

Abstract
Bacterial diversity among environmental samples is commonly assessed with PCR-amplified 16S rRNA gene (16S) sequences. Perceived diversity, however, can be influenced by sample preparation, primer selection, and formation of chimeric 16S amplification products. Chimeras are hybrid products between multiple parent sequences that can be falsely interpreted as novel organisms, thus inflating apparent diversity. We developed a new chimera detection tool called Chimera Slayer (CS). CS detects chimeras with greater sensitivity than previous methods, performs well on short sequences such as those produced by the 454 Life Sciences (Roche) Genome Sequencer, and can scale to large data sets. By benchmarking CS performance against sequences derived from a controlled DNA mixture of known organisms and a simulated chimera set, we provide insights into the factors that affect chimera formation such as sequence abundance, the extent of similarity between 16S genes, and PCR conditions. Chimeras were found to reproducibly form among independent amplifications and contributed to false perceptions of sample diversity and the false identification of novel taxa, with less-abundant species exhibiting chimera rates exceeding 70%. Shotgun metagenomic sequences of our mock community appear to be devoid of 16S chimeras, supporting a role for shotgun metagenomics in validating novel organisms discovered in targeted sequence surveys.
The analysis of 16S rRNA (16S) genes has become an essential component of the microbial ecologist's tool kit to evaluate the microbial composition of diverse habitats such as soils, oceans, and our own bodies. The high-sequence conservation of 16S genes among diverse bacteria allows for the phylogenetic analysis of organism diversity and the identification of new taxa. The majority of bacterial phyla are known only from 16S surveys and have no cultured representatives (Rappe and Giovannoni 2003; Wu et al. 2009). Several online resources host large, curated collections of 16S sequences, including GreenGenes (DeSantis et al. 2006a), the Ribosomal Database Project (RDP) (Cole et al. 2009), SILVA (Pruesse et al. 2007), and EZ-Taxon (Chun et al. 2007). Despite efforts by the curators to remove low-quality sequences from survey data, it is likely that many of these reference sequences reflect sequencing artifacts rather than real biological diversity.
A common source of 16S sequence artifacts is the formation of chimeric sequences during PCR amplification of the 16S genes (Fig. 1). Prior studies have indicated that ∼5% of the sequences within curated collections are anomalous or suspect, with chimeras accounting for the majority of problematic sequences (Ashelford et al. 2005). Individual sequence libraries vary greatly in sequence quality and contain few to more than 45% chimeric sequences (Huber et al. 2004; Ashelford et al. 2005, 2006; Quince et al. 2009). Experimental measurements of chimera formation during PCR coamplification of 16S rRNA sequences from cloned 16S genes or from mixed bacterial genomic DNA have indicated chimera formation rates of over 30% (Wang and Wang 1996, 1997). Multiple factors including pairwise sequence identity between 16S rRNA genes, number of PCR cycles, and relative abundance of gene-specific PCR templates have been shown to influence chimera formation (Wang and Wang 1996, 1997; Thompson et al. 2002; Acinas et al. 2005; Lahr and Katz 2009).
Figure 1.

Formation of chimeric sequences during PCR. An aborted extension product from an earlier cycle of PCR can function as a primer in a subsequent PCR cycle. If this aborted extension product anneals to and primes DNA synthesis from an improper template, a chimeric molecule is formed.
Although chimera formation rates can be lowered experimentally, no method has been shown to eliminate these artifacts entirely. Hence, the ability to recognize chimeric sequences is critical in using 16S sequences to profile microbial communities. Several computational methods have been used to identify chimeric sequences: The two algorithms most widely used for 16S chimera detection are Pintail (Ashelford et al. 2005), used by both the RDP (Cole et al. 2009) and SILVA (Pruesse et al. 2007), and Bellerophon (DeSantis et al. 2006a), used by the GreenGenes 16S rRNA sequence collection (DeSantis et al. 2006a). The 16S-specific Bellerophon algorithm developed at GreenGenes differs from the more general Bellerophon algorithm published earlier by Huber et al. (2004) and is referred to herein as BellerophonGG. Although Pintail is a more general 16S anomaly detection tool rather than a chimera detection tool, most anomalies detected by Pintail are chimeras (Ashelford et al. 2005). Although these utilities have been widely used, their accuracy for chimera detection has not been rigorously examined, particularly with respect to chimeras between closely related genes. Critically, their effectiveness when applied to data generated using newer sequencing technologies such as 454 Life Sciences (Roche) pyrosequencing has not been examined.
Unprecedented diversity in a range of samples has been reported using pyrosequencing, and has been interpreted as evidence of an important and pervasive “rare biosphere” (Sogin et al. 2006). However, these technologies may exacerbate the problem of differentiating between true, novel 16S gene sequences and sequence artifacts. For example, the combination of rigorous chimera checking and eliminating errors from flowgram interpretation have reduced diversity estimates based on pyrosequencing by a factor of 10 (Quince et al. 2009; Caporaso et al. 2010; Huse et al. 2010; Turnbaugh et al. 2010). Because next-generation sequencing technologies are increasingly used for community surveys, it is essential to determine how well these chimera-detection tools perform on these datasets.
We introduce a new chimera-detection algorithm, Chimera Slayer (CS), which can be applied to large datasets, performs well on short sequences, and is sensitive to chimeras between closely related 16S genes. We have benchmarked CS and existing tools using a carefully constructed set of simulated chimeric 16S sequences, testing the performance of each algorithm as a function of the diversity and length of the sequences. Using CS, we explore characteristics of experimentally derived chimeras from PCR-amplified 16S sequences leveraging traditional Sanger sequencing of cloned full-length PCR products and direct 454 FLX Titaninum pyrosequencing of PCR-amplified windows of the 16S gene. In applying our methods to a defined mixture of DNA representing 20 bacterial and one archaeal species we were able to assess the effects of sequence abundance, cross-taxonomic sequence similarity, and PCR conditions on the frequency and nature of experimentally derived chimeras.
Results
Evaluation of chimera detection accuracy
We evaluated the accuracy of chimera detection algorithms against a simulated set of near full-length chimeras generated from reference 16S gene sequences believed to be largely free of interspecies chimeric sequences, i.e., type strain sequences and 16S gene sequences extracted directly from sequenced bacterial genomes (see Methods). Simulated chimeras were generated from pairs of reference sequences to create a set of chimeras that ranged from 1% to 25% global sequence alignment divergence between parental pairs of reference sequences (henceforth referred to as chimera-pair divergence). One hundred chimeras were generated at each 1% chimera-pair divergence interval with single breakpoints for each pair positioned randomly. We applied each algorithm to the simulated chimera set and evaluated the sensitivity of each method by noting the percent of true-positive (TP) chimeric sequences identified as being chimeric. False-positive (FP) rates were estimated by applying the algorithms to the nonchimeric reference sequences, where predicted chimeras represented a FP event.
Published implementations of the Pintail and GreenGenes Bellerophon (BellerophonGG) algorithms were either not accessible for evaluation as part of this work or were not designed for high-throughput automated execution. Therefore, we reimplemented the algorithms based on published descriptions and evaluated our own implementations (see Methods). Our reimplemented versions of these tools perform similarly to the original tools (Supplementary Fig. S1). WigeoN is our reimplementation of Pintail.
We developed and evaluated two additional algorithms, KmerGenus and Chimera Slayer (see Methods). KmerGenus computed a catalog of exact 50-mers unique to each genus within a reference 16S sequence set. Query sequences found to contain genus-unique 50-mers matching multiple taxa were flagged as chimeras.
Chimera Slayer (CS) involved the following series of steps that operate to flag chimeric 16S sequences: (1) the ends of a query sequence (30% of the length from each end) were searched against a database of reference chimera-free 16S sequences to identify potential parents of a chimera. The top matching reference sequences were retrieved in NAST (DeSantis et al. 2006b) multiple alignment format; (2) candidate parents of a chimera were selected as those that form a branched best-scoring alignment to the NAST-formatted query sequence; (3) the NAST alignment of the query sequence was improved in a “chimera-aware” profile-based NAST realignment to the selected reference parent sequences; and (4) an evolutionary framework was used to flag query sequences found to exhibit greater sequence homology to an in silico chimera formed between any two of the selected reference parent sequences (complete details in Methods).
Different chimera-checking methods have markedly different detection accuracy, especially for chimeras between closely related sequences
All tested methods identified simulated chimeras derived from highly divergent 16S sequences (e.g., >15% divergence) with high sensitivity (Fig. 2A). As the pairwise divergence of the sequences leading to a chimera decreased, however, differences in the sensitivity of chimera detection became apparent. The sensitivity of BellerophonGG was limited to chimeras with the highest chimera-pair sequence divergence, requiring at least 13% chimera-pair divergence to flag at least 50% of the chimeric sequences as chimeras. The sensitivity of WigeoN largely mirrored that of KmerGenus and both were intermediate between CS and BellerophonGG. CS exhibited the best overall sensitivity, recognizing the most divergent chimeras while retaining high sensitivity for chimeras with minimal chimera-pair divergence. CS recognized >87% of chimeras with a minimum of 4% chimera-pair divergence. In addition, the FP rate remained low at only 1.6%. The sensitivity of these algorithms to the level of chimera divergence correlated with their ability to detect chimeras formed at different taxonomic levels (Fig. 2B). All methods have excellent sensitivity for intra-phylum, intra-class, and intra-order chimeras. All but BellerophonGG maintained high sensitivity for intra-family chimeras, but CS was especially effective for detecting intra-genus chimeras. Because BellerophonGG exhibited relatively low sensitivity and a high FP rate (7.1%), we did not pursue it further. KmerGenus was incapable of detecting intra-genus chimeras due to design constraints. Sequence variations due to simulated sequencing error or sequence evolution adversely impacted chimera detection accuracy, but both WigeoN and ChimeraSlayer were largely robust to these effects (Supplemental Text S1; Supplemental Figs. S2–S6). In contrast, the accuracy of KmerGenus rapidly deteriorated with diverged query sequences, exhibiting 57% TP and 26% FP at 5% sequence divergence (Supplemental Fig. S2). Simple taxon-specific Kmer methods thus become unreliable when sequencing increasingly novel diversity in biological samples that is not represented in the reference set, or when sequencing errors are frequent.
Figure 2.

Comparison of chimera detection sensitivity among methods. (A) Chimera detection sensitivity as a function of chimera divergence; (B) chimera detection sensitivity according to the shared level of taxonomy between the proposed parental sequences. Cumulative false-positive rates were as follows: CS, 1.6%; WigeoN, 0.67%; BellerophonGG, 7.13%; KmerGenus, 0%.
It was possible that some sequences flagged as chimeras in our reference set, and presently designated false-positives, represent genuine 16S sequences that had recombinant origins or otherwise unusual evolutionary histories. Of the 4769 presumed nonchimeric, reference sequences evaluated by CS, 77 were flagged as putative chimeras, distributed as 40 intra-genus, 28 intra-family, seven intra-order, one intra-class, and one intra-phylum chimeras. However, upon close inspection, some of these 77 putative chimeras appeared to reflect recombined sequences. Of the 40 intra-genus chimeras, 19 corresponded to Actinobacteria. For example, the Mycobacterium pulveris 16S sequence (S000004105) appears to be a chimera between the 16S sequences of Mycobacterium elephantis (S000002743) and Mycobacterium rhodesiae (S000015160) (Supplemental Fig. S7). We could not rule out the possibility that these sequences were genuine chimeras, since chimeric/recombinant 16S genes do occur in nature (Boucher et al. 2004; Harth et al. 2007). If some of these “false-positives” were genuine chimeras, the specificity of CS and the other tools evaluated here may be higher than estimated, and predicted intra-genus chimeras among certain taxonomic groups such as the Actinobacteria would warrant further attention.
Leveraging a controlled community to study effects of 16S chimeras
One difficulty in analyzing sequences from environmental samples is that it is not possible to discriminate a priori and with high confidence between novel but genuine sequences and anomalous sequences. By sequencing known species assemblages, however, we could quantify and characterize the performance of a tool in chimera detection. Thus, we applied these tools to a synthetic (also known as mock) microbial community created from purified genomic DNA of bacteria for which finished genome sequences were available (see Methods). This mock community contained equivalent concentrations of 16S genes for each included species (eMC [even composition mock community]).
Organisms were chosen to represent a broad range of phylogenetic distances, genome sizes, and GC content. We subjected this community to 16S profiling by both traditional Sanger (Supplemental Text S3, S4) and 454 pyrosequencing methods (described below) and assessed the frequency of chimeric and anomalous sequences. Each sequencing effort involved four technical replicates, with each replicate performed by four sequencing centers at Baylor College of Medicine, the Broad Institute, the J. Craig Venter Institute, and Washington University.
Evaluation of chimera content in 454 pyrosequencing surveys
Because read lengths using 454 FLX Titanium pyrosequencing were limited to ∼500 bp, only a portion of the 16S gene could be targeted for 454 sequencing. Detection of chimeras among these shorter sequences was crucial for obtaining accurate diversity results (Quince et al. 2009). Although WigeoN provided effective chimeric sequence detection with full-length sequences, it lacked sensitivity at shorter sequence lengths (Supplemental Text S5; Supplemental Fig. S8). However, CS retained near maximal chimera detection accuracy for sequences with length at least 400 bases (Supplemental Fig. 8A), and therefore was suitable for application to 454 sequencing reads.
Although several different regions of the 16S gene have been targeted for 16S surveys via 454 pyrosequencing, most studies (e.g., Liu et al. 2007, 2008; Wang et al. 2007) suggest that several regions are each adequate for community comparison and taxonomy assignment. We have used CS to determine whether the rates of chimera formation differed when these separate regions were PCR amplified. These comparisons had not been previously performed, in part because of the lack of effective methods for detecting chimeras in large numbers of short reads. Using our eMC DNA as a template, we amplified three separate 16S windows, V1–V3, V3–V5, and V6–V9 (Supplemental Fig. S9).
The amplification of these shorter windows showed differential bias resulting in non-uniform species abundance estimates (Supplemental Fig. S10). Only the V3–V5 primer set yielded a detectable number of sequences corresponding to Methanobrevibacter, with ∼100-fold fewer detected than the other organisms in our mock community.
Sequencing of technical replicate samples demonstrated consistently high chimera rates ranging from ∼15% to over ∼20% (Supplemental Fig. S11A). The cumulative chimera rates for the V6–V9 region were slightly (∼3%) greater than the V1–V3 and V3–V5 regions. Relative chimera pair abundance estimates were similar across the V-regions; notable exceptions included the high prevalence of Acinetobacter/Staphylococcus pairs in the V6–V9 window and Deinococcus/Staphylococcus pairs in the V1–V3 window (Supplemental Fig. S11B). The distribution of chimera breakpoints observed in full-length clones did not readily explain the higher frequency for certain organism pairs and windows. For example, the Deinococcus/Staphylococcus pairs in full-length data had breakpoints enriched in the V6 region (Supplemental Text S4) while in the 454-sequenced windows, chimera pairs in the V1–V3 window were almost twofold higher than those obtained with V3–V5 or V6–V9. Further, among the organisms in our mock community, we do not find evidence for differential CS sensitivity in detecting chimera abundance by organism pair or region. However, two notable chimera pairs eluded detection due to insufficient sequence variation: Staphylococcus aureus/Staphylococcus epidermidis in windows V3–V5 and V6–V9, and Streptococcus agalactiae/Streptococcus pneumoniae in window V6–V9 (Supplemental Fig. S12).
More similar 16S genes clearly form chimeras more readily. When we mitigated sequence abundance effects by considering only cases where a less-abundant species formed chimeras with a more abundant species, we observed a strong positive correlation between the percent identities shared by the 16S sequence of chimera pair species and the percent of chimeras observed. This correlation (R2 = 0.90–0.94) was best demonstrated with Staphylococcus, Acinetobacter, and Listeria, each of which had a wide range of sequence identity to alternate organisms within the mock community (Fig. 3A). The total number of chimeric sequences observed for a given genus showed a strong positive correlation (R2 = 0.87) with the total sequence abundance corresponding to that genus (Fig. 3B). Thus, the abundance of chimeras corresponding to a given genus appeared to be a reflection of both the degree of 16S sequence identity and the abundance of sequences from organisms within the genus.
Figure 3.

Correlation of chimera content with sequence homology and organism abundance. (A) Percent of other organism abundance corresponding to chimeras with the indicated more abundant species (y-axis), plotted according to percent identity (x-axis) between homologous 16S genes. (B) Number of chimeric sequences corresponding to a given genus were plotted as a function of total genus-level classified reads for the even (eMC) and staggered (sMC) mock community. Total read counts were based on best BLASTN match (E ≤ 10−10) to reference sequences for nonchimeras in addition to the genus representation within the CS-predicted chimeras. (C) Percent of sequences that correspond to chimeras for each genus plotted according to genus-level sequence abundance. Error bars correspond to standard error from the mean based on four technical replicates.
To further test the hypothesis that more abundant organisms form chimeras more readily, we used another mock community (sMC [staggered mock community]) containing the same species, but with 16S template concentrations staggered across four orders of magnitude (Supplemental Fig. S13). The strong positive correlation between organism abundance and number of chimeras in the sMC is much more apparent (R2 = 0.97) (Fig. 3B). The eMC data exhibited a range from ∼10% to 53% chimeras in each genus (Enterococcus, avg. 46%) with the cumulative chimera content of the eMC at <20% (Fig. 3C). In contrast, the sMC, with its expanded range of species sequence abundance, exhibited greater disparity in the amount of chimeras detected in each genus, exceeding 70% (Enterococcus, avg. 65%) of sequences in a given genus represented by chimeras.
Interestingly, the same chimera often appeared in multiple, independent amplifications. For example, we observed a chimera between Streptococcus and Staphylococcus 16S sequences generated during PCR across the V3–V5 region (Fig. 4). This chimera pair was generated in each of four experimental replicates, and exemplifies the reproducibility of chimera formation and breakpoint occurrence across multiple PCR reactions. Often the appearance of “novel” sequences in multiple independent 16S libraries is viewed as confirmation of the validity of such sequences (Kunin et al. 2010), but our results cast doubt on this practice.
Figure 4.

Alignment of sequences corresponding to chimeras between Streptococcus and Staphylococcus 16S rRNA genes. Only columns from the NAST multiple alignment containing nonidentical nucleotides between the reference sequences (top and bottom) are shown. Nucleotides matching Streptococcus sequences are colored red. Sequence prefixes correspond to the four experimental replicates A–D.
Exploration of 16S chimeras within 454 whole-genome shotgun metagenomics
Chimeras are clearly a hindrance to the accurate discovery of novel organisms in PCR-based 16S surveys. An alternative to targeted PCR-based surveys is whole-genome shotgun (WGS) metagenomics. These surveys randomly sample every DNA sequence present, and sequences corresponding to 16S can be retrieved and analyzed separately. Although methods involving WGS metagenomic sequencing can involve PCR amplification steps, they are not directed to specific gene targets, and so chimera formation would be expected to be minimal.
To explore 16S chimeras in WGS metagenomic surveys, we performed 454 WGS metagenomic sequencing on our eMC DNA (SRR072233). Approximately 1.4 million reads were generated from which 4273 reads (0.31%) were found to correspond to 16S based on a BLASTN search of the mock community reference 16S sequences (E ≤ 10−10), representing each of the included organisms (Supplemental Fig. S14). These 16S reads were examined using CS and no reads were flagged as potential chimeras.
Discussion
The essential function of the 16S gene, and its highly conserved sequence and structure, has made it the molecule of choice for studies of microbial evolution and ecological surveys (Pace 1997; Tringe and Hugenholtz 2008). The many highly conserved regions spanning the length of the gene enable the amplification of sequences from a broad range of species. These same highly conserved regions, however, contribute to cross-hybridization and mispriming events during amplification that create chimeric sequences. Although the majority of chimeras form between closely related sequences, organisms across different phyla can form chimeras, and these are most likely to be classified as novel organisms if not properly identified as aberrant.
Properly identifying chimeric 16S sequences is a challenging computational problem. In evaluating chimera detection accuracy of the widely utilized Pintail and BellerophonGG algorithms, we found them to vary considerably, with BellerophonGG capable of recognizing chimeras mostly restricted to the most divergent sequence pairs. Recently, attention has turned toward sequence surveys that sample shorter regions of the 16S gene and/or are applying next-generation sequencing technologies that are currently limited to short sequence lengths. Although the Pintail algorithm has excellent chimera detection capabilities in full-length sequences, it has little sensitivity for detecting chimeras in shorter sequences. Our new CS tool is the only method currently capable of sensitive chimera detection in short 16S sequence reads. However, perfect chimera detection is still an unsolved problem. Although CS is largely robust to varying sequence characteristics including divergence and length, detection accuracy does begin to degrade with increasing divergence to reference sequences. This underscores the importance of obtaining and validating sequences that represent novel bacterial diversity and continuing to expand upon the reference database leveraged by CS and additional analysis tools. Also, CS is designed to detect only the simplest form of chimeras, involving two homologous parental sequences. More complex chimeras and sequence anomalies may evade detection. Given that chimeric sequences can be rare and diverse, the problem of identifying rare species correlated with disease or other important microbial ecosystem function remains challenging.
Shorter PCR products targeted to windows of the 16S gene were surprisingly rife with chimeras. Experiments with our synthetic mock community indicated higher chimera rates (∼15%–20%) as compared with our observations with Sanger-sequenced clones of full-length PCR products, with <10% chimeras (Supplemental Text S3). Although breakpoints among chimeras in full-length PCR products appeared to show bias toward the V6 region for multiple species pairs, chimeric content of shorter PCR products spanning the V6 region was not significantly greater than with products spanning the V1–V3 or V3–V5 regions (Welch two sample t-test, P > 0.05). The high chimera rates within short PCR products targeted to next-generation DNA-sequencing technologies indicates the continued importance of chimera screening in such sequence surveys and the need for tools such as CS that are capable of detecting chimeras in short reads.
Cumulative chimera rates, as often cited in previous studies, grossly understate the magnitude of the chimera problem. Cumulative rates can be heavily biased toward the most abundant species in the sample. Although a cumulative chimera rate of ∼20% may be observed with our 454 FLX Titanium sequences, >70% of sequences representing particular genera in the sample can be chimeric.
Sequence reads from previously known organisms tend to be well classified by existing methods (Wang et al. 2007), and these methods continue to perform accurately in the presence of chimeras. However, many new taxa are incorrectly “discovered” due to chimeric sequences. By restricting evaluation of sample diversity to those sequences classified at high confidence, chimeras appeared to minimally affect estimates of diversity via taxonomic binning (Supplemental Text S6, S7), with the caveat that low-abundance taxa should be treated with skepticism.
Chimera formation between highly divergent species is not rare; it occurs reproducibly and over both long (∼1500 bp) and short (∼500 bp) PCR amplicons. This implies that the often-suggested criterion for trusting a novel sequence—that it appear in multiple samples or experiments (Kunin et al. 2010)—may not be sufficiently stringent. Even when applying PCR to harvest 16S sequences from clonal species, one must be very careful in analyzing such sequences, since even low levels of contaminating microbes can result in chimeric PCR products (data not shown).
The goal of chimeric 16S detection tools should be to identify likely unnatural artifacts, such as chimeras resulting from PCR amplification, and to avoid flagging sequences that correctly represent biology and evolution. Including such naturally occurring chimeric sequences in the reference set ensures that query sequences with best alignments to naturally chimeric reference sequences are not flagged inappropriately. Since the reference collection of 16S sequences does not represent all of the bacterial diversity, putative intra-genus chimeras identified in sequence surveys should be treated with skepticism since many may represent genuine sequence diversity and naturally occurring chimeras. The predicted intra-taxon chimera type is reported in the output of CS so that researchers can make informed decisions regarding the types of chimeras that may deserve special attention. For example, retaining intra-genus chimeras for subsequent analyses such as taxonomic binning may be warranted, but defining new organisms based on sequence clustering should proceed with caution, especially given that chimeras reproducibly form across multiple experiments.
In addition to pursuing advancements in detection of chimeras once they are formed, there is a need to identify experimental conditions that are least conducive to chimera formation (Wang and Wang 1996; Thompson et al. 2002; Lahr and Katz 2009). Our investigation into the effects of multiple PCR conditions on the observed prevalence of chimeras among 454 pyrosequences and Sanger-sequenced clones supports a dominant effect of amplification cycle number (Supplemental Text S8). By limiting the number of amplification cycles to the fewest number needed to produce yields required for sequencing, one can mitigate the relative yield of chimeric sequences. Although we detect minimal chimeras formed at 20 cycles, earlier studies observed near peak chimeras formed at 20 cycles (Wang and Wang 1996). Capturing the amplification product at a time where yield is maximized and chimeras are minimized will likely depend on the PCR protocol utilized. Further exploration of PCR conditions, such as by leveraging single molecule amplification in oil emulsions, could prove highly advantageous (Williams et al. 2006); our preliminary investigation into emulsion PCR targeting 16S genes suggests this may be a promising avenue (data not shown).
We were unable to detect chimeric 16S sequences in our 454 pyrosequencing WGS experiment, suggesting that WGS is relatively chimera free. However, the concentration of 16S reads in this data set was very low (0.31% of total reads), which likely minimized the opportunity for cross-hybridization among 16S sequences. Ultimately, the small number of 16S sequences generated by the WGS approach suggests that pursuing WGS methods as an alternative to directed 16S sequence surveys to specifically mine 16S data is neither efficient nor cost effective. Perhaps, as costs of sequencing continue to plummet, WGS methods will become a viable alternative to directed 16S sequence surveys. Until then, optimizing PCR conditions to mitigate chimera amplification and leveraging tools such as CS to flag suspect sequences should help minimize the impact of such artifacts on related microbiota research.
It is also important to note that chimeras are only one source of diversity artifacts. Even with filtering of chimeras, the appearance of unique sequence clusters occurs at a high rate when compared with known sample diversity. This is particularly true for reads generated using 454 pyrosequencing as compared with the Sanger-generated reads; thus, the effects of sequencing error and other anomalies cannot be ignored (Quince et al. 2009; Kunin et al. 2010). Additional studies leveraging controlled mock communities should help clarify insights into the true diversity represented within the rare biosphere.
Methods
PCR, cloning, sequencing, and analysis of Sanger-sequenced 16S sequences are described in the Supplemental Methods section of the Supplemental Text.
Mock communities
The organisms for the mock community included a variety of different genera commonly found on/within the human body. The bacterial DNAs were collected from the American Type Culture Collection (ATCC), the Deutsche Sammlung von Mikroorganism und Zellkulturen (DSMZ), and our internal repository, with contributions from collaborating scientists. The selection of mock organisms and preparation of genomic DNAs are to be described as part of a separate HMP consortium manuscript (J.F. Petrosino et al., in prep.). Information describing the mock community contents are available on the HMP Data Analysis and Coordination Center website (http://www.hmpdacc.org/). The 16S gene content from each DNA preparation was assayed by qPCR to calculate the concentration of 16S gene copies. To generate the even and staggered mock communities, DNA from each organism was mixed according to the calculated 16S concentration. In the even community, the 16S concentration from all organisms was normalized so that each organism contributed a calculated number of 100,000 16S molecules to each amplification reaction. In the staggered mock community, species were present in one of four concentrations calculated to contribute either 103, 104, 105, or 106 16S molecules per reaction. The strains and the molecules per staggered reaction are as follows: 103 16S molecules per reaction (Actinomyces odontolyticus ATCC17982, Bacteroides vulgatus ATCC8482, Deinococcus radiodurans ATCC20539, Enterococcus faecalis ATCC7077, Streptococcus pneumoniae ATCC BAA-334), 104 molecules per reaction (Acinetobacter baumannii ATCC17978, Helicobacter pylori ATCC700392, Lactobacillus gasseri ATCC20243, Listeria monocytogenes ATCC BAA-679, Neisseria meningitidis ATCC BAA-335, Propionibacterium acnes DSM16379), 105 molecules per reaction (Bacillus cereus ATCC10987, Clostridium beijerinckii ATCC51743, Pseudomonas aeruginosa ATCC47085, Staphylococcus aureus ATCC BAA-1718, Streptococcus agalactiae ATCC BAA-611), and 106 molecules per reaction (Escherichia coli ATCC700926, Methanobrevibacter smithii ATCC35061, Rhodobacter sphaeroides ATCC17023 Staphylococcus epidermidis ATCC12228, Streptococcus mutans ATCC700610). Candida albicans ATCC SC5314 was included as a negative control, but limited to only 103 18S copies (calculated) per microliter.
Amplification and 454 sequencing of targeted 16S gene variable regions
Amplification primers were designed with FLX Titanium adapters (A adapter sequence: 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′; B adapter sequence: 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-3′) and a sample barcode sequence where applicable directly on the 5' end of the 16S primer sequence: Forward primers contained the B adapter and the reverse primers contained the A. The 16S-specific sequence with 454 adapters were as follows: V1–V3 primers: 454B_27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 454A_534R (5′-ATTACCGCGGCTGCTGG-3′); V3–V5 primers: 454B_357F (5′-CCTACGGGAGGCAGCAG-3′) and 454A_926R (5′-CCGTCAATTCMTTTRAGT-3′); V6–V9: 454B_U968F (5′-AACGCGAAGAACCTTAC-3′) and 454A_1492R-MP (5′-TACGGYTACCTTGTTAYGACTT-3′) (Lane 1991; Yu and Morrison 2004; Hamady et al. 2008). Polymerase chain reaction (PCR) mixtures (25 μL) contained 10 ng of template, 1× Easy A reaction buffer (Stratagene), 200 mM of each dNTP (Stratagene), 200 nM of each primer, and 1.25 U of Easy A cloning enzyme (Stratagene). The cycling conditions for the V1–V3 amplicon consisted of an initial denaturation of 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 40 sec, annealing at 56°C for 30 sec, extension at 72°C for 1 min and a final extension at 72°C for 7 min. The cycling conditions for the V3–V5 and V6–V9 amplicons consisted of an initial denaturation of 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 40 sec, annealing at 50°C for 30 sec, extension at 72°C for 1 min, and a final extension at 72°C for 7 min. The PCR products were purified with QIAquick PCR purification kit (QIAGEN) according to the manufacturer, and size was selected on a 1% agarose gel. The gel bands were purified with QIAquick gel extraction kit (QIAGEN) according to the manufacturer's instructions with one modification: The gel bands were dissolved at room temperature on a Dynal Bioteck Rotator (Model RKDYNAL, setting 30, Invitrogen, Life Technologies) for 15 min. DNA was eluted in 25 μL of 1× low TE buffer (pH 8.0). The DNA was quantified on an Agilent Bioanalyzer 2100 DNA 1000 chip (Agilent Technologies). The number of molecules for each sample was calculated using size (bp) and concentration (ng/mL) data from the Agilent. All three PCR products were normalized to the same molecule concentration (1.0 × 109 molecules/μL), pooled in equal volumes, and diluted to an emulsion PCR working concentration of 2.0 × 106 molecules/μL. Emulsion PCR and sequencing were performed according to the manufacturer's specifications.
Processing of raw sequence data
454 FLX Titanium pyrosequence processing
Pyrosequences were processed using a combination of MOTHUR (Schloss et al. 2009) and custom PERL scripts. Sequences were removed from the analysis if they were <200 nt or >600 nt, had a read quality score <25, contained ambiguous characters, had a nonexact barcode match, or did show more than four mismatches to one of the three used reverse primer sequences (534R, 926R, and 1492R). Remaining sequences were assigned to samples based on barcode matches, after which barcode and primer sequences were trimmed and reads were oriented such that all sequences begin with the 5′ end according to standard sense strand conventions. Because of sequencing bias likely due to hairpin formations with the adapter and forward 16S primer, we restricted our analyses to sequences derived from the reverse 16S primer. Counts of pyrosequenced reads analyzed are included in Supplemental Table S1. Sequence data were deposited under NCBI Genome Project ID 48465 as SRA project SRP002443. Processed data partitioned according to replicate and 16S region (V1–V3, V3–V5, and V6–V9) are provided as downloadable FASTA files at http://microbiomeutil.sf.net.
Fixed-width alignment of 16S sequences using NAST-iEr
NAST-formatted alignments (DeSantis et al. 2006b) were generated using a variant of Needleman–Wunsch dynamic programming (Needleman and Wunsch 1970). A query sequence was aligned to a NAST-formatted reference sequence (or set of NAST-formatted reference sequences), and gap insertion was restricted to the query sequence in generating the global optimal alignment. End-gaps in the aligned query sequence were not penalized (because the subject sequences were usually partial), and regions of the query sequence that extended beyond the boundaries of the NAST-formatted reference sequence(s) were excluded in order to maintain the fixed width; this was particularly useful in the case where the query included unaligned vector or low-quality sequence at its ends, which in many cases became excluded from the resulting alignment. When a query was aligned to a set of multiple reference sequences, a profile was constructed based on the multiple reference sequences, and alignment scores were computed by summing all match and mismatch scores within a position of the alignment. Pre-existing gap characters in the NAST-formatted reference sequences were not penalized when aligned to a gap inserted in the query. The global dynamic programming algorithm with a fixed width profile P and unaligned query sequence Q was defined by the following recursion:
 |
The optimal scoring alignment was chosen as max[F(i,j)], where i was the position of the last position in the NAST alignment profile.
Reference NAST alignments were selected by searching a FASTA formatted database of reference 16S sequences using MEGABLAST. Those reference sequences with a BLAST E-value ≥ 10−50 and having a BLAST alignment score within 80% of the value of the top match were selected (maximum of 10 sequences) and an alignment profile was constructed, tabulating the residue types (including gaps) at each column of the multiple alignment. The query sequence was aligned to this profile as described above. The NAST-iEr alignment algorithm was written in the C language, and wrapped by a PERL script that performed the BLAST search against the unaligned reference sequences and extracted the corresponding reference sequences from the NAST-formatted database. Generating a NAST alignment for a single query sequence, including performing the reference sequence database search, takes on the order of one second per sequence on an average desktop computer.
Obtaining a database of chimera-free reference 16S sequences
A database of what was expected to be mostly chimera-free sequences was compiled from two sources: 5165 full-length 16S sequences corresponding to type strains were obtained from the RDP website (http://rdp.cme.msu.edu/), and 4218 16S genes were identified from complete and high-quality draft bacterial genomes. All available bacterial genomes were downloaded from GenBank and 16S genes were identified using RNAmmer (Lagesen et al. 2007). A large overlap exists between the sequences derived from these two sources, and so CD-HIT (Li and Godzik 2006) was used to retrieve the longest nonredundant reference sequence (requiring 99.5% identity), yielding 5408 sequences. Sequences found to contain greater than 2% “N” characters were excluded (eliminating 196 sequences). The remaining sequences were aligned using NAST-iEr against the GreenGenes “core” NAST alignment database (http://greengenes.lbl.gov/Download/Sequence_Data/Fasta_data_files/core_set_aligned.fasta). Those sequences with <90% of their length represented within the confines of the NAST alignment were removed (eliminating 31 sequences). The resulting reference database consisted of 5181 sequences, 4468 corresponding to type strains, and the remaining 713 derived from complete or draft genome sequences. The complete taxonomy of each sequence, including domain, phylum, class, order, family, and genus was predicted using the RDP Bayesian classifier (Wang et al. 2007). The NAST alignments for this reference database were iteratively improved by leveraging NAST-iEr in rounds of realignments until the alignments stabilized (Supplemental Figure S15).
Construction of a database of simulated 16S chimeras for evaluation of detection methods
Simulated chimeric 16S sequences were constructed by joining two immediately adjacent segments of a pair of NAST-formatted reference sequences. A random breakpoint was selected from the range of the NAST alignment (7682 columns) between the positions corresponding to 200 and 1200 in the E. coli unaligned reference sequence. At least 50 nucleotide characters (G, A, T, or C) were required on each side of the breakpoint. Sequence divergence between the pair of reference sequences on each side of the breakpoint was required to differ by <10 % of the global sequence divergence between the two selected reference sequences. The disparate sequence regions from each side of the breakpoint were joined to create a simulated chimera. The pair of reference sequences from which the chimera was derived is referred to as the parents. The divergence between the parents is referred to as the chimera-pair divergence. Pairs of parental reference sequences to be joined into a chimera were randomly selected based on differences at each level of their taxonomy (intra-phylum chimeras down to intra-genus chimeras). Smaller length simulated chimeras were constructed similarly according to the targeted unaligned sequence lengths.
Simulated sequence divergence was performed by randomly selecting a position within the NAST-formatted chimera sequence and introducing a mismatch, insertion, or deletion, as specified. Point mutations were applied until reaching the targeted level of sequence divergence, disallowing multiple mutations at the same site. Mutated positions were selected based on a uniform random distribution provided by the rand() function in PERL, thus effectively using the Jukes-Cantor one-parameter model of molecular sequence evolution with no heterogeneity of rates across sites.
Detection of chimeric 16S sequences
GreenGenes Bellerophon
The GreenGenes Bellerophon algorithm (DeSantis et al. 2006a) is currently available only in the form of a web service offered by the GreenGenes website (http://greengenes.lbl.gov/cgi-bin/nph-bel3_interface.cgi). It was not possible for us to examine the accuracy of the GreenGenes Bellerophon web service with our test regime due to its special formatting requirements, such as requiring NAST alignments and associated data generated by the webserver as a prerequisite to chimera checking. Instead, we reimplemented a GreenGenes Bellerophon utility based on the published algorithm description and set parameters according to default settings on the GreenGenes website. An abridged set of sequences from the test regime was submitted to the web service for processing and the results were highly comparable to our reimplemented version (Supplemental Fig. S1A).
The GreenGenes Bellerophon algorithm was reimplemented as follows: A whole query sequence was searched against only the reference 16S database using BLASTN. The top 10 reference database sequences were retrieved in NAST format. The query was NAST aligned using NAST-iEr. Each pair of the top 10 matching reference sequences were considered as potential parents of the candidate chimeric query sequence. First, the GreenGenes-provided lane mask (http://greengenes.lbl.gov/Download/Sequence_Data/Fasta_data_files/lanemask.fasta) was applied to conceal hyper-variable positions in the alignment. The NAST-formatted query and each pair of potential parents were examined separately using the GreenGenes Bellerophon algorithm: The columns of the NAST multiple alignment of the three sequences (two parents and query) that exclusively contain gap characters were first removed. A pair of adjacent windows (left and right window) each of 300 columns of the resulting alignment was slid from 5′ to 3′ across the multiple alignment with a step length of 10 columns, each time repositioning a putative chimera breakpoint.
Given a candidate chimeric query sequence Q and two putative parents of the chimera (P1 and P2) and a putative chimeric breakpoint with 300-bp windows to the left Wl and right Wr, the percent identities were computed between each pair of sequences within each window. At the position of a breakpoint, two chimeric products between the parents were possible: {(P1,Wl), (P2,Wr)} and {(P2,Wl), (P1,Wr)}.
A divergence ratio was computed as the average percent identity (PerID) between the two windows corresponding to the query and a putative chimera, divided by the percent identity between the two nonchimeric parents:
 |
If, at any step, the divergence ratio meets a minimum threshold of 1.1 (default value at GreenGenes), the query sequence was flagged as a potential chimera.
WigeoN (reimplemented Pintail)
The publicly available version of the Pintail chimera detection software is a graphical interface-driven software intended for manual analysis of potentially chimeric sequences. It was not designed for use in a high-throughput setting. In addition, the available software was not suited for use with NAST-formatted alignments. To evaluate the Pintail algorithm and to obtain a version of the software that was both compatible with NAST-alignments and for use in a high-throughput environment, we reimplemented the algorithm as previously described (Ashelford et al. 2005). A query sequence was searched against the reference 16S database using MEGABLAST. The top matching reference sequence and the query sequence, both in NAST format, were compared using the Pintail algorithm, using our implementation that we named WigeoN. A mask was applied to the NAST alignment to include only those columns that correspond to residues in the E. coli reference sequence. The global sequence divergence between the resulting reference and query alignment was computed. A window of 300 columns of the multiple alignments was slid from left to right with a step of 25 columns, and the sequence divergence within each window was calculated. The standard deviation of sequence divergence among all windows was computed as the deviation from expected (DE) value. The distribution of DE values for nonanomalous 16S sequences at a given interval of global sequence divergence was computed a priori by performing an all-vs.-all WigeoN analysis of sequences in the 16S reference database and binning DE values at every 1% average sequence divergence interval between 1% and 30%. The DE value computed from the query and reference sequence comparison was compared with the distribution of known reference DE values at that global sequence divergence, and if it exceeded the 99th percentile of known values, it was flagged as a potential anomalous sequence. To ensure a proper reimplementation of the Pintail software, we compared DE values for simulated chimeras between Pintail and WigeoN and found the values to be nearly perfectly correlated (R2 = 0.993, Supplemental Fig. S1B).
Taxon-specific Kmers
All overlapping 50 mers (Kmers of length 50) were extracted from each of the reference sequences in the database corresponding to those sequences with validated taxonomic predictions and those used for synthetic chimera construction (as described earlier). Those Kmers that were identified as unique to a genus were cataloged as genus-specific Kmers. Given a query sequence, all overlapping 50 mers were examined and those matching taxon-specific Kmers were identified. If multiple taxon-specific Kmers are identified in the query sequence and the second most abundant set of taxon-specific Kmers comprised at least 10% of all genus-specific Kmers, the query was flagged as a potential chimera.
ChimeraSlayer (CS)
Detection of chimeric 16S sequences by CS occurred in several stages outlined below:
Search query sequence termini to identify nearest neighbors. The terminal regions of the query sequence, each corresponding to 30% of the query length, were independently searched against the reference 16S database using MEGABLAST. The top 15 matches from each search were extracted in NAST format.
-
Identification of chimera parent candidates. Potential parents of a candidate chimeric sequence were identified such that an in silico chimera among multiple parent reference 16S sequences existed that had a higher scoring pairwise alignment to the query than did any individual 16S reference sequence across the length of the entire alignment. In the context of the existing NAST multiple alignment of reference sequences chosen above in step 1, the highest-scoring alignment of the query to reference sequences allowing for multiple breakpoints (chimerization events) was computed. This best alignment was computed using a dynamic programming alignment algorithm, conceptually similar to the algorithm implemented in CHECK_CHIMERA (Komatsoulis and Waterman 1997), penalizing mismatches and breakpoints, like so:
Given a NAST alignment for each of the i top matching reference sequence and NAST alignment position j:
where s(i,j) corresponds to the score between the query sequence at position j with the NAST-formatted reference sequence i at position j, valued as a match (+5), mismatch (−4), or zero in the case where two gaps are aligned. F(i,j) corresponds to the maximum alignment score between the query and reference sequence i between NAST alignment positions 1..j, allowing for breakpoints. To minimize overzealous branching of the alignments (which, given a low breakpoint penalty, could occur to circumvent most mismatches in the alignment), the breakpoint penalty was computed at runtime as described below. CS used the concept of a minimum divergence ratio (minDivR), computed as the minimum value of the percent identity between a query sequence and putative chimera (C) divided by the percent identity between the query (Q) and either of the parents (P1 or P2):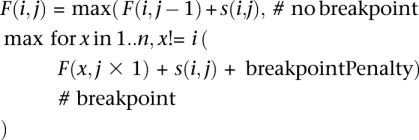
The default value of 1.007 required that if a query was to be flagged as a chimera, an alignment between a query and one of the parents should be, at most, 99.3% identical when the alignment between the query and a chimera was a perfect alignment. The breakpoint penalty was set based on this premise. The breakpoint penalty corresponds to the minimum value required to exceed the cost of the minimal number of mismatches allowed between a query sequence and a nonchimeric parent, according to the minDivR.
A best alignment that lacked branching (and hence, included only a single reference sequence) was reported as nonchimeric. The branched alignments, having one or more breakpoints including two or more reference sequences, continued on to the next stage of the chimera detection pipeline. The output of this parent selection step included the neighboring regions of the alignment that corresponded to the multiple reference sequences separated by putative breakpoints, and their local percent identity compared with the global percent identity between the query and each reference sequence.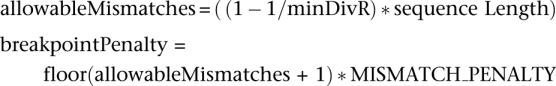
Chimera-aware NAST realignment of the query to the selected parents. Accurate NAST alignments of chimeric sequences required a proper set of reference NAST-formatted sequences to align to. In the standard NAST-iEr alignment approach, the best matching 16S sequences were chosen. In the case of a chimeric sequence, an optimal NAST alignment would require representatives for each corresponding homologous region of the chimera. Depending on the level of chimera divergence or breakpoint chosen, the top matching database hits may not contain each of the most informative sequences required for an accurate chimeric NAST alignment. However, the reference sequences identified by parent selection step above provided a minimal set of sequences to represent the putative regions of a chimera. To generate such a chimera-aware alignment, selected putative parent sequences were extracted in NAST format, and NAST-iEr was used to realign the query sequence against a profile based on these candidate parents.
- Chimera prediction in an evolutionary framework. The realigned NAST-formatted query and the candidate parents were next examined in an evolutionary framework for final chimera prediction. Given a pair of candidate parents and the single query sequence in NAST format, the three-sequence multiple alignment was removed of all columns containing a gap or non-{G,A,T,C} character, thus yielding a multiple alignment where each cell of each column contained a nucleotide (GATC). The multiple alignment was divided into two parts (left and right) by a breakpoint, requiring a minimum of 50 unaligned bases from each end. The breakpoint was slid from left to right across the multiple alignment with a step of five bases. At each breakpoint position, the parents were examined to determine whether a chimera between the two parents formed at that breakpoint was more similar to the query sequence than either of the parents. Note that, unlike Bellerophon, the entire sequences on both sides of the breakpoint were analyzed (the corresponding windows extend to each end of the sequence). If such a putative chimera existed, a bootstrapping operation was performed to compute a measure of confidence in the chimera relationship at that breakpoint. Bootstrapping was performed as follows: Columns of the multiple alignment containing nonidentical nucleotides and not neighboring a gap were identified from each side of the breakpoint; for 100 iterations, 10% of the mismatch-containing columns were sampled with replacement and examined in support of the chimera relationship, as defined using familiar terms:
where percent identity was based on those columns of the multiple alignment corresponding to nonidentical residues that were sampled with replacement. In either case, for the relationship to hold, the query sequence must have been more similar to a chimera between the two parents than to either of the parents separately.
In addition to computing bootstrap support, the minimum divergence ratio was computed at each breakpoint corresponding to PerID(Q,C)/max(PerID(Q,A), PerID(Q,B)). The breakpoint having the highest bootstrap support followed by the highest divergence ratio was selected as the best evidence for the chimera. If the maximally scoring breakpoint had at least 90% bootstrap support and the minimum divergence ratio exceeds the set threshold (1.007), the query was flagged as a chimera.
CS, in its current implementation, takes ∼10 sec per execution on pyrosequencing reads of approximately 500 bases, and 20–30 sec per full-length (∼1200 bases) 16S query sequence on an average desktop computer.
Acknowledgments
We thank Qiandong Zeng and Jared White for compiling the bacterial genome-based 16S data set, Robert Edgar and Eric Alm for helpful discussions regarding chimera detection algorithms, and Julie Segre and Sean Conlan for helpful comments on the manuscript. We acknowledge NIH for funding this project with awards to the Baylor College of Medicine (grants U54-HG003273 and U54-HG004973), the Broad Institute (grant U54-HG004969 and NIAID contract HHSN27220090018C), the J. Craig Venter Institute (NIAID contract N01-AI30071 and grant U54-AI084844), Washington University (grants U54-HG003079 and U54-HG004968), and NIH common fund contract U01-HG004866, a Data Analysis and Coordination Center for the Human Microbiome Project to Gary Andersen (Lawrence Berkeley National Laboratory).
Footnotes
[Supplemental material is available for this article. The sequence data from this study have been submitted to the NCBI Entrez Genome Project database (http://www.ncbi.nlm.nih.gov/genomeprj) under ID nos. 48465, 48471, 53501, and 60767. Software tools and data sets are freely available at http://microbiomeutil.sourceforge.net.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.112730.110.
References
- Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF 2005. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microbiol 71: 8966–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ 2005. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71: 7724–7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ 2006. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol 72: 5734–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher Y, Douady CJ, Sharma AK, Kamekura M, Doolittle WF 2004. Intragenomic heterogeneity and intergenomic recombination among haloarchaeal rRNA genes. J Bacteriol 186: 3980–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YW 2007. EzTaxon: A web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57: 2259–2261 [DOI] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, et al. 2009. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL 2006a. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ Jr, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL 2006b. NAST: A multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34: W394–W399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R 2008. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5: 235–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harth E, Romero J, Torres R, Espejo RT 2007. Intragenomic heterogeneity and intergenomic recombination among Vibrio parahaemolyticus 16S rRNA genes. Microbiology 153: 2640–2647 [DOI] [PubMed] [Google Scholar]
- Huber T, Faulkner G, Hugenholtz P 2004. Bellerophon: A program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20: 2317–2319 [DOI] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12: 1889–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsoulis GA, Waterman MS 1997. A new computational method for detection of chimeric 16S rRNA artifacts generated by PCR amplification from mixed bacterial populations. Appl Environ Microbiol 63: 2338–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Engelbrektson A, Ochman H, Hugenholtz P 2010. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12: 118–123 [DOI] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW 2007. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35: 3100–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahr DJ, Katz LA 2009. Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase. Biotechniques 47: 857–866 [DOI] [PubMed] [Google Scholar]
- Lane D 1991. 16S/23S rRNA sequencing. John Wiley & Sons Ltd, Chichester, United Kingdom [Google Scholar]
- Li W, Godzik A 2006. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659 [DOI] [PubMed] [Google Scholar]
- Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R 2007. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35: e120 doi: 10.1093/nar/gkm541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, DeSantis TZ, Andersen GL, Knight R 2008. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res 36: e120 doi: 10.1093/nar/gkn491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453 [DOI] [PubMed] [Google Scholar]
- Pace NR 1997. A molecular view of microbial diversity and the biosphere. Science 276: 734–740 [DOI] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO 2007. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT 2009. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods 6: 639–641 [DOI] [PubMed] [Google Scholar]
- Rappe MS, Giovannoni SJ 2003. The uncultured microbial majority. Annu Rev Microbiol 57: 369–394 [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci 103: 12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Marcelino LA, Polz MF 2002. Heteroduplexes in mixed-template amplifications: Formation, consequence and elimination by ‘recond7itioning PCR’. Nucleic Acids Res 30: 2083–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe SG, Hugenholtz P 2008. A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol 11: 442–446 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F, Affourtit J, Egholm M, Henrissat B, Knight R, et al. 2010. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci 107: 7503–7508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GC, Wang Y 1996. The frequency of chimeric molecules as a consequence of PCR co-amplification of 16S rRNA genes from different bacterial species. Microbiology 142: 1107–1114 [DOI] [PubMed] [Google Scholar]
- Wang GC, Wang Y 1997. Frequency of formation of chimeric molecules as a consequence of PCR coamplification of 16S rRNA genes from mixed bacterial genomes. Appl Environ Microbiol 63: 4645–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R, Peisajovich SG, Miller OJ, Magdassi S, Tawfik DS, Griffiths AD 2006. Amplification of complex gene libraries by emulsion PCR. Nat Methods 3: 545–550 [DOI] [PubMed] [Google Scholar]
- Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. 2009. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462: 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Morrison M 2004. Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Appl Environ Microbiol 70: 4800–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]