor dyes, enable one to choose the most appropriate excitation wavelength to reduce unwanted direct excitation of the acceptor and thus increase FRET efficiency. Moreover, because the emission spectrum of the QD donor is narrow, symmetric, and has no red tail, the overlap with the fluorescence spectrum of the acceptor is minimized. FRET efficiency can also be enhanced because multiple acceptors can interact at the surface of a single QD [76]. One such example made use of two probes, a reporter probe labeled with an organic fluorophore and a capture probe labeled with biotin. In the presence of the target DNA, these two probes hybridized as a sandwich hybrid that could be captured by streptavidin-coated QDs [82]. This nanoassembly enabled FRET to occur from the QD to the fluorophores in close proximity, and the system had major advantages such as amplified or multiple FRET response (colocalization of two fluorescent signals) or reduced S/B ratio (unhybridized probes do not participate in FRET background). Performance of the QD-labeled MB can be further improved by the presence of graphene oxide (GO) [83, 84]. The ability of GO to differentiate hairpin and dsDNA structures [85], is a new approach to discriminating between intact (free) MB and open (hybridized) MB forms. The strong interaction between free QD-labeled MB and GO led to the fluorescence quenching of QDs. On recognition of the target (double-stranded hybrid formation), the interaction between target-bound MB and GO became weaker, and the distance between the QDs and GO increased, which significantly hindered the FRET and, thus, increased the fluorescence of QDs [84]. Another promising improvement of MB performance relies on the application of the luminescent lanthanide chelates as MB labels. Recently, Krasnoperov et al. reported ultrasensitive detection of nucleic acids by MB labeled with Eu(III) chelates of quinolone derivatives [77]. Time-resolved acquisition of the luminescent signal that occurs on hybridization of the probe to the target enabled elimination of short-lived background fluorescence, significantly enhancing the sensitivity of detection, which was less than 1 pmol L−1 [77].
Another approach to improving MB sensitivity and S/B ratio was based on modification of MB platform by incorporation of the third fluorophore into the hairpin structure [78–81]. The MB system reported by Li et al. [81] consisted of three distinct fluorophores, FAM, TAMRA, and Cy5. FAM as the primary donor was attached to one arm of MB, whereas the primary energy acceptor/secondary donor (TAMRA) and the secondary acceptor (Cy5) were located at the other terminus of the MB. In the absence of target DNA or RNA, the MB exists in the stem-closed form. Excitation of FAM initiates an energy transfer cascade to TAMRA and further to Cy5 generating unique fluorescence signatures defined as the ratio of the emission from each of the three fluorophores. In the presence of the complementary target DNA, the MB opens and hybridizes with the target, separating FAM and the TAMRA–Cy5 pair by a large distance, so that energy transfer from FAM to TAMRA is blocked.
Beside disadvantages related to imperfect spectral characteristics of MB probes, there are other difficulties related to proper design of a stem sequence of MB. Its stability must be finely tuned to ensure close proximity of the dyes in the native form and, at the same time, enable efficient hybridization with the target. Additionally, MB should not take part in unintended target hybridization, which might affect selectivity, or interfere with formation of the hairpin structure, for example by binding to the stem sequence, which may also lead to an increase in background or loss of signal intensity. To avoid incomplete FRET or quenching in a hairpin form, Haner et al. reported MB design in which signal control is accomplished by formation of a donor–acceptor (D–A) complex [86]. The stem contains incorporated pairs of pyrene or perylenediimide moieties that can interact by interstrand stacking. In the native structure this leads to efficient quenching of pyrene fluorescence, whereas the hybridized form is characterized by an excimer signal produced from two adjacent moieties. Additionally, the formation of a stable D–A complex helps to minimize the number of natural bases in the stem, thus reducing the chances of unwanted base-pairing interactions.
The excimer monomer switching (EMS) approach with pyrene fluorophore has been widely used in developing MB probes. The first EMS MB was reported by Fujimoto et al. [38]. In this approach, two pyrene groups were attached at both termini of the MB. When the Py-MB is in the hairpin conformation, the pyrene groups are close to one another and excimer emission can be observed. In contrast, in the presence of the target, the pyrene groups are at opposite sites of the strand and only monomer emission is produced. An isoemissive point was observed indicating the presence of only two fluorescent species, the nonhybridized and hybridized probes. The EMS MB approach enhances the fluorescence output and enables a time-resolved signaling option for improved background discrimination. Several such probes have been synthesized; these differ in the chemistry used to attach pyrene to the oligonucleotide strand, the length of the linker chain, and the strand position where the pyrene group is attached [46, 87–90]. It has also been shown that the intensity of the excimer emission and base-pair mismatch discrimination can be enhanced by labeling one arm of the MB with a bis-pyrene group, contrary to the classical MB approach with pyrene attached to both termini of the MB. Strong excimer fluorescence was observed for MB labeled with bis-pyrene in the stem-loop form, whereas for the hybrid with a target monomer fluorescence was the major spectral component [46]. Conlon et al. [88] reported a multiple-labeled MB probe that signaled hybridization by quenching of pyrene excimer emission. The 5′ end of the MB was modified with 2–4 pyrene molecules using dendrimer linkers, and the quencher DABCYL was attached on the 3′ end. A stem-closed conformation resulted in quenching of the pyrene excimer fluorescence. In the presence of the target, the beacon switched to a stem-open conformation, which separated the pyrene labels from the quencher molecule and restored the excimer emission signal in proportion to the target concentration. Steady-state fluorescence or time-resolved assays enabled a subnanomolar detection of the target in buffer or in cell-growth medium, respectively. The excimer emission intensity could be scaled by increasing the number of pyrene monomers conjugated to the 5′ terminal [88].
An ethynylpyrene group was used by Kim et al. to modify the nucleobases adenosine (AP) and uridine (UP) [42, 89]. These modified nucleotides were incorporated into the middle positions of hairpin stem to form MB stabilized by stacking interactions between modified opposite bases [89]. The close location of the nucleobase (rigid ethynyl bridge) seriously affected pyrene spectral behavior, with UP being nonfluorescent and AP with much higher fluorescence quantum yield. Moreover, UP quenched fluorescence of AP when they formed a stacking complex (UPAP) within the MB stem, but excimer-like red-shifted emission was observed for the system with the APAP pair. Hairpins with the APAP and APUP pairs were proposed as efficient MB that change the fluorescence properties on forming their perfectly matched duplexes [89]. Another approach with pyrene moieties inserted into the stem of MB was described by Matsumoto et al. [90] who incorporated into the MB stem one or two guanosines conjugated with pyrene through C8 alkylamino linkers. These flexible connectors enabled the pyrene moieties to intercalate into the stem of the MB, which resulted in efficient quenching of the pyrene fluorescence in a stem-closed configuration (probably because of an electron-transfer mechanism [27]). This quencher-free MB probe emitted monomer or excimer emission after hybridization with the target, depending on the number of pyrene labels attached.
Other probes
Molecular beacons and binary probes are excellent tools for sequence-specific detection of nucleic acids. However, real-time monitoring of amplification reactions, as in quantitative polymerase chain reaction (QPCR) systems, sometimes require more sophisticated probe designs [11, 91]. The QPCR technique enables detection and quantification of minute numbers of copies of target nucleic acid sequences isolated from biological material. The basis of the PCR technique is the ability of DNA polymerase to extend an oligodeoxyribonucleotide primer that is specifically hybridized to a single-stranded DNA template. PCR consists of three thermally different steps that constitute one amplification cycle. Multiple rounds of thermal cycling result in exponential accumulation of PCR products. For example, in 30 thermal cycles 108-fold amplification of the target sequence can be achieved. There is a quantitative relationship between the number of target nucleic acid strands initially present in a sample and the number of product strands synthesized at any given thermal cycle. Therefore, real-time monitoring of amplification progress in closed tubes is required; this can be achieved by use of fluorescence-based hybridization probes or primers. Beside binary probes and molecular beacons widely used in QPCR analysis, a hydrolysis probe has been developed [92, 93] that is based on the inherent 5′ → 3′ exonuclease activity of the Taq DNA polymerase [94]. This hydrolysis probe (known also as a TaqMan probe) is a dual-labeled fluorogenic short oligonucleotide. One end is labeled with a donor, and an acceptor fluorophore is attached on the other end of the probe so that they interact with each other by FRET, because the oligonucleotide is short enough [93] (Fig. 3a). Probes that are free in solution transfer energy from the donor fluorophore to the acceptor, resulting in a low donor fluorescence signal (and/or FRET emission of the acceptor). The probe is designed to hybridize to the target strand at the same time as the PCR primer. When Taq DNA polymerase extends the primer, it encounters the probe, and, because of its 5′-nuclease activity, it cleaves the probe. Cleavage of the probe results in separation of the donor and the acceptor from each other, leading to an increase in the intensity of the fluorescence signal from the donor fluorophore. With each cycle of amplification, additional fluorophores are liberated from their probes and the increasing fluorescence intensity is monitored during the extension step of each PCR cycle. Another hydrolysis probe, a minor groove-binding (MGB) probe, has been reported [95]. MGBs are short oligonucleotides characterized by the conjugation of minor groove binders, for example dihydrocyclopyrroloindole, at one end. This chemical modification increases the stability of the hybridized probe and facilitates highly specific binding to the targeted sequence at the minor groove of the DNA helix [95].
Fig. 3.

Schematic representation of TacMan hydrolysis probe (a) and fluorescent primers Scorpions (b) and Amplifluor (c) used for monitoring QPCR amplification progress
Another class of QPCR hybridization probes contains single-labeled oligonucleotides, fluorescence of which is quenched by the oligonucleotide component of the probe by base–dye stacking and electron-transfer. When these probes hybridize to target sequences, there is an increase in probe fluorescence emission without a significant change in probe structure. The HyBeacon and Light-up probes are examples of such design. HyBeacons are single-stranded fluorescent oligonucleotides that lack a secondary structure, but the fluorophore is quenched by internal nucleotides [96]. When the probe binds to a nucleic acid, its conformation changes and, as a result, the fluorescence intensity of the fluorophore increases. Light-up probes consists of a thiazole orange derivative linked to a PNA oligomer [97]. On hybridization with a target a duplex structure is formed in which the thiazole orange moiety intercalates and, similarly to HyBeacons, the probe becomes brightly fluorescent. Because of their PNA backbone, Light-up probes are highly specific: a single base mismatch in the target sequence is sufficient to prevent probe binding, as indicated by the lack of fluorescence increase [98].
It is also possible to use QPCR detection systems based on modified primers that are able to generate fluorescence signals on target amplification. Examples of such detection systems include Scorpion primers and Amplifluor primers [99–101]. The Scorpions method developed by Whitcombe et al. [99] combines, within a single molecule, both a PCR primer and a sequence-specific probe (labeled with donor and acceptor FRET labels). These two sequence moieties are separated by a non-PCR amplifiable linker (Fig. 3b). In the absence of target, the donor fluorophore and the quencher (acceptor) are in close proximity, resulting in quenching of the fluorophore emission. When the probe sequence hybridizes to its target sequence, PCR occurs with the primer moiety and the probe loops forward and binds to the PCR product strand upstream of the primer, thus restoring fluorescence. This is a unimolecular reaction which occurs extremely quickly and efficiently, and is accompanied by the dissociation of a fluorophore–quencher pair. Nazarenko et al. developed similar labeled hairpin primers, called Amplifluor probes [100, 101], that are directly incorporated into amplification products (Fig. 3c). The hairpin structure contains a fluorophore and quencher moiety in the hairpin stem. Hairpin primers that are not incorporated into an amplification product do not generate a fluorescence signal, because the reporter and quencher stay in close proximity. During amplification, hairpin primers are incorporated into the double-stranded amplification products and generate a fluorescence signal because they are linearized by further amplification. Further simplification of this approach was achieved with single-labeled hairpin primers [102]. Similar to HyBeacons and Light-up probes, these fluorogenic primers contain a single fluorophore that is intrinsically quenched by nucleobases (mainly guanosine) in a hairpin form. They increase fluorescence intensity when incorporated into the double-stranded PCR products. All these systems are less expensive than probe-based detection formats and may provide an attractive alternative, especially if multiple targets have to be detected simultaneously by multiplex reactions [102, 103]. A disadvantage of this approach is that hairpin primers can generate “false” amplicons or primer–dimers, resulting in false-positive fluorescence signals.
Aptamer affinity probes
The flexibility offered by nucleic acids (stability, ease of chemical modifications and labeling) have stimulated innovative applications of these species in bioanalysis. It was demonstrated recently that, like antibodies, nucleic acids with defined sequences and structures can recognize specific nonnucleic acid targets. Such single-stranded nucleic acid sequences are called aptamers.
Selection of aptamers
Most aptamers are obtained by the SELEX method [21, 22, 104]. In a typical DNA aptamer selection, a large single-stranded DNA library (up to 1018 random sequences) is incubated with a target molecule to enable the binding reaction to occur. The bound and unbound DNAs are subsequently separated. For small-molecule targets, separation is usually achieved by use of affinity columns, by covalently attaching the target molecule to the column and washing away the unbound DNA. Other separation formats include filters, magnetic beads [105], gel and capillary electrophoresis [106], surface plasmon resonance [107], and atomic force and fluorescence microscopy [108].
Aptamers can be regarded as nucleic acid analogs of antibodies. Although both have comparable specificity and affinity, aptamers are more flexible for the design of novel bioanalytical sensors. A striking feature of aptamers (RNA and DNA) is the broad range of targets that can be recognized. Not considering metal cations [109, 110], aptamers have been engineered for such a small target as ethanolamine [111] and for many protein targets with high molecular weights [112, 113]. A comprehensive list of aptamer targets has been reported [114], including proteins, organic molecules, ions, viruses, and even organelles. In addition, aptamers are easily synthesized and suitable for chemical modification. Aptamers maintain or restore their functions when exposed to the harsh environmental conditions. The most interesting aspect of adapting aptamers to act as biosensors or molecular probes is their structural flexibility, which means that binding of a target usually results in structural alteration or rearrangement of the aptamer [115, 116]. Even if the target binding has negligible effect on the structural characteristics of the aptamer, the system is still suitable for bioanalytical applications, for example, in competition assays with a fluorescent derivative of the target [117], or the fluorescent aptamer may serve as a reporter sensor for detection of target molecules in chromatographic or electrophoretic separations [118]. However, in many cases the molecular motions exerted by recognition of the target, although small, are sufficient to generate an analytical signal, as in the anti-adenosine aptamer labeled with fluorescein [119]. On the other hand conformational changes can be significant, leading to global structure rearrangements, as observed on G-quadruplex formation [13, 109]. Aptamers that adopt a tetraplex structural motif are among the most extensively exploited probes for biosensing purposes.
G-quadruplex structures
G-quadruplexes or G4 DNA have the tetraplex structure and can be formed by single-stranded fragments of nucleic acids with guanine-rich sequences [109, 120]. G-quadruplexes have recently received much attention because G-rich sequences are often found in genomes and because of their potential links to mechanisms that relate to cancer, AIDS, and other diseases [121, 122].
The G-quadruplexes have four-stranded structures (DNA tetraplexes ) containing one or more nucleic acid strands, in parallel or antiparallel orientations [123]. Four guanines on a plane, interacting via Hoogsteen bonding, form a G-quartet as shown in Fig. 4a. Typically, two, three, or four G-quartets are stacked within a quadruplex and held together by π–π nonbonded attractive interactions (Fig. 4b–e). The most interesting feature of the quadruplex structure is the presence of a central channel formed by stacking of guanine tetrads that can accommodate some cations selectively. The ion-binding site is thought to be a cavity between two G-quartets that is flanked with eight O-6 atoms from guanines participating in the precise coordination of the cation [123]. The structural polymorphism and stability of G4 structures depend on many factors including strand stoichiometry and polarity, glycosidic torsion angle, connecting loops, and the nature of coordinated metal cation [123, 124]. For example, the sodium quadruplex formed by the human telomeric DNA sequence, d[AG3(T2AG3)3], has an antiparallel strand orientation with the basket-type structure (c in Fig. 4) [125], whereas the potassium complex has a variety of G-quadruplex structures ranging from antiparallel basket- or chair-type structures (b and c in Fig. 4) to parallel and hybrid conformations (d and e in Fig. 4) [123, 124, 126]. In contrast, the thrombin-binding aptamer (5′-d(GGTTGGTGTGGTTGG)-3′) forms only one G4 structure, a chair-type quadruplex (b in Fig. 4), irrespective of the metal cation coordinated [127]. Knowledge of the structural details of a particular G-quadruplex is crucial for the rational design of FRET probes if one considers different orientations of both termini of an oligonucleotide. Beside metal cations, several promising small organic molecules have been devised to selectively promote the formation and/or stabilization of guanine-quadruplex structures, ranging from derivatives of anthraquinones to porphyrins, acridines, and others planar compounds [128]. Another interest in G-quadruplexes is connected with their biological significance. Beside involvement in the regulation of telomerase activity, G-quadruplexes have been linked to proteins that either specifically bind to or promote the folding of quadruplex structures [113, 129]. Thrombin-binding aptamer (TBA) is a good example of an oligonucleotide that exploits quadruplex motif for thrombin-binding [113].
Fig. 4.

Structure of G-tetrad showing hydrogen bonds between four guanines and the interactions with a cation (a) and schematic representation of G-quadruplex structures: an antiparallel “chair-type” G-quadruplex with all lateral loops (b), an antiparallel “basket-type” G-quadruplex with one diagonal and two lateral loops (c), a hybrid-type quadruplex with parallel-antiparallel loops orientation (d), and a parallel quadruplex with all loops positioned alongside the grooves (e)
Intramolecular folding of a flexible single-stranded DNA molecule into a compact G-quadruplex is a structural transition leading to closer proximity of its 5′ and 3′ ends. Thus, labeling both ends of a DNA strand with the donor and acceptor fluorophores enables monitoring of the quadruplex-formation process by means of the FRET signal. Simonsson and Sjoback reported the first example of a FRET system based on tetraplex DNA, showing intramolecular folding of oligonucleotides with sequences corresponding to human c-myc protooncogene labeled with fluorescein (FAM) and tetramethylrhodamine (TAMRA) at the 5′ and 3′ ends, respectively [130]. The pyrene excimer monomer-switching approach has been also exploited in the monitoring of G-quadruplex folding [40]. Examples of fluorescence-based probes with the G-quadruplex motif are described in subsequent sections.
Detection of proteins
Aptamers with sequences characteristic of particular protein-binding regions can be used for protein recognition [131]. Tan et al. have used the molecular beacon strategy to study protein–DNA interactions [132]. In the presence of single-stranded DNA binding protein (SSB), the beacon was opened and fluorescence enhancement was observed, similar to that induced by the complementary DNA target.
Anti-thrombin aptamer is an example of an oligonucleotide that exploits a quadruplex motif for thrombin-binding [113]. This 15-mer oligonucleotide, with the sequence 5′-d(GGTTGGTGTGGTTGG)-3′, forms an intramolecular quadruplex with two G-tetrads that strongly binds to thrombin. Different strategies have been proposed for the design of thrombin-signaling systems [133–137]. Hamaguchi et al. reported a molecular-beacon approach with the thrombin sequence extended by addition of five nucleotides to the 5′ end to obtain an MB with a stem-loop structure [133]. A fluorophore–quencher pair (fluorescein–DABCYL) attached to the 5′ and 3′ ends, respectively, of the stem was responsible for signaling thrombin-dependent conformational changes. In the absence of thrombin, the aptamer formed a beacon (stem-loop) conformation with the fluorescence signal quenched. Addition of thrombin shifted the equilibrium toward the quadruplex (thrombin-binding conformation) that destroyed the hairpin structure and caused a change in the distance between the fluorophore and the quencher, resulting in a dequenching effect (Fig. 5a). The system could detect less than 10 nmol L−1 thrombin, with a maximum fluorescence increase of 2.5-fold.
Fig. 5.

Schematic representation of generation of fluorescence with different aptamer affinity probes
A simpler approach for a thrombin sensor was presented by Tan et al. [134]; fluorescent reporter groups were attached directly to the 5′ and 3′ ends of the 15-mer thrombin aptamer via one-base spacers (Fig. 5b). Two types of thrombin probe were tested:
a quenching type (labeled with a fluorescein-DABCYL pair); and
a FRET probe (coumarin–fluorescein as a donor–acceptor pair).
For both sensors sensitivity was rather modest, with 60% quenching of the fluorescence signal observed for the quenching probe and a twofold increase in the energy-transfer signal with the same concentration of thrombin as for the FRET-based aptamer. The FRET-based assay was adapted to a microarray format and the fluorescence intensity was recorded with a digital camera. Ratiometric imaging was found to increase the signal-to-background ratio, and thus afforded greater sensitivity in thrombin imaging.
A more general approach for preparing fluorescent signaling aptamers was presented by Nutiu and Li [135]. Three synthetic oligonucleotides were used to obtain a two-stem duplex assembly (Fig. 5c). The first strand was modified with a fluorophore at the 5′ end (F-labeled strand); the second contained a quencher at the 3′ end (Q-labeled strand) and the third unmodified strand contained regions complementary to the F and Q-strand sequences and a thrombin-binding domain. When the target (thrombin) was introduced, the duplex structure transformed into the thrombin–quadruplex complex with release of Q-labeled strand and enhancement of the fluorescence signal (dequenching). The fluorescein–DABCYL system was used as a fluorophore–quencher pair. The disadvantage of this approach was a relatively slow response at room temperature.
Aptamer beacons have also been constructed by splitting aptamers into two fragments. Yamamoto et al. reported such an aptamer beacon for HIV tat protein [136]. The aptamer sequence was divided into two parts, with one part in the hairpin conformation (labeled to form an MB system) and the other as a separated unlabeled strand (Fig. 5d). In the presence of the target analyte (Tat protein of HIV); the two parts self-assembled to bind the analyte and thus separated the quencher from the fluorophore, resulting in increased fluorescence. In the presence of 200 nmol L−1 Tat protein a ∼14-fold increase in fluorescence intensity was observed.
Most protein assays are based on recognition of protein by one aptamer. Heyduk et al. [137] introduced an assay using two MBs. The assay involved protein-induced hybridization of two aptamers, recognizing two distinct epitopes of the thrombin. Thrombin has two known aptamers that bind to two different sites on thrombin. An essential condition of this assay was stronger bivalent binding of both aptamers at the same time compared with binding of aptamers one at a time. The designed aptamers were covalently linked to the “signaling” 7-bp oligonucleotides with donor or acceptor fluorophores. In the absence of thrombin, donor and acceptor aptamers were randomly distributed in solution and an intramolecular FRET signal was not observed. On addition of the target protein, the binding of both aptamers to the same thrombin molecule brought the donor and acceptor fluorophores close to each other, resulting in generation of a FRET signal. By using fluorescein and Cy5 as a FRET pair, they were able to detect as little as 50 pmol L−1 thrombin [137].
The pyrene excimer monomer switching (EMS) approach has been used by Tan and co-workers to design a wavelength-shifting aptamer probe by labeling a platelet derived growth factor (PDGF)-binding DNA aptamer with two pyrene molecules [39]. The aptamer was optimized such that the two ends were far away in the absence of PDGF but close to each other in the presence of target (a working principle similar to that shown in Fig. 5b). On PDGF binding, the aptamer switched its fluorescence emission from 400 nm (pyrene monomer, with a fluorescence lifetime of ∼5 ns) to 485 nm (pyrene excimer, with a lifetime of ∼40 ns). The wavelength-shifting and time-resolved measurement capabilities make the system useful for sensing in complex sample matrixes, for example cell media, in which strong background fluorescence often makes intensity-based detection difficult. A FRET assay for PDGF was developed by the same group by appending a fluorophore and a quencher to the termini of the anti-PDGF aptamer [138]. In the absence of PDGF, the aptamer was largely denatured and its ends were far apart. Fluorescence quenching was observed when two termini were brought together as result of the structural transition of the aptamer imposed by PDGF.
Most research on aptamer probes has been performed with organic fluorophores in the visible region of the spectrum. To overcome problems with high background of the biological matrix, Zhang et al. designed a near-infrared (NIR) fluorescent aptamer probe that could sense NF-kB p50 protein-transcription factor [139]. The sensor had donor–acceptor NIR fluorophores with an emission excitation spectral overlap suitable for the FRET approach. Detection of aptamer–protein interactions in vivo by NIR dyes has an advantage over visible fluorescence labels because of much lower light scattering and absorption in the tissue. However, the choice of such fluorophores is limited.
Nanoparticles (NPs) have been also used in the development of aptamer sensors for proteins. Zhao and co-workers used AuNPs as quenchers for designing structure-switching aptamer sensors for thrombin detection [140]. Under their experimental conditions, recognition of thrombin by the hybrid of a TAMRA-labeled DNA strand and anti-thrombin aptamer was immobilized on gold NPs was possible. In another approach, Ellington et al. used quantum dots (QDs) to construct sensors based on the structure-switching aptamer design [141]. A thrombin aptamer was attached to QDs, and a complementary DNA with a quencher was hybridized to the aptamer, resulting in quenched QDs fluorescence. Addition of thrombin released the quencher and an 14-fold emission increase was observed.
Fluorescence anisotropy measurements have also been used as transduction mechanism for aptamer sensors. These assays can be performed with a single labeled aptamer probe in homogeneous solutions without the need for target separation or purification. Tan et al. labeled the 5′ end of the PDGF aptamer with fluorescein [31] and increased anisotropy on addition of PDGF was observed with a detection limit of 0.22 nmol L−1. Similarly, anisotropy-based assays have been performed on the IgE aptamer [142], the thrombin aptamer [143], and aptamers for amyloid β-peptides [144].
Small molecule detection
Many aptamers designed for recognition of biologically important small molecules have been modified into fluorescence sensors [145]. Careful studies have been devoted to the direct labeling of aptamers with fluorescence reporter groups to optimize the performance and sensing ability of such aptamers. Ellington and co-workers first explored covalent labeling of fluorophores to aptamers by a rational approach to sensor design [131]. On the basis of the structure of the RNA aptamer for ATP, an acridine fluorophore was attached at the site close to the binding pocket but not directly involved in binding. This aptamer resulted in ATP-dependent fluorescence enhancement, while labeling with fluorescein at the same position or at the terminus of the aptamer failed to generate a signal change. Interestingly, the same design strategy applied on the ATP DNA aptamer and labeling with fluorescein resulted in ATP-dependent signal changes, but the fluorescence increase was less than onefold. Yamana et al. synthesized a bis-pyrene fluorophore and attached it to a number of positions on the ATP DNA aptamer [146]. Only labeling at particular positions resulted in ATP-dependent fluorescence changes. They also attached a pyrene moiety at the 2′-ribose positions on the ATP aptamer. Of the seven positions tested, two gave an ATP-dependent fluorescence increase [147]. Similar labeling has been reported by Weeks and co-workers, who placed a Bodipy-FL fluorophore at the 2′-ribose of the cytosine residues on three DNA aptamers [148]. For the ATP aptamer and argininamide aptamer, among the three cytosine residues tested, only single positions generated fluorescence enhancement. In contrast, all three positions tested on a tyrosinamide aptamer resulted in an increase in fluorescence [148].
These studies demonstrated that it is possible to rationally design aptamer sensors by strategically attaching fluorophores to aptamers. This method often requires knowledge of aptamer structures and a few trial-and-error experiments. The fluorescence increase is relatively small (<fourfold). Placing a fluorophore too close to the binding site may disrupt binding, and fluorophores too far away from the binding site may not result in fluorescence changes.
Therefore, design of small-molecule probes based on MB or other approaches similar to those used for detection of nucleic acids and proteins were more widely used. For example, MB design was reported by Morse for a tobramycin sensor [149]. The RNA aptamer was modified to adopt a hairpin structure and the two ends of the aptamer were labeled with a fluorophore and a quencher to construct sensors (Fig. 5a). However, in the two sequences tested, the fluorescence increase in the presence of saturating tobramycin concentration was only ∼10%, and a detection limit of 30 μmol L−1 was reported. Some aptamer beacons have been designed on the basis of the fluorophore–quencher approach but without the formation of an initial hairpin structure. The cocaine RNA aptamer has been proposed as a means of constructing such a sensor, because it has the binding structure of a short hairpin flanked by two single-stranded regions [150]. A fluorophore and a quencher were labeled at the two ends, and high fluorescence intensity was initially observed in the absence of cocaine (the aptamer ends were separated in a single-stranded form). In the presence of cocaine, the compact structure was formed and the fluorophore was brought close to the quencher (Fig. 5b). Fifty percent quenching was observed with 1 mmol L−1 cocaine. Even in blood serum, 10 μmol L−1 cocaine could be detected. Similar probes with a fluorophore and a quencher on the two ends of an oligonucleotide have been developed using the DNA aptamer for l-argininamide [151] and adenosine aptamer [152]. A detection limit of ∼5 μmol L−1 was reported for the adenosine probe with quenching efficiency up to 67% [152]. An aptamer splitting approach has also been used by Stojanowic et al. to construct cocaine and ATP sensors [153]. Aptamers were split into two halves. One half was labeled with a fluorophore and the other with a quencher. In the absence of target molecule these two strands existed in solution as monomers and a strong fluorescence signal was observed. Target-dependent association of the two halves of the aptamer brought the fluorophore and the quencher together and quenching of fluorescence was observed (Fig. 5e). These sensors were able to detect cocaine from 10 to 1250 μmol L−1 and ATP from 8–2000 μmol L−1. Another design of ATP detection system was reported by Li et al. who developed a general strategy for signaling aptamer binding based on the structure-switching properties of the aptamer [135, 154, 155]. Competitive binding of target or complementary DNA constitutes a structure-switching process between the two states. The DNA competitor(s) hybridizes with the aptamer but enables dehybridization when the aptamer binds to the target molecule. Two DNA competitors labeled with a fluorophore and a quencher were designed in such a way that they were brought close to each other on hybridizing to the anti-ATP aptamer, resulting in quenched fluorescence [135]. In the presence of ATP, the aptamer fragment switched to the target-binding structure. As a result, the number of base pairs between the quencher-labeled DNA and the aptamer DNA was reduced, so that the quencher-labeled DNA competitor dissociated in solution, giving an increased fluorescence signal (Fig. 5c). Sensing of ATP was demonstrated with a detection limits of 10 μmol L−1 and a very high fluorescence increase (over 15-fold) [135]. Alternatively, a system with only one DNA competitor was constructed (Fig. 5f), in which the aptamer carried a fluorophore and a DNA strand was labeled with a quencher [154] or the DNA strand was labeled with both the fluorophore and the quencher and adopted an MB structure after dehybridization [155]. In a recent design reported by the Tan group, the aptamer and complementary DNA strand labeled with a fluorophore and a quencher, respectively, were linked by a PEG chain to form a hairpin structure of an MB type [156]. In the presence of the ATP target, increased fluorescence was observed.
Aptamers can be conjoined with nucleic acid enzymes (ribozymes or deoxyribozymes) to generate a new class of catalyst, so called aptazymes [3]. Aptazymes are nucleic acid sequences that catalyze chemical reactions, for example cleavage of nucleic acid targets, and can be switched on (or shut off) by interaction of its recognition part (aptamer) with an analyte. Fluorescent signal generation can be achieved by cleaving a FRET-labeled substrate (Fig. 6a). Srinivasan et al. identified ADP-specific aptazymes that could be used as sensors of kinase activity [157]. On activation by ADP, the hammerhead aptazyme cleaved an oligonucleotide substrate, resulting in fluorescence signal generation. Achenbach and colleagues have recently described design of an ATP-dependent aptazyme by joining the 8-17 deoxyribozyme to the ATP-binding DNA aptamer [158]. This aptazyme remained inactive in the absence of the target, but when ATP was introduced, the oligonucleotide was released and the activity of the deoxyribozyme was restored (Fig. 6a). Nutiu and Li have designed an assay that exploits a fluorescent DNA aptamer to report the activity of alkaline phosphatase (ALP) [159]. The fluorescence of the aptamer was higher for adenosine than for AMP.
Fig. 6.

Schematic representation of generation of fluorescence with aptazyme sensors for ATP (a) and Pb2+ ion (b)
Metal cation sensors
Some metal ions, for example lead and mercury, are harmful environmental contaminants with a health risk to humans. Sensors for easy detection of these metals are therefore highly desirable. Advances in many different design principles for aptazymes have led to development of DNAzyme-based sensors for metal ions. These probes have been reported for such metal ions as Cu2+, Zn2+, Pb2+, Hg2+, UO2+2, and Ca2+ [160–171]. The first such probe was reported by Li and Lu who designed a DNAzyme-based lead sensor (Fig. 6b) [160]. The catalytic DNA used was 17E, a variant of the 8-17 deoxyribozyme mentioned in the previous section. In the absence of lead ions, the 17E had very weak activity towards a fluorogenic RNA-containing substrate labeled with FRET partners. When lead was present, however, 17E rapidly cleaved the substrate, leading to a strong fluorescence signal [160]. The signaling properties of this design have been improved by adding another acceptor molecule to the other end of the substrate [165]. The signaling of this new probe was enhanced (five-times higher signal) because of background suppression [165]. A similar arrangement of FRET labels was used for an uranium probe with 17-fold fluorescence signal enhancement in the presence of uranium ion [166]. Perrin and co-workers have engineered a mercury sensor based on a deoxyribozyme whose activity could be specifically inhibited by Hg(II) [162]. Li and co-workers have reported several RNA-cleaving fluorescence-signaling deoxyribozymes that were dependent on one or more transition divalent metal ions, for example Mn(II), Cd(II), and Ni(II) [163, 164]. Two strategies in the design of metal ion-dependent DNAzymes can be distinguished. One utilizes a molecular beacon consisting of two specific regions related to a DNAzyme and a substrate. In the absence of target ion, the MB has an intact structure with the fluorescence signal quenched. When the target ion is bound, the fluorophore-labeled substrate quenched by a quencher-modified DNAzyme is irreversibly cleaved and released to produce a fluorescent signal [160–169]. Wang et al. [168] recently proposed the DNAzyme for lead ion monitoring by applying a unimolecular design with a leaving substrate DNA strand labeled with a fluorophore linked to a hairpin 8-17 DNAzyme sequence labeled with a quencher. The hairpin structure and the substrate were connected using poly T, which brings the quencher into close proximity with the fluorophore in the inactive state. In the presence of Pb2+, however, the leaving substrate fragment was cleaved by the enzyme, releasing a fluorescent fragment. Liu and Lu reported a highly sensitive and selective catalytic probe for mercury that was based on a uranium-specific DNAzyme containing thymine–thymine mismatches [169]. The Hg2+ ions enhanced the DNAzyme activity through allosteric interactions. In the absence of Hg2+ ions, the DNAzyme was incapable of binding UO2+2 ions because the active secondary structure could not form. Addition of Hg2+ ions stabilized the T–T mismatches and, restoring the stem-loop structure, activated the DNAzyme to cleave the substrate, releasing the fluorophore-labeled leaving strand and resulting in increased fluorescence. The same strategy of Hg2+ binding between two thymine bases in DNA, which stabilizes the T–T mismatch, has been used by Ono et al. in the fluorescent sensor for Hg2+ detection [170]. The two ends of a thymine-rich DNA were labeled with a fluorophore and a quencher. Addition of Hg2+ led to the formation of a DNA hairpin that caused fluorescence quenching. A detection limit of 40 nmol L−1 was reported. Another probe design uses the conformation alteration of the probe that results from cleavage of the substrate by the DNAzyme. In this case, the horseradish peroxidase (HRP)-mimicking DNAzyme is activated by a cleavage process, thus generating colorimetric or chemiluminescence readout signals [171]. Yin et al. reported such a dual-DNAzyme unimolecular probe with a simple, label-free design [171]. The probe was a combination of a DNA substrate, a cleaving DNAzyme1 and an HRP-mimicking DNAzyme2. In the absence of the target metal ion, these three domains acted cooperatively in the DNA-cleaving active state as a result of strong intramolecular interactions, and the resulting structure revealed higher stability than the G-quadruplex structure responsible for activity of HRP-like DNAzyme2. When the target Cu(II) ion was present, cleavage of a substrate by DNAzyme1 disturbed the intramolecular DNA conformation, and this event resulted in an allosteric transformation from the active state of DNAzyme1 to the active state of HRP-like DNAzyme2 with a tetraplex structure, which in turn gave a colorimetric signal [171].
The tetraplex structural motif found in HRP-like DNAzymes has been also used to develop fluorescence probes for potassium ion or pH monitoring [29, 40, 172–176]. The common idea behind these sensors was to exploit the metal cation binding properties of quadruplex-forming DNA sequences. After labeling with reporter fluorescent groups, the oligonucleotides preserved their abilities to fold into a quadruplex structure and to interact with metal cations with binding affinity and selectivity comparable with that of unmodified oligonucleotides [29, 40, 172, 173]. The preferential ability of potassium to stabilize guanine tetraplexes is commonly observed. Potassium has an ionic radius of approximately 1.3 Å, and is believed to fit exceptionally well in the cavities between guanine tetrads. Three different transduction processes shown in Fig. 7 have been proposed for construction of potassium fluorescent probes with the use of quadruplex-forming oligonucleotides:
FRET between two fluorophores attached to the both termini of the telomeric oligonucleotide (Fig. 7a) [29, 172, 173];
excimer emission of the pyrene-labeled thrombin aptamer (Fig. 7b) [40, 174]; and
FRET between the cationic conjugated polymer and the fluorescein-labeled quadruplex (Fig. 7c) [176].
Fig. 7.

Schematic illustration of strategies used to develop oligonucleotide sensors for potassium ion: a FRET-based probe (a), an excimer emission sensor based on pyrene-labeled thrombin binding aptamer (b), and a FRET probe sensitive to the electrostatic interactions between cationic conjugated polymer (CCP) and G-quadruplex labeled with fluorescein (c)
In the first strategy, the G-rich oligonucleotides were labeled with fluorophores suitable to serve as FRET partners and the probes obtained were called PSO (potassium-sensing oligonucleotide). Two FRET systems were reported, PSO-1 and PSO-2, with the telomere sequence from human G3(TTAG3)3 and Oxytricha nova G4(T4G4)3, respectively, and a common FRET pair, FAM-TAMRA [29, 172, 173]. The selectivity ratio of PSO-1 for K+ against Na+ was 43,000; for PSO-2 this value approaches 150 only [173]. Factors responsible for the extremely high value of K/Na selectivity of PSO-1 included inherent preferences of this quadruplex for K+ binding, a stabilizing effect of stacking interactions between fluorophores (a chair-type or hybrid K+ quadruplex vs. a basket-type Na+ form), and different FRET efficiency, which is a function of interflurophore distance (quadruplex structure). General observations regarding FRET in PSO-1 and PSO-2 systems with metal cations were:
FRET signal (sensitized fluorescence of TAMRA) appeared only for K(I) and Na(I) complexes;
a much lower concentration of potassium ion was required to generate the FRET signal;
the intensity of sensitized fluorescence of TAMRA finally attained was larger for Na+ than that for K+ ions; and
multivalent metal cations (Mg2+) caused quenching of the donor fluorescence (FAM) only, without sensitization of the TAMRA emission [172, 173].
Systematic studies have been carried out to assess the effect of complexation of metal cation by PSO-1 on the spectral properties of FRET donor (FAM) and acceptor (TAMRA) [29]. Three different fluorescent DNA oligonucleotides with human telomere sequence were investigated—a dual-labeled PSO-1 probe and two reference telomeric 21-mers, FAM-21 and 21-TAMRA, single-labeled at the 5′ and 3′ ends, respectively. Steady-state fluorescence, FRET, and anisotropy measurements were carried out in the presence of selected cations (Et4N+, Li+, Na+, K+, Mg2+). Fluorescence intensity of FAM-21 was sensitive to the nature and concentration of cations. Anisotropy of FAM-21 (but not 21-TAMRA) seemed to be sensitive to the G-quadruplex formation, with a significant increase with increasing cation concentration, and indicated some restrictions in rotational depolarization of FAM. FRET experiments revealed that although all tested cations caused quenching of FAM fluorescence in PSO, only Na+ and K+ ions induced sensitized emission of the TAMRA acceptor. Fluorimetric detection of K+ with PSO-1 was possible at submicromolar concentrations under optimum conditions, with a linear calibration graph in a wide concentration range of K+ (1–1000 μmol L−1) [172].
An attempt has been made to construct potassium FRET probes using the thrombin binding aptamer (TBA) sequence and FAM or TAMRA fluorescence labels at either terminus, with spacer DNA sequences of TmA (m = 2, 4, or 6) introduced between FAM and the 5′ end [175]. The reference probe (no spacer) did not result in sensitized emission of the acceptor (TAMRA) in the presence of K+, because of the quenching of TAMRA fluorescence by dye–dye interactions with FAM. When the spacer was introduced, the fluorescence of TAMRA was increased, because of prevention of the dye–dye interaction. Both donor quenching and sensitized emission of the acceptor could be used as sensitive signals to monitor folding processes of quadruplex DNA. These FRET results unambiguously confirmed that addition of a spacer significantly reduced contact quenching and improved the performance of FRET probes [175].
In contrast with the FRET approach, stacking interactions between fluorophores are advantageous in the production of efficient excimer emission when pyrene moieties serve as fluorophores [37, 40, 174]. Therefore, another potassium probe has been developed (denoted as PSO-py), which exploited the pyrene excimer emission for transduction of cation-binding (probe b in Fig. 7) [40, 174]. Two pyrene moieties were attached to the thrombin biding aptamer (TBA) termini. TBA is known to form a chair-type quadruplex structure on binding to thrombin protein, and this quadruplex incorporates K+ with a 1:1 stoichiometry [127]. Therefore, binding of potassium ion organized the 5′ and 3′ termini of TBA in such a way that two attached pyrene molecules were arranged face-to-face (Fig. 7b). Owing to this spatial orientation of pyrene rings, the PSO-py/K+ complex emitted excimer fluorescence whereas the random coil structure of PSO-py in the absence of K+ gave only monomer emission. Consequently, the intensity of excimer emission by PSO-py depended on the K+ concentration. The presence of other metal cations caused minor interferences, because TBA forms weak complexes with Na+, Mg2+, and Ca2+ ions [127]. A modification of this assay with improved signal-to-background ratio has been recently proposed by Shi and co-workers [176]. A hairpin oligonucleotide containing a sequence complementary to TBA, was modified with pyrene moieties and hybridized with TBA aptamer. The hybrid gave only monomer emission in the absence of K+, because the duplex rigid structure protected the pyrene moieties from stacking interactions. In the presence of K+, the complementary Py-labeled oligonucleotide was displaced from the aptamer, which adopted the G-quadruplex structure. This structural transition was accompanied by excimer fluorescence of pyrene because the self-hairpin structure of the complementary oligonucleotide brought the pyrene moieties into close proximity. Under optimum conditions, the relative fluorescence intensity of pyrene was proportional to the concentration of K+ in the range 0.6–20 mmol L−1.
Recently, we have reported lifetime measurements for G-quadruplexes formed between metal cations (K+, Na+, Sr2+) and pyrene-modified oligonucleotides with sequences of thrombin binding aptamer (Py–TBA–Py) and human telomeric sequence (Py–Htelom–Py) [48]. All decays of investigated systems could be characterized by a tri-exponential rate. Calculated lifetimes and their fractional distributions depended on the sequence of attached oligonucleotide and the nature of the metal cation that occupied a tetraplex internal cavity. The monomer emission data reflected the extent of quenching of the pyrene excited state on interaction with nucleobases or with a second pyrene molecule. The long-wavelength emission was ascribed to pyrene excimer formation, which proved pyrene–pyrene interactions. On the other hand, the structure of the quadruplex seriously affected label–label and label–nucleobase interactions. The quadruplex topology that enables efficient label–label interactions, and thus generation of excimer emission with very long lifetime, seemed to be a chair-type structure formed by the Py–TBA–Py probe. Next, the hybrid and propeller-type structures of Py–Htelom–Py quadruplexes facilitated deactivation of the excited state of pyrene, because of stacking interactions between pyrene and guanine tetrads. Finally, the Htelom quadruplex with the basket-type structure and a diagonal TTA loop that disturbs both pyrene–pyrene interactions and pyrene–guanine tetrad stacking resulted in lengthening of the monomer pyrene lifetime.
He et al. [177] suggested use of another FRET sensor based on quadruplex formation for potassium detection. As oligonucleotide probe they used a TBA quadruplex labeled at the 5′ terminus with fluorescein that served as FRET acceptor. Unlike other oligonucleotide FRET-labeled systems [172], these authors used a cationic conjugated polymer (CCP) as an external energy donor (Fig. 7c). Amplification of the FRET signal of the acceptor (fluorescein) was expected as a result of excitation energy transfer from a large number of fluorophore units in CPP (donor). In the absence of K+ (random coil conformation), the probe resulted in inefficient FRET whereas formation of a G-quadruplex on addition of KCl caused an increase in the FRET signal (ca. 16-fold). Good performance of the system was explained by the charge density around the DNA that controlled the extent of association with CPP, and thus the efficiency of FRET from the CCP to the fluorescein label. This approach has the potential for high sensitivity, because of the light-harvesting properties of the energy donor (a conjugated polymer).
The tetraplex structure of DNA has also been reported to be suitable for pH monitoring in the physiological range. Cytosine-rich oligonucleotides associate under acidic conditions to form a tetraplex i-motif consisting of two parallel-stranded C–H–C+ base-paired duplexes that are mutually intercalated in an antiparallel orientation [178]. Krishnan et al. [179] constructed a nanomachine called an “I-switch” that consisted of three oligonucleotides O1, O2, and O3, in which O1 and O2 were hybridized on to sites adjacent to O3, leaving a one-base gap. The O1 and O2 probes had single-stranded cytosine-rich overhangs designed such that each overhang formed one-half of a bimolecular i-motif. At acidic pH, these overhangs were protonated and the assembly could fold to form an intramolecular i-motif. The system adopted an extended conformation at pH 7.3, whereas at pH 5 the single-stranded overhangs formed an i-motif, yielding a “closed state” and I-switching action. The I-switch, fluorescently labeled at its 3′ and 5′ termini with Alexa-488 and Alexa-647 on O1 and O2, respectively, gave a FRET signal at pH ∼5, with efficiency of 54–60%. No significant FRET was observed in the open state at pH 7.3. At pH 7.3 the donor lifetime of the probe was ∼3.5 ns, whereas at pH ∼5 it was shortened to 2.1 ns. Moreover, the I-switch could switch reversibly between the “closed” and “open” states on variation of pH. To demonstrate its ability to function inside living cells, the I-switch was used to map spatial and temporal pH changes associated with the endosome maturation, by use of fluorescence microscopy [179].
Concluding remarks
This review mainly focuses on the molecular recognition and sensing aspects of fluorescent oligonucleotide probes. Nucleic acids are generally believed to be able to target any analyte of choice. Enormous progress in the “omics” fields and in chemical biology has been possible because of the development of new rapid, accurate and cost-effective methods for analysis of nucleic acids, proteins, and other molecules, including metabolites. Various efficient and practical signaling strategies have been successfully developed to transduce nucleic acid recognition into physically detectable fluorescence signals. In addition, the rapid development of oligonucleotide-based sensing is also a result of superior properties of nucleic acids as excellent sensors, including high stability, low cost, and ease of synthesis or modification. Although many assays have been successfully proved in real sample applications (especially hybridization probes in FISH or QPCR), there is still a long way to go from proving experimental feasibility to demonstrating real-world applications of aptamer probes. Most sensing work using oligonucleotide probes has been performed on model systems, and few clinically relevant targets have actually been tested. Most sensors have, in addition. been developed and tested in buffer systems under controlled laboratory settings. For real medical diagnosis and environmental monitoring, significant matrix effects must be carefully evaluated. he performance of reported probes, especially in aptamer sensors, could decrease substantially in serum or plasma samples. Therefore, it will take more research and technological effort to optimize the sensors and eliminate matrix effects for practically useful sensitivity and specificity. Although pyrene probes are good proof of concept, their excitation wavelength (<360 nm) might be a problem in studies of biological samples. Other molecules with longer fluorescence lifetimes may improve the time resolution capabilities of the probes even further. One alternative is the use of transition-metal complexes or lanthanide chelates, which can have lifetimes of the order of milliseconds.
Another important issue is that current methods have mostly been developed for in-vitro detection. To accomplish in-vivo sensing, novel methods and materials are needed, for example, nucleic acid–protein conjugates, new fluorophores, for example QDs and other nanomaterials, or innovative transduction techniques. With the advent of new time-resolved fluorescence microscopes and with the recent developments in fluorescence lifetime imaging microscopy (FLIM), these kinds of probes will find their place in the study of cell processes in vivo. Other transduction techniques, for example two-photon excitation and multiple FRET labeling, are also good prospects for detection purposes.
When a sufficient number of sensors has been obtained, the next significant advance in this sensing field would be the development of sensor arrays for more sophisticated target analysis. The benefits of the sensor arrays come from the possibility of simultaneous detection of multiple targets (the capability of high-throughout analysis of complex samples). The selectivity of a single sensor may, however, be insufficient for reliable target identification. Similarly, one target may induce different responses from sensors on an array, thus forming a “fingerprints” signal.
In addition to detection of target analytes, oligonucleotide-based fluorescence sensors can also be used as tools for many research fields and practical applications, for example high throughput drug screening, nanotechnology, and materials science. Assuming an increasing demand for convenient and effective sensors, one can expect continued and rapid development and advances in nucleic acid-based sensing in the future.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Published in the special issue Focus on Analytical Science in Poland (VIIIth Polish Conference on Analytical Chemistry) with Guest Editor Pawel Koscielniak.
References
- 1.Thompson MB, editor. Fluorescence sensors and biosensors. Boca Raton: Taylor and Francis; 2006. [Google Scholar]
- 2.Marti AA, Jockusch S, Stevens N, Ju J, Turro NJ. Acc Chem Res. 2007;40:402–409. doi: 10.1021/ar600013q. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Cao Z, Lu Y. Chem Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 5.Mergny J-L, Boutorine AS, Garestier T, Belloc F, Rougée M, Bulychev NV, Koshkin AA, Bourson J, Lebedev AV, Valeur B, Thuong NT, Hèléne C. Nucleic Acids Res. 1994;22:920–928. doi: 10.1093/nar/22.6.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sei-Iida Y, Koshimoto H, Kondo S, Tsuji A. Nucleic Acids Res. 2000;28:e59. doi: 10.1093/nar/28.12.e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsuji A, Koshimoto H, Sato Y, Hirano M, Sei-Iida Y, Kondo S, Ishibashi K. Biophys J. 2000;78:3260–3274. doi: 10.1016/S0006-3495(00)76862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang XH, Mi YM, Li JWJ, Tan W. Cell Biochem Biophys. 2002;37:71–81. doi: 10.1385/CBB:37:2:071. [DOI] [PubMed] [Google Scholar]
- 9.Yang CJ, Medley CD, Tan W. Curr Pharm Biotechnol. 2005;6:445–452. doi: 10.2174/138920105775159322. [DOI] [PubMed] [Google Scholar]
- 10.Peng XH, Cao ZH, Xia JT, Carlson GW, Lewis MM, Wood WC, Yang L. Cancer Res. 2005;65:1909–1917. doi: 10.1158/0008-5472.CAN-04-3196. [DOI] [PubMed] [Google Scholar]
- 11.Marras SAE, Tyagi S, Kramer FR. Clin Chim Acta. 2006;363:48–60. doi: 10.1016/j.cccn.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 12.Marti AA, Li X, Jockusch S, Li Z, Raveendra B, Kalachikov S, Russo JJ, Morozowa I, Putthanveettil SV, Ju J, Turro NJ. Nucleic Acids Res. 2006;34:3161–3168. doi: 10.1093/nar/gkl406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juskowiak B, Takenaka S. In: Methods and molecular biology vol. 335. Didenko VV, editor. Totowa: Humana Press Inc; 2006. [Google Scholar]
- 14.Chen HW, Kim Y, Meng L, Mallikaratchy P, Martin J, Tang Z, Shangguan D, O’Donoghue M, Tan W. In: Functional nucleic acids for analytical applications. Li Y, Lu Y, editors. New York: Springer; 2009. [Google Scholar]
- 15.Orum H, Nielsen PE, Egholm M, Berg RH, Buchardt O, Stanley C. Nucleic Acids Res. 1993;21:5332–5336. doi: 10.1093/nar/21.23.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egholm M, Buchardt O, Nielsen PE, Berg RH. J Am Chem Soc. 1992;114:1895–1897. [Google Scholar]
- 17.Svanvik N, Westman G, Wang D, Kubista M. Anal Biochem. 2000;281:26–35. doi: 10.1006/abio.2000.4534. [DOI] [PubMed] [Google Scholar]
- 18.Kumar R, Singh SK, Koshkin AA, Rajwanshi VK, Meldgaard M, Wengel J. Bioorg Med Chem Lett. 1998;8:2219–2222. doi: 10.1016/s0960-894x(98)00366-7. [DOI] [PubMed] [Google Scholar]
- 19.Jepsen JS, Sorensen MD, Wengel J. Oligonucleotides. 2004;14:130–146. doi: 10.1089/1545457041526317. [DOI] [PubMed] [Google Scholar]
- 20.Costa J-M, Ernault P, Olivi M, Gaillon T, Arar K. Clin Biochem. 2004;37:930–932. doi: 10.1016/j.clinbiochem.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 21.Elington AD, Szostak JW. Nature (Lond) 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 22.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 23.Tyagi S, Bratu DP, Kramer FR. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 24.Clegg RM, Murche AIH, Zechel A, Carlberg C, Diekmann S, Llley DMJ. Biochemistry. 1992;31:4846–4856. doi: 10.1021/bi00135a016. [DOI] [PubMed] [Google Scholar]
- 25.Sauer M, Drexhage KH, Liberwirth U, Muller R, Nord S, Zander C. Chem Phys Lett. 1998;284:153–163. [Google Scholar]
- 26.Nazarenko I, Pires R, Lowe B, Obaidy M, Rashtchian A. Nucleic Acids Res. 2002;30:2089–2195. doi: 10.1093/nar/30.9.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manoharan M, Tivel KL, Zhao M, Nafisi K, Netzel TL. J Phys Chem. 1995;99:17461–17472. [Google Scholar]
- 28.Crockett AO, Wittwer CT. Anal Biochem. 2001;290:89–97. doi: 10.1006/abio.2000.4957. [DOI] [PubMed] [Google Scholar]
- 29.Juskowiak B, Galezowska E, Zawadzka A, Gluszynska A, Takenaka S. Spectrochim Acta A. 2006;64:835–843. doi: 10.1016/j.saa.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 30.Sueda S, Ihara T, Juskowiak B, Takagi M. Anal Chim Acta. 1998;365:27–34. [Google Scholar]
- 31.Fang X, Cao ZC, Beck T, Tan W. Anal Chem. 2001;73:5752–5757. doi: 10.1021/ac010703e. [DOI] [PubMed] [Google Scholar]
- 32.Kwok PY. Human Mutant. 2002;19:315–323. doi: 10.1002/humu.10058. [DOI] [PubMed] [Google Scholar]
- 33.Knemeyer J-P, Marme N, Sauer M. Anal Chem. 2000;72:3717–3724. doi: 10.1021/ac000024o. [DOI] [PubMed] [Google Scholar]
- 34.Hoefelschweiger B, Wolfbeis OS. J Fluoresc. 2008;18:413–421. doi: 10.1007/s10895-007-0281-2. [DOI] [PubMed] [Google Scholar]
- 35.Clegg RM. Meth Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 36.Lakowicz JR. Principles of fluorescence spectroscopy. 3. New York: Springer; 2006. [Google Scholar]
- 37.Winnik FM. Chem Rev. 1993;93:587–614. [Google Scholar]
- 38.Fujimoto K, Shimizu H, Inouye M. Org Chem. 2004;69:3271–3275. doi: 10.1021/jo049824f. [DOI] [PubMed] [Google Scholar]
- 39.Yang CJ, Jockusch S, Vicens M, Turro NJ, Tan W. Proc Natl Acad Sci USA. 2005;102:17278–17283. doi: 10.1073/pnas.0508821102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagatoishi S, Noima T, Juskowiak B, Takenaka S. Angew Chem Int Ed. 2005;44:5067–5070. doi: 10.1002/anie.200501506. [DOI] [PubMed] [Google Scholar]
- 41.Yamana K, Zako H, Asazuma K, Iwase R, Nakano H, Murakami A. Angew Chem Int Ed. 2001;40:1104–1106. doi: 10.1002/1521-3773(20010316)40:6<1104::aid-anie11040>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Hwang GT, Seo YJ, Kim BH. Tetrahedron. 2005;46:1475–1477. [Google Scholar]
- 43.Wanninger-Weiss C, Valis L, Wagenknecht H-A. Bioorg Med Chem. 2008;16:10–106. doi: 10.1016/j.bmc.2007.04.064. [DOI] [PubMed] [Google Scholar]
- 44.Yamana K, Iwai T, Ohtani Y, Sato S, Nakamura M, Nakano H. Bioconjug Chem. 2002;13:1266–1273. doi: 10.1021/bc025530u. [DOI] [PubMed] [Google Scholar]
- 45.Okamoto A, Ichiba T, Saito I. J Am Chem Soc. 2004;126:8364–8365. doi: 10.1021/ja049061d. [DOI] [PubMed] [Google Scholar]
- 46.Yamana K, Ohshita Y, Fukunaga Y, Nakamura M, Maruyama A. Bioorg Med Chem. 2008;16:78–83. doi: 10.1016/j.bmc.2007.04.053. [DOI] [PubMed] [Google Scholar]
- 47.Kosuge M, Kubota M, Ono A. Tetrahedron Lett. 2004;45:3945–3947. [Google Scholar]
- 48.Dembska A, Juskowiak B. J Photochem Photobiol A. 2010;212:36–42. [Google Scholar]
- 49.Heller MJ, Morrison LE, Prevatt WD, Akin C. European Patent Application. 1983;070:685. [Google Scholar]
- 50.Morrison LE, Halder TC, Stols LM. Anal Biochem. 1989;183:231–244. doi: 10.1016/0003-2697(89)90473-9. [DOI] [PubMed] [Google Scholar]
- 51.Cardullo RA, Agrawal S, Flores C, Zamecnik PC, Wolf DE. Proc Natl Acad Sci USA. 1988;85:8790–8794. doi: 10.1073/pnas.85.23.8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marras SAE, Kramer RF, Tyagi S. Nucleic Acids Res. 2002;30:e122. doi: 10.1093/nar/gnf121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martí AA, Li X, Jockusch S, Stevens N, Li Z, Raveendra B, Kalachikov S, Morozova I, Russo JJ, Akins DL, Ju J, Turro NJ. Tetrahedron. 2007;63:3591–3600. doi: 10.1016/j.tet.2006.08.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ebata K, Masuko M, Ohtani H, Kashiwasake-Jibu M. Photochem Photobiol. 1995;62:836–839. doi: 10.1111/j.1751-1097.1995.tb09144.x. [DOI] [PubMed] [Google Scholar]
- 55.Masuko M, Ohtani H, Ebata K, Shimadzu A. Nucleic Acids Res. 1998;26:5409–5416. doi: 10.1093/nar/26.23.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindegaart D, Medsen AS, Astakhowa IV, Malakhov AD, Babu BR, Korshun VA, Wengel J. Bioorg Med Chem. 2008;16:94–99. doi: 10.1016/j.bmc.2007.04.056. [DOI] [PubMed] [Google Scholar]
- 57.Liu C, Wang L, Jiang W. Talanta. 2010;81:597–601. doi: 10.1016/j.talanta.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 58.Morrison LE, Stols LM. Biochemistry. 1993;32:3095–3104. doi: 10.1021/bi00063a022. [DOI] [PubMed] [Google Scholar]
- 59.Yang CJ, Martinez K, Lin H, Tan W. J Am Chem Soc. 2006;128:9986–9987. doi: 10.1021/ja0618346. [DOI] [PubMed] [Google Scholar]
- 60.Zhang P, Bect T, Tan W. Angew Chem Int Ed. 2001;40:402–405. doi: 10.1002/1521-3773(20010119)40:2<402::AID-ANIE402>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 61.Santangelo PJ, Nix B, Tsourkas A, Bao G. Nucleic Acids Res. 2004;32:e57. doi: 10.1093/nar/gnh062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim Y, Sohn D, Tan W. Int J Clin Exp Pathol. 2008;1:105–116. [PMC free article] [PubMed] [Google Scholar]
- 63.Tsourkas A, Behlke MA, Bao G. Nucleic Acids Res. 2002;30:5168–5174. [PMC free article] [PubMed] [Google Scholar]
- 64.Seitz O. Angew Chem Int Ed Engl. 2000;39:3249–3252. doi: 10.1002/1521-3773(20000915)39:18<3249::aid-anie3249>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 65.Wang L, Yang CYJ, Medley CD, Benner SA, Tan WH. J Am Chem Soc. 2005;127:15664–15665. doi: 10.1021/ja052498g. [DOI] [PubMed] [Google Scholar]
- 66.Perlette J, Tan WH. Anal Chem. 2001;73:5544–5550. doi: 10.1021/ac010633b. [DOI] [PubMed] [Google Scholar]
- 67.Kim JH, Morikis D, Ozkan M. Sens Actuators B. 2004;102:315–319. [Google Scholar]
- 68.Kushon SA, Ley KD, Bradford K, Jones RM, McBranch D, Whitten D. Langmuir. 2002;18:7245–7249. [Google Scholar]
- 69.Dubertret B, Calame M, Libchaber AJ. Nat Biotechnol. 2001;19:365–370. doi: 10.1038/86762. [DOI] [PubMed] [Google Scholar]
- 70.Yang CYJ, Lin H, Tan WH. J Am Chem Soc. 2005;127:12772–12773. doi: 10.1021/ja053482t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sandhya S, Chen W, Mulchandani A. Anal Chim Acta. 2008;614:208–212. doi: 10.1016/j.aca.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 72.Li Y, Zhou X, Ye D. Biochem Biophys Res Commun. 2008;373:457–461. doi: 10.1016/j.bbrc.2008.05.038. [DOI] [PubMed] [Google Scholar]
- 73.Vet JA, Marras SA. Meth Mol Biol. 2005;288:273–290. doi: 10.1385/1-59259-823-4:273. [DOI] [PubMed] [Google Scholar]
- 74.Santangelo PJ. WIREs Nanomed Nanobiotechnol. 2010;2:11–19. doi: 10.1002/wnan.52. [DOI] [PubMed] [Google Scholar]
- 75.Massey M, Algar WR, Krull UJ. Anal Chim Acta. 2006;568:181–189. doi: 10.1016/j.aca.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 76.Algar WR, Krull UJ. Anal Bioanal Chem. 2008;391:1609–1618. doi: 10.1007/s00216-007-1703-3. [DOI] [PubMed] [Google Scholar]
- 77.Krasnoperov LN, Marras SAE, Kozlow M, Wirpsza L, Mustaev A. Bioconjug Chem. 2010;21:319–327. doi: 10.1021/bc900403n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tyagi S, Marras SA, Kramer FR. Nat Biotechnol. 2000;18:1191–1196. doi: 10.1038/81192. [DOI] [PubMed] [Google Scholar]
- 79.Zhang P, Beck T, Tan W. Angew Chem Int Ed. 2001;40:402–405. doi: 10.1002/1521-3773(20010119)40:2<402::AID-ANIE402>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 80.Tong AK, Jockusch S, Li Z, Zhu H-R, Akins DL, Turro NJ, Ju J. J Am Chem Soc. 2001;123:12923–12924. doi: 10.1021/ja016904h. [DOI] [PubMed] [Google Scholar]
- 81.Li X, Li Z, Martí AA, Jockusch S, Stevens N, Akins DL, Turro NJ, Ju J. Photochem Photobiol Sci. 2006;5:896–902. doi: 10.1039/b605936h. [DOI] [PubMed] [Google Scholar]
- 82.Zhang C-Y, Yeh H-C, Kuroki MT, Wang T-H. Nat Mater. 2005;4:826–831. doi: 10.1038/nmat1508. [DOI] [PubMed] [Google Scholar]
- 83.Li F, Huang Y, Yang Q, Zhong Z, Li D, Wang L, Song S, Fan C. Nanoscale. 2010;2:1021–1026. doi: 10.1039/b9nr00401g. [DOI] [PubMed] [Google Scholar]
- 84.Dong H, Gao W, Yan F, Ji H, Ju H. Anal Chem. 2010;82:5511–5517. doi: 10.1021/ac100852z. [DOI] [PubMed] [Google Scholar]
- 85.Lu CH, Yang HH, Zhu CL, Chen X, Chen GN. Angew Chem Int Ed. 2009;48:4785–4787. doi: 10.1002/anie.200901479. [DOI] [PubMed] [Google Scholar]
- 86.Haner R, Biner SM, Langeregger SM, Meng T, Malinovskii VL. Angew Chem Int Ed. 2010;49:1227–1230. doi: 10.1002/anie.200905829. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y, Yang CJ, Wu Y, Conlon P, Kim Y, Lin H, Tan W. Chembiochem. 2008;9:355–359. doi: 10.1002/cbic.200700542. [DOI] [PubMed] [Google Scholar]
- 88.Conlon P, Yang CJ, Wu Y, Chen Y, Martinez K, Kim Y, Stevens N, Marti AA, Jockusch S, Turro NJ, Tan W. J Am Chem Soc. 2008;130:336–342. doi: 10.1021/ja076411y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seo YJ, Hwang GT, Kim BH. Tetrahedron Lett. 2006;47:4037–4039. [Google Scholar]
- 90.Matsumoto K, Shinihara Y, Bag SS, Takeuchi Y, Morii T, Saito Y, Saito I. Bioorg Med Chem Lett. 2009;19:6392–6395. doi: 10.1016/j.bmcl.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 91.Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, Sindelka R, Sjoback R, Sjogreen B, Strombom L, Stahlberg A, Zoric N. Mol Aspects Med. 2006;27:95–125. doi: 10.1016/j.mam.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 92.Holland PM, Abramson RD, Watson R, Gelfand DH. Proc Natl Acad Sci USA. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee LG, Connell CR, Bloch W. Nucleic Acids Res. 1993;21:3761–3766. doi: 10.1093/nar/21.16.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Innis MA, Myambo KB, Gelfand DH, Brow MA. Proc Natl Acad Sci USA. 1988;85:9436–9440. doi: 10.1073/pnas.85.24.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, Singer MJ, Walburger DK, Lokhov SG, Gall AA, Dempcy R, Reed MW, Meyer RB, Hedgpeth J. Nucleic Acids Res. 2000;28:655–661. doi: 10.1093/nar/28.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.French DJ, Archard CL, Brown T, McDowell DG. Mol Cell Probes. 2001;15:363–374. doi: 10.1006/mcpr.2001.0384. [DOI] [PubMed] [Google Scholar]
- 97.Svanvik N, Stahlberg A, Sehlstedt U, Sjoback R, Kubista M. Anal Biochem. 2000;287:179–182. doi: 10.1006/abio.2000.4824. [DOI] [PubMed] [Google Scholar]
- 98.Isacsson J, Cao H, Ohlsson L, Nordgren S, Svanvik N, Westman G, Kubista M, Sjoback R, Sehlstedt U. Mol Cell Probes. 2000;14:321–328. doi: 10.1006/mcpr.2000.0321. [DOI] [PubMed] [Google Scholar]
- 99.Whitcombe D, Theaker J, Guy SP, Brown T, Little S. Nat Biotechnol. 1999;17:804–807. doi: 10.1038/11751. [DOI] [PubMed] [Google Scholar]
- 100.Uehara H, Nardone G, Nazarenko I, Hohman RJ. Biotechniques. 1999;26:552–558. doi: 10.2144/99263pf01. [DOI] [PubMed] [Google Scholar]
- 101.Nazarenko I, Bhatnagar SK, Hohman RJ. Nucleic Acids Res. 1997;25:2516–2521. doi: 10.1093/nar/25.12.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nazarenko I, Lowe B, Darfler M, Ikonomi P, Schuster D, Rashtchian A. Nucleic Acids Res. 2002;30:e37. doi: 10.1093/nar/30.9.e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Johnson SC, Sherrill CB, Marshall DJ, Moser MJ, Prudent JR. Nucleic Acids Res. 2004;32:1937–1941. doi: 10.1093/nar/gkh522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wilson DS, Szostak JW. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 105.Stoltenburg R, Reinemann C, Strehlitz B. Anal Bioanal Chem. 2005;383:83–91. doi: 10.1007/s00216-005-3388-9. [DOI] [PubMed] [Google Scholar]
- 106.Mendonsa SD, Bowser MT. J Am Chem Soc. 2005;127:9382–9383. doi: 10.1021/ja052406n. [DOI] [PubMed] [Google Scholar]
- 107.Misono TS, Kumar PKR. Anal Biochem. 2005;342:312–317. doi: 10.1016/j.ab.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 108.Peng L, Stephens BJ, Bonin K, Cubicciotti R, Guthold M. Microsc Res Tech. 2007;70:372–381. doi: 10.1002/jemt.20421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Neidle S, Balasubramanian S, editors. Quadruplex nucleic acids. Cambridge: RSC Publishing; 2006. [Google Scholar]
- 110.Ciesiolka J, Yarus M. RNA. 1996;2:785–793. [PMC free article] [PubMed] [Google Scholar]
- 111.Mann D, Reinemann C, Stoltenburg R, Strehlitz B. Biochem Biophys Res Commun. 2005;338:1928–1934. doi: 10.1016/j.bbrc.2005.10.172. [DOI] [PubMed] [Google Scholar]
- 112.Yamamoto R, Katahira M, Nishikawa S, Baba T, Taira K, Kumar PK. Genes Cells. 2000;5:371–388. doi: 10.1046/j.1365-2443.2000.00330.x. [DOI] [PubMed] [Google Scholar]
- 113.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Nature (Lond) 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 114.Silverman SK. In: Functional nucleic acids for analytical applications. Li Y, Lu Y, editors. New York: Springer; 2009. [Google Scholar]
- 115.Westhof E, Patel DJ. Curr Opin Struct Biol. 1997;7:305–309. doi: 10.1016/s0959-440x(97)80044-9. [DOI] [PubMed] [Google Scholar]
- 116.Ramos A, Gubser CC, Varani G. Curr Opin Struct Biol. 1997;7:317–323. doi: 10.1016/s0959-440x(97)80046-2. [DOI] [PubMed] [Google Scholar]
- 117.Ito Y, Fujita S, Kawazoe N, Imanishi Y. Anal Chem. 1998;70:3510–3512. doi: 10.1021/ac9800816. [DOI] [PubMed] [Google Scholar]
- 118.German I, Buchanan D, Kennedy RT. Anal Chem. 1998;70:4540–4545. doi: 10.1021/ac980638h. [DOI] [PubMed] [Google Scholar]
- 119.Huizenga DE, Szostak JW. Biochemistry. 1995;34:56–665. doi: 10.1021/bi00002a033. [DOI] [PubMed] [Google Scholar]
- 120.Guschlbauer W, Chantotand JF, Thiele D. J Biomol Struct Dyn. 1990;8:491–511. doi: 10.1080/07391102.1990.10507825. [DOI] [PubMed] [Google Scholar]
- 121.Neidle S, Parkinson G. Nat Rev Drug Des. 2002;1:383–393. doi: 10.1038/nrd793. [DOI] [PubMed] [Google Scholar]
- 122.Mergny JL, Riou JF, Mailliet P, Teulade-Fichou MP, Gilson E. Nucleic Acids Res. 2002;30:839–865. doi: 10.1093/nar/30.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Simonsson T. Biol Chem. 2001;382:621–628. doi: 10.1515/BC.2001.073. [DOI] [PubMed] [Google Scholar]
- 124.Neidle S, Parkinson G. Curr Opin Struct Biol. 2003;13:275–283. doi: 10.1016/s0959-440x(03)00072-1. [DOI] [PubMed] [Google Scholar]
- 125.Wang Y, Patel DJ. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 126.Xu Y, Noguchi Y, Sugiyama H. Bioorg Med Chem. 2006;14:5584–5591. doi: 10.1016/j.bmc.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 127.Macaya RF, Schultze P, Smith FW, Roe JA, Feigon J. Proc Natl Acad Sci USA. 1993;90:3745–3749. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kerwin SM. Curr Pharm Des. 2000;6:441–478. doi: 10.2174/1381612003400849. [DOI] [PubMed] [Google Scholar]
- 129.Oliver AW, Bogdarina I, Kneale GG. J Mol Biol. 2000;301:575–584. doi: 10.1006/jmbi.2000.3991. [DOI] [PubMed] [Google Scholar]
- 130.Simonsson T, Sjoback R. J Biol Chem. 1999;274:17379–17383. doi: 10.1074/jbc.274.24.17379. [DOI] [PubMed] [Google Scholar]
- 131.Jhaveri S, Kirby R, Conrad R, Maglott E, Bowser M, Kennedy RT, Glick G, Ellington AD. J Am Chem Soc. 2000;122:2469–2473. [Google Scholar]
- 132.Li JJ, Fang X, Schuster SM, Tan W. Angew Chem Int Ed. 2000;39:1049–1052. doi: 10.1002/(sici)1521-3773(20000317)39:6<1049::aid-anie1049>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 133.Hamaguchi N, Ellington A, Stanton M. Anal Biochem. 2001;294:126–131. doi: 10.1006/abio.2001.5169. [DOI] [PubMed] [Google Scholar]
- 134.Li JJ, Fang X, Tan W. Biochem Biophys Res Commun. 2002;292:31–40. doi: 10.1006/bbrc.2002.6581. [DOI] [PubMed] [Google Scholar]
- 135.Nutiu R, Li Y. J Am Chem Soc. 2003;125:4771–4778. doi: 10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- 136.Yamamoto R, Baba T, Kumar PK. Genes Cells. 2000;5:389–396. doi: 10.1046/j.1365-2443.2000.00331.x. [DOI] [PubMed] [Google Scholar]
- 137.Heyduk E, Heyduk T. Anal Chem. 2005;77:1147–1156. doi: 10.1021/ac0487449. [DOI] [PubMed] [Google Scholar]
- 138.Fang X, Sen A, Vicens M, Tan W. Chembiochem. 2003;4:829–834. doi: 10.1002/cbic.200300615. [DOI] [PubMed] [Google Scholar]
- 139.Zhang S, Metelev V, Tabatadze D, Zamecnik P, Bogdanov A., Jr Proc Natl Acad Sci USA. 2008;105:4156–4161. doi: 10.1073/pnas.0800162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang WJ, Chen CL, Qian MX, Zhao XS. Anal Biochem. 2008;373:213–219. doi: 10.1016/j.ab.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 141.Levy M, Cater SF, Ellington AD. Chembiochem. 2005;6:2163–2166. doi: 10.1002/cbic.200500218. [DOI] [PubMed] [Google Scholar]
- 142.Gokulrangan G, Unruh JR, Holub DF, Ingram B, Johnson CK, Wilson GS. Anal Chem. 2005;77:1963–1970. doi: 10.1021/ac0483926. [DOI] [PubMed] [Google Scholar]
- 143.Potyrailo RA, Conrad RC, Ellington AD, Hieftje GM. Anal Chem. 1998;70:3419–3425. doi: 10.1021/ac9802325. [DOI] [PubMed] [Google Scholar]
- 144.Takahashi T, Tada K, Mihara H. Pept Sci. 2006;42:483–484. [Google Scholar]
- 145.Nutiu R, Li Y. Chem Eur J. 2004;10:1868–1876. doi: 10.1002/chem.200305470. [DOI] [PubMed] [Google Scholar]
- 146.Yamana K, Ohtani Y, Nakano H, Saito I. Bioorg Med Chem Lett. 2003;13:3429–3431. doi: 10.1016/s0960-894x(03)00799-6. [DOI] [PubMed] [Google Scholar]
- 147.Kamekawa N, Shimomura Y, Nakamura M, Yamana K. Chem Lett. 2006;35:660–661. [Google Scholar]
- 148.Merino EJ, Weeks KM. J Am Chem Soc. 2005;127:12766–12767. doi: 10.1021/ja053189t. [DOI] [PubMed] [Google Scholar]
- 149.Morse DP. Biochem Biophys Res Commun. 2007;359:94–101. doi: 10.1016/j.bbrc.2007.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stojanovic MN, de Prada P, Landry DW. J Am Chem Soc. 2001;123:4928–4931. doi: 10.1021/ja0038171. [DOI] [PubMed] [Google Scholar]
- 151.Ozaki H, Nishihira A, Wakabayashi M, Kuwahara M, Sawai H. Bioorg Med Chem Lett. 2006;16:4381–4384. doi: 10.1016/j.bmcl.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 152.Urata H, Nomura K, Wada S, Akagi M. Biochem Biophys Res Commun. 2007;360:459–463. doi: 10.1016/j.bbrc.2007.06.075. [DOI] [PubMed] [Google Scholar]
- 153.Stojanovic MN, de Prada P, Landry DW. J Am Chem Soc. 2000;122:1547–1548. doi: 10.1021/ja0022223. [DOI] [PubMed] [Google Scholar]
- 154.Nutiu R, Li Y. Angew Chem Int Ed. 2005;44:5464–5467. doi: 10.1002/anie.200501214. [DOI] [PubMed] [Google Scholar]
- 155.Li N, Ho CM. J Am Chem Soc. 2008;130:2380–2381. doi: 10.1021/ja076787b. [DOI] [PubMed] [Google Scholar]
- 156.Tang Z, Mallikaratchy P, Yang R, Kim Y, Zhu Z, Wang H, Tan W. J Am Chem Soc. 2008;130:11268–11269. doi: 10.1021/ja804119s. [DOI] [PubMed] [Google Scholar]
- 157.Srinivasan J, Cload ST, Hamaguchi N, Kurz J, Keene S, Kurz M, Boomer RM, Blanchard J, Epstein D, Wilson C, Diener JL. Chem Biol. 2004;11:499–508. doi: 10.1016/j.chembiol.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 158.Achenbach JC, Nutiu R, Li Y. Anal Chim Acta. 2005;534:41–51. [Google Scholar]
- 159.Nutiu R, Yu JM, Li Y. Chembiochem. 2004;5:1139–1144. doi: 10.1002/cbic.200400026. [DOI] [PubMed] [Google Scholar]
- 160.Li J, Lu Y. J Am Chem Soc. 2000;122:10466–10467. [Google Scholar]
- 161.Carmi N, Breaker RR. Bioorg Med Chem. 2001;9:2589–2600. doi: 10.1016/s0968-0896(01)00035-9. [DOI] [PubMed] [Google Scholar]
- 162.Thomas JM, Ting R, Perrin DM. Org Biomol Chem. 2004;2:307–312. doi: 10.1039/b310154a. [DOI] [PubMed] [Google Scholar]
- 163.Liu Z, Mei SH, Brennan JD, Li Y. J Am Chem Soc. 2003;125:7539–7545. doi: 10.1021/ja035208+. [DOI] [PubMed] [Google Scholar]
- 164.Chiuman W, Li Y. J Mol Biol. 2006;357:748–754. doi: 10.1016/j.jmb.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 165.Liu JW, Lu Y. Anal Chem. 2003;75:6666–6672. doi: 10.1021/ac034924r. [DOI] [PubMed] [Google Scholar]
- 166.Liu J, Brown AK, Meng X, Cropek DM, Istok JD, Watson DB, Lu Y. Proc Natl Acad Sci USA. 2007;104:2056–2061. doi: 10.1073/pnas.0607875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Peracchi A. J Biol Chem. 2000;275:11693–11697. doi: 10.1074/jbc.275.16.11693. [DOI] [PubMed] [Google Scholar]
- 168.Wang H, Kim Y, Liu H, Zhu Z, Bamrungsap S, Tan W. J Am Chem Soc. 2009;131:8221–8228. doi: 10.1021/ja901132y. [DOI] [PubMed] [Google Scholar]
- 169.Liu J, Lu Y. Angew Chem Int Ed. 2007;46:7587–7590. doi: 10.1002/anie.200702006. [DOI] [PubMed] [Google Scholar]
- 170.Ono A, Togashi H. Angew Chem Int Ed. 2004;43:4300–4302. doi: 10.1002/anie.200454172. [DOI] [PubMed] [Google Scholar]
- 171.Yin BC, Ye B-C, Tan W, Xie C-C. J Am Chem Soc. 2009;131:14624–14625. doi: 10.1021/ja9062426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ueyama H, Takagi M, Takenaka S. J Am Chem Soc. 2002;124:14286–14287. doi: 10.1021/ja026892f. [DOI] [PubMed] [Google Scholar]
- 173.Takenaka S, Ueyama H, Nojima T, Takagi M. Anal Bioanal Chem. 2003;375:1006–1010. doi: 10.1007/s00216-003-1799-z. [DOI] [PubMed] [Google Scholar]
- 174.Hayashida H, Paczesny J, Juskowiak B, Takenaka S. Bioorg Med Chem. 2008;16:9871–9881. doi: 10.1016/j.bmc.2008.08.035. [DOI] [PubMed] [Google Scholar]
- 175.Nagatoishi S, Nojima T, Galezowska E, Gluszynska A, Juskowiak B, Takenaka S. Anal Chim Acta. 2007;581:125–131. doi: 10.1016/j.aca.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 176.Shi C, Gu H, Ma C. Anal Biochem. 2010;400:99–102. doi: 10.1016/j.ab.2009.12.034. [DOI] [PubMed] [Google Scholar]
- 177.He F, Tang Y, Wang S, Li Y, Zhu D. J Am Chem Soc. 2005;127:12343–12345. doi: 10.1021/ja051507i. [DOI] [PubMed] [Google Scholar]
- 178.Gehring K, Leroy J-L, Gueron M. Nature. 1993;363:561–565. doi: 10.1038/363561a0. [DOI] [PubMed] [Google Scholar]
- 179.Modi S, Swetha MG, Goswami D, Gupta GD, Mayor S, Krishnan Y. Nat Nanotechnol. 2009;4:325–330. doi: 10.1038/nnano.2009.83. [DOI] [PubMed] [Google Scholar]