

Abstract
Saccharomyces cerevisiae Kre6 is a type II membrane protein with amino acid sequence homology with glycoside hydrolase and is essential for β-1,6-glucan synthesis as revealed by the mutant phenotype, but its biochemical function is still unknown. The localization of Kre6, determined by epitope tagging, is a matter of debate. We raised anti-Kre6 rabbit antiserum and examined the localization of Kre6 and its tagged protein by immunofluorescence microscopy, subcellular fractionation in sucrose density gradients, and immunoelectron microscopy. Integration of the results indicates that the majority of Kre6 is in the endoplasmic reticulum; however, a small but significant portion is also present in the secretory vesicle-like compartments and plasma membrane. Kre6 in the latter compartments is observed as strong signals that accumulate at the sites of polarized growth by immunofluorescence. The truncated Kre6 without the N-terminal 230-amino acid cytoplasmic region did not show this polarized accumulation and had a severe defect in β-1,6-glucan synthesis. This is the first evidence of a β-1,6-glucan-related protein showing the polarized membrane localization that correlates with its biological function.
Keywords: Cell Wall; Endoplasmic Reticulum (ER); Membrane Trafficking; Plasma Membrane; Yeast; β-1,6-Glucan; Saccharomyces cerevisiae
Introduction
The fungal cell wall physically supports the cell by its rigid structure and contributes to communication with the environment. It is the most specific and effective target of anti-fungal chemotherapy because it is essential for fungal cells, but human cells do not have some of its constituents.
The cell wall of the budding yeast Saccharomyces cerevisiae is composed of mannoproteins and three kinds of polysaccharides: β-1,3-glucan, β-1,6-glucan, and chitin. β-1,6-Glucan has an essential role to connect the other constituents covalently. β-1,3-Glucan and chitin are synthesized at the plasma membrane (PM)2 by their synthetases that polymerize the unit sugars from the cytoplasmic UDP-sugars. These polymerases have catalytic subunits with multiple membrane spanning domains. However, the polymerase of β-1,6-glucan comprised of ∼350 glucose residues is not uncovered yet (1).
Studies using anti-β-1,6-glucan antibody showed that β-1,6-glucan is detectable only at the outside of the PM without any indication of intracellular production (2), and new β-1,6-glucan is produced most actively at the site of polarized growth in small buds (3). The in vitro β-1,6-glucan synthesis system using the yeast membrane fraction was reported by Vink et al. (4), but Aimanianda et al. (5) recently reported that the membrane fraction was unable to synthesize β-1,6-glucan, but the permeabilized semi-intact cells prepared by osmotic shock could synthesize β-1,6-glucan that is covalently bound to alkali-insoluble β-1,3-glucan from UDP-glucose.
The genes whose products participate in β-1,6-glucan synthesis were discovered as kre mutants resistant to the yeast K1 killer toxin that requires β-1,6-glucan for its adsorption and subsequent formation of lethal pores in the PM, and several additional genes that affect the β-1,6-glucan contents were found later (6, 7). Most of their gene products were suggested to localize in the intracellular secretory pathway from the ER to the PM. Kre9 is a secreted protein, and Kre1 is a glycosylphosphatidylinositol anchor protein on the outer surface of PM, and it is impossible for them to directly participate in the reaction using cytoplasmic UDP-glucose as the substrate (6). The candidates with possible reactions concerning glycosides are both reported to be in the ER, i.e. Kre5 with homology to UDP-glucose glucosyltransferase (8) and Kre6 with homology to family 16 glycoside hydrolase (9), but there still is some debate. The biggest difficulty in the study of β-1,6-glucan synthesis is that although β-1,6-glucan can be detectable on the outside of PM, there is no PM protein that is likely to synthesize β-1,6-glucan using UDP-glucose and whose loss shows a decrease in the β-1,6-glucan content (6).
Kre6 is an important candidate for an enzyme that may directly be related to β-1,6-glucan synthesis because it is homologous to family 16 glycoside hydrolase with a UDP-glucose binding domain at the C terminus (2, 10). It was first reported in the early Golgi by the observation of the tagged protein produced by 2-μm plasmid (3, 11). We reported that Kre6 is a resident in the ER by the observation of Kre6-6myc produced by the CEN plasmid that is in less of an artificial condition than the multicopy production. The double-ring ER profiles were clearly observed, and its interaction with ER-resident essential protein Keg1 supported this localization of Kre6 (9). Takeuchi et al. (12) showed that an ER chaperon Rot1 and ubiquitin ligase Ubc7 affects the stability of Kre6 and suggested that Kre6 recycles between the ER and Golgi based on the results of sucrose gradient cell fractionation.
We raised anti-Kre6 antiserum and detected the intrinsic Kre6 in the wild-type cells. By integration of the immunofluorescence microscopy, cell fractionation, and immunoelectron microscopy, here we report that Kre6 is present in the PM and secretory vesicle (SV)-like compartment in addition to the ER. This localization is apparently required for β-1,6-glucan synthesis.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Media
S. cerevisiae strains used in this study are listed in Table 1. Tagging of Kre6 with three copies of the HA epitope at their C termini was done by homologous recombination between an appropriate fragment on the plasmid and the chromosome as described previously (13). The expression units of N-terminal-truncated Kre6 derivatives were constructed by joining the PCR products of the 300-bp 5′-upstream region of KRE6, truncated KRE6 ORFs with extraneous start codon and BamHI site, and 3HA-TDH1terminator, and these were integrated at the chromosomal ura3-52 locus of KTY604 (kre6Δ::kanMX4). The nucleotide sequences were confirmed. The YPT1promoter-GFP-LIP1 (a generous gift of Dr. Keisuke Sato) and TPIpromoter-GFP-SNC1 (a generous gift of Dr. Hugh R. B. Pelham (14)) genes were integrated at the chromosomal ura3-52 locus to make KSY466 and YKY218, respectively.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype and plasmid | Origin |
|---|---|---|
| BY4741 | MATa, his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Euroscarf |
| BY4742 | MATα, his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Euroscarf |
| Y05574 | as BY4741, kre6Δ::kanMX4 | Euroscarf |
| Y04773 | as BY4741, skn1Δ::kanMX4 | Euroscarf |
| YKY213 | as BY4741, ura3-52 | This study |
| YKY276 | as BY4742, ura3-52 | This study |
| SAY181 | MATa his3Δ leu2Δ trp1Δ ura3Δ ypt11Δ::LEU2 OCH1-3HA::HIS3 pRS73 (YPT11 TRP1 CEN) | Arai et al. (28) |
| KSY466 | as YKY276, ura3-52::(YPT1promoter-GFP-LIP1 URA3) | This study |
| YKY218 | as YKY213, ura3-52::(TPIpromoter-GFP-SNC1 URA3) | This study |
| KTY393 | as YKY276, ura3-52::(YPT1promoter-GFP-KEG1 URA3) | This study |
| KTY634 | as YKY213, ura3-52::(YPT1promoter-GFP-KEG1 URA3), keg1Δ::kanMX4 | This study |
| KTY284 | as BY4741, KRE6-3HA LEU2 | This study |
| KTY604 | as YKY213, kre6Δ::kanMX4 | This study |
| KTY626 | as KTY604, ura3-52::(KRE6promoter-KRE6-3HA URA3) | This study |
| KTY628 | as KTY604, ura3-52::(KRE6promoter-KRE6Δ137-3HA URA3) | This study |
| KTY632 | as KTY604, ura3-52::(KRE6promoter-KRE6Δ230-3HA URA3) | This study |
| KTY630 | as KTY604, ura3-52::(KRE6promoter-KRE6Δ248-3HA URA3) | This study |
| NCYC232 | K1 killer strain | ATCC60782 |
Yeast cells were grown in YPD (1% Bacto yeast extract (BD Biosciences), 2% Bacto peptone (BD Biosciences), and 2% glucose) or SD (0.17% yeast nitrogen base without amino acids (BD Biosciences), 0.5% ammonium sulfate, 2% glucose, and appropriate supplements) medium at 30 °C. Solid media were made with 1.5% agar. The medium used to test the sensitivity to K1 killer or Calcofluor white was described (9).
Escherichia coli KTY417 (BL21 (F−, ompT hsdSB gal dcm), pKT73 (PLac-GST-KRE61–84aa)) was used to produce the antigen to prepare the anti-Kre6 rabbit antiserum. E. coli was grown in an LB (1% Bacto Tryptone (BD Biosciences), 0.5% Bacto yeast extract (BD Biosciences) and 0.5% NaCl) medium, and the production of GST- Kre61–84aa was induced by adding 1 mm isopropyl β-thiogalactoside.
Antibodies and Immunoblotting
Antisera against Scs2 and Kar2 were kindly provided by Dr. Satoshi Kagiwada (Nara Women's University, Nara, Japan) and Dr. Masao Tokunaga (Kagoshima University, Kagoshima, Japan), respectively. Anti-HA (12CA5, Roche Diagnostics) monoclonal antibody was purchased. Rabbit polyclonal antibody to Gas1 was described previously (15). For Western blotting, anti-Kre6 rabbit antiserum was used at a dilution of 1/500, and the other antisera were used at a dilution of 1/1000. After incubation with peroxidase-conjugated secondary antibodies (KPL, Gaithersburg, MD), detection was performed using enhanced chemiluminescence (Thermo Scientific, Waltham, MA).
Indirect Immunofluorescence
The localizations of HA-tagged proteins were observed by indirect immunofluorescence as described previously (13). Anti-Kre6 rabbit antiserum and anti-HA mouse mAb diluted to 1/200 were used as primary antibodies. Alexa 488-, 546-, or 567-conjugated goat antibodies to rabbit or mouse immunoglobulin G (Molecular Probes, Eugene, OR) were diluted to 1/200 and used as a secondary antibody.
Sucrose Density Gradient Fractionation
Cells of 10 A600 nm units were suspended in an ice-cold lysis buffer (10 mm HEPES, 12.5% (w/v) sucrose, pH 7.4, containing protease inhibitors; 1 μg/ml each of chymostatin, aprotinin, leupeptin, pepstatin A, antipain, 1 mm benzamidine, and 1 mm phenylmethylsulfonyl fluoride) either with 1 mm MgCl2 or 10 mm EDTA and disrupted by vortexing with glass beads 4 times for 1 min with 1-min intervals at 4 °C between each burst. Unbroken cells were removed by centrifugation at 400 × g for 5 min. The cell lysate was fractionated on a sucrose density gradient composed of 1.0 ml each of 60, 54, 50, 46, 42, 38, 34, 30, 26, 22, and 18% sucrose solution in 10 mm HEPES, pH 7.4, either with 1 mm MgCl2 or 10 mm EDTA as described (16). After a centrifugation for 2.5 h at 114,000 × g (in the presence of 1 mm MgCl2) or 19 h at 155,000 × g (in the presence of 10 mm EDTA) in an SW40 Ti rotor (Beckman Coulter, Fullerton, CA), 12 fractions of 1 ml were sequentially collected from the top of the gradient. Aliquots of each fraction were analyzed by SDS-PAGE followed by immunoblotting using anti-Kre6, anti-Scs2, and anti-Gas1 antibodies. Enhanced chemiluminescence signals were captured by an image analyzer equipped with a cooled charge-coupled-device camera (LAS-1000plus; Fuji Film, Tokyo, Japan), and digital images were quantified using Image J software and graphed on Excel (Microsoft, Radmond, WA).
Cell Cycle Synchronization
Synchronization was done according to Amberg et al. (17). The cell cycle of an early log-phase random culture of BY4741 was blocked by 2 μg/ml α-factor (GenScript, Piscataway, NJ) for 3 h. After confirming that there were no cells with buds, the culture was released from the block by washing and suspending the cells in fresh YEPD. After 105–115 min, when more than half of the cells had buds smaller than 20% of the mother cell size, the synchronized cells were collected and processed for sucrose density gradient fractionation.
Topology Analysis by Protease Protection
The topology of the Kre6-3HA protein was examined by the protease protection analysis as described (18).
Immunoelectron Microscopy
Freezing and freeze substitution of yeast cells were performed as described previously (19, 20). Immunoelectron microscopy was performed as described previously (21, 22) with anti-HA mAb (1:25 dilution) and goat antibody to mouse IgG labeled with 1-nm colloidal ultra-small gold (1:100 dilution; GAM IgG GP-US; Aurion, Wageningen, The Netherlands). The ultra-small gold particles were enhanced by silver deposition using a silver enhancement kit (Aurion RGent SE-EM, Aurion) (20, 21). The stained cells were examined using a transmission electron microscope JEM 1200 EXS (JEOL Ltd., Tokyo, Japan) at 80 kV.
RESULTS
Chromosome-encoded Kre6-3HA Is Strongly Detected at the Site of Polarized Growth by Immunofluorescence Microscopy
We observed the ER-characteristic double ring images of Kre6-6myc encoded by a CEN plasmid by immunofluorescence microscopy (see Fig. 4 in Ref. 9) and corrected the early Golgi localization based on the observation of overproduced tagged Kre6 (3, 11). However, because this Kre6-6myc did not fully recover the Calcofluor white sensitivity of the Δkre6 null mutant to the wild-type level (see Fig. 6C in Ref. 9), we consider the localization of Kre6 should be re-examined further. We added 3HA epitope coding sequence at the 3′ end of the chromosomal KRE6 gene. The resulting haploid yeast, KTY284, having only the tagged KRE6-3HA gene in the genome, grew as well as the wild type, and no difference in sensitivity to both Calcofluor white and the K1 killer toxin was found by comparing the parent BY4741 (supplemental Fig. 1).
FIGURE 4.

Sucrose density gradient fractionation of Kre6-containing compartments. The cell lysate of the KRE6 wild-type was fractionated on an 18–60% (w/v) sucrose density gradient in the absence (A and C) or presence (B and D) of 10 mm EDTA. Cells were collected from an early-log phase random culture (A and B). The experiment was repeated using a synchronized culture in which more than half of the cells had buds smaller than 20% of the mother cell size (C and D). After centrifugation, 12 fractions were collected from the top of the gradient, and aliquots were analyzed by SDS-PAGE and immunoblotting using anti-Kre6, anti-Scs2, and anti-Gas1 antibodies. The signal intensity of indicated proteins was quantified with ImageJ software and graphed using Microsoft Excel. The top of the gradient corresponds to fraction 1.
FIGURE 6.

Immunofluorescent images of Kre6 in comparison with the SV/PM-marker Snc1. The fixed cells with the chromosomal TPIpromoter-GFP-SNC1 gene were observed by immunofluorescent staining with anti-Kre6 serum and compared with fluorescent images of GFP-Snc1 of the same cells. A, cells in various cell cycle stages are shown. B, Section is the serial confocal slice image of each 0.25-μm depth from the surface to the slide glass, and Stacked is the sum of signals.
When the localization of the HA epitope was observed by immunofluorescence microscopy, brilliant fluorescent signals were observed in the cells with buds (Fig. 1A). This profile is different from that of the ER we previously reported (9) or that of the early Golgi shown by Och1-3HA (Fig. 1B). In the budding cells, the fluorescence signals were concentrated in the daughter cells, with the brightest signals at the growing tips. Some of the large cells are probably at the cell cycle stage just before the bud emergence, and a bright signal was found at one tip of the cell, which may be the incipient bud. Those signals of Kre6-3HA are similar to the immunofluorescent staining images of secretory vesicles that are in transit from the late Golgi to the PM (23). The fluorescence signal of Kre6-3HA was found in the mother cells, and some ER-like structures were found in Fig. 1A, although the signals were much less intensive than the polarized bright signals.
FIGURE 1.

Localization of wild-type Kre6 and the tagged Kre6–3HA proteins expressed from the chromosomal genes. Kre6–3HA (A) and the early-Golgi marker Och1–3HA (B) were expressed from the chromosomal genes and detected by immunofluorescent staining with mouse anti-HA mAb. The cells of the Δkre6 null mutant (C) and KRE6 wild-type (D) were stained with rabbit anti-Kre6 antiserum and observed by fluorescent microscopy and Nomarski images. E, Kre6–3HA was visualized by double immunofluorescent staining with anti-Kre6 antiserum and anti-HA mAb. The bar indicates 10 μm.
Preparation of Anti-Kre6 Antibody and Its Specificity
Even though Kre6-3HA was produced from the chromosomal single-copy gene with the transcription and translation information of authentic KRE6, the localization and behavior of the tagged protein is not certified to be identical to those of the authentic Kre6. Therefore, we prepared an anti-Kre6 antibody to detect the intrinsic Kre6 in the wild-type cells. There is a paralog of KRE6 named SKN1 in S. cerevisiae. The Δskn1 null mutant shows not so significant defects as the Δkre6 mutant, but the Δskn1 Δkre6 double disruption is lethal. So these genes have a functional overlap, and Kre6 plays a major role (3, 11, 24). The amino acid identity is as high as 86% in the C-terminal 472 amino acids that are predicted to be the luminal domain of the type II membrane protein. On the contrary, the cytoplasmic N-terminal 137-amino acid region has 32% identity (3). We prepared a GST fusion protein of N-terminal 84-amino acid polypeptide of Kre6 in E. coli and used it to immunize rabbits after affinity purification.
A 140-kDa protein was detected by a rabbit antiserum in Western blots of lysates of the wild-type and Δskn1 cells but not the Δkre6 cells (Fig. 2). The calculated molecular weight of Kre6 is 80,122, but the detected Kre6 is significantly larger than the expected size, probably because of N-glycosylation and phosphorylation (3, 12). We concluded a specific anti-Kre6 antiserum was obtained that specifically recognizes Kre6 but not Skn1. There are several nonspecific signals including the most significant 43-kDa protein, but most of them are soluble cytoplasmic proteins (data not shown).
FIGURE 2.
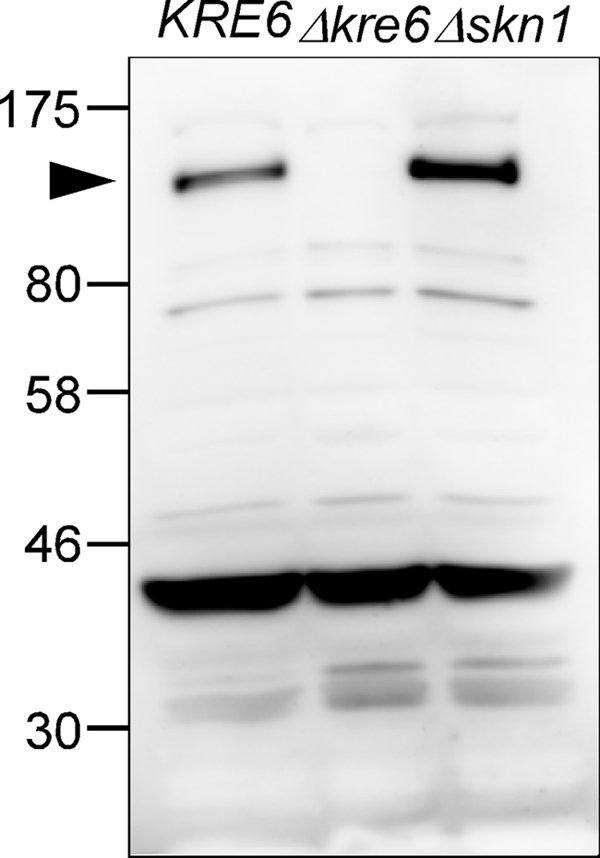
Specificity of rabbit anti-Kre6 antiserum. The total lysate of KRE6, Δkre6, and Δskn1 cells were analyzed by SDS-PAGE and Western blotting using an anti-Kre61–84 aa antiserum. The arrowhead indicates Kre6 protein.
Indirect Immunofluorescence Images of the Wild-type and KRE6-3HA Cells by Anti-Kre6 Antiserum
When the Δkre6 cells were processed for indirect immunofluorescence microscopic observation using the anti-Kre6 rabbit antiserum, a small number of dots with strong fluorescence were observed. These should represent nonspecific signals of this antiserum (Fig. 1C). The wild-type S. cerevisiae cells processed in the same way using anti-Kre6 antiserum had fluorescent signals very similar to those observed in the chromosomal KRE6-3HA cells using anti-HA mAb (Fig. 1D); brilliant foggy fluorescence was found in the daughter cells and at the incipient buds. The fluorescence has a gradient to increase or concentrate at the bud tips. The presence of a small number of dispersed bright dots was significant in the cells, which are likely to be the nonspecific signals found in the Δkre6 disruptant cells using anti-Kre6 antiserum.
Next we observed the KTY284 cells that have the single tagged KRE6-3HA gene in the chromosome by double immunofluorescence staining with anti-Kre6 antiserum and anti-HA mAb (Fig. 1E). The foggy fluorescence that has a gradient to concentrate at the bud tip was similarly observed by both antibodies, which agrees well with the prediction that these signals should represent two different epitopes in the same polypeptide. Small numbers of bright dots that were only observed by anti-Kre6 antiserum should be the nonspecific signals that appeared in Δkre6 cells. From these results, the chromosomally encoded Kre6-3HA is indistinguishable from the intrinsic Kre6 either in the activity and localization and will be an excellent material because it is detectable without nonspecific background signals.
Topology of the Kre6 Polypeptide
The type II membrane protein topology of Kre6 with the cytoplasmic N terminus and luminal C terminus is a prediction based on amino acid sequence characteristics (10), and no experimental data have been reported yet. We analyzed the topology of the Kre6-3HA polypeptide of KTY284 by the protease protection method as described (18).
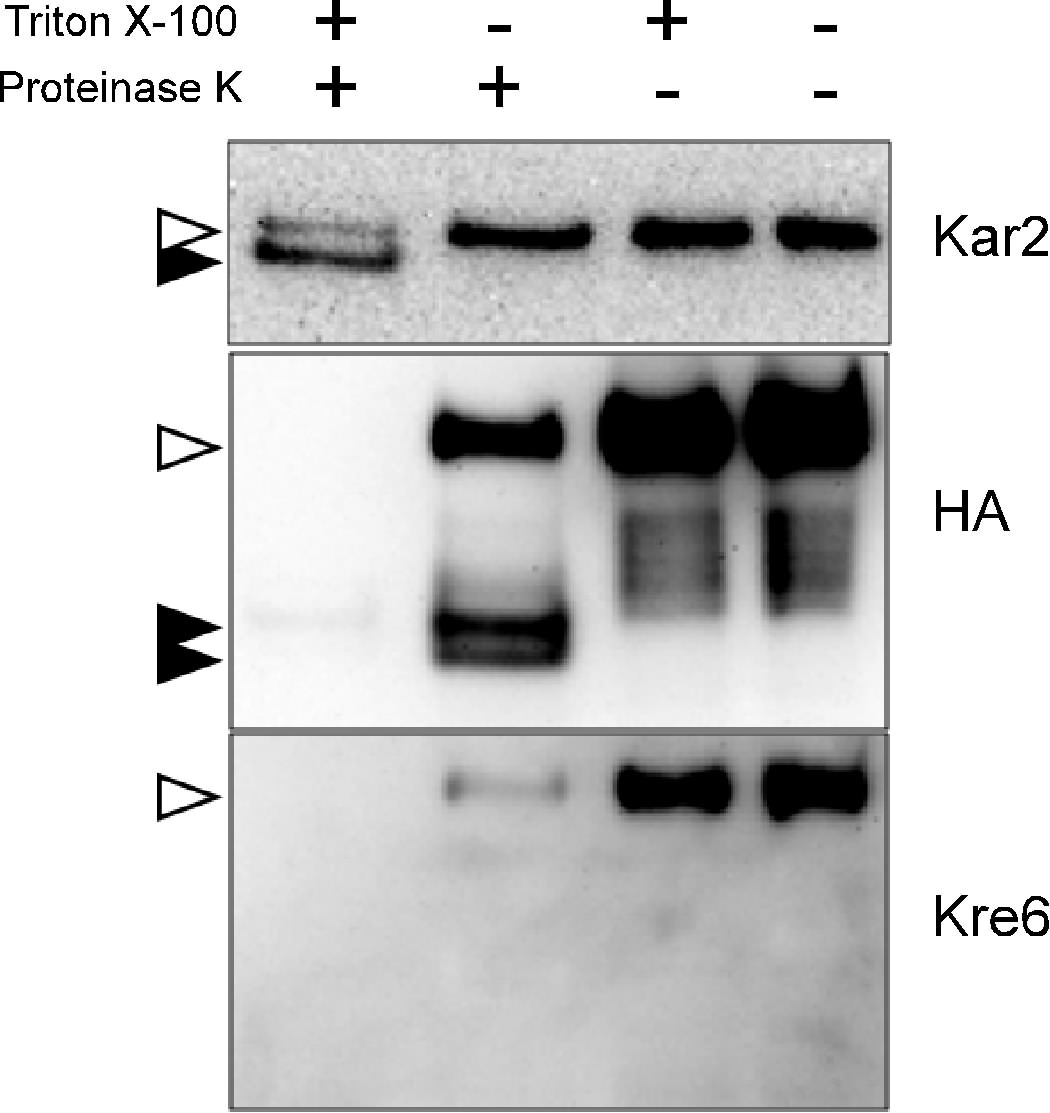
As the Western blots in Fig. 3 clearly show, the epitope in the N-terminal 84 amino acids was digested and became undetectable after proteinase K treatment both in the presence or absence of the protective membrane. On the contrary, whereas the C-terminal HA epitope was undetectable after proteinase K digestion in the presence of detergent Triton X-100, the intensity of the full-length Kre6-3HA decreased, and two fragment signals appeared after digestion in the absence of Triton X-100. Two bands are likely to be the membrane-protected Kre6-3HA polypeptides with different degrees of modification. These HA-containing C-terminal polypeptides should be protected from proteinase K digestion in the presence of the sealed membrane. The remaining full-length Kre6-3HA might be somehow covered by membrane fragments during cell disruption and protected from digestion. We concluded the Kre6 is a type II membrane protein as previously predicted from the amino acid sequence.
FIGURE 3.
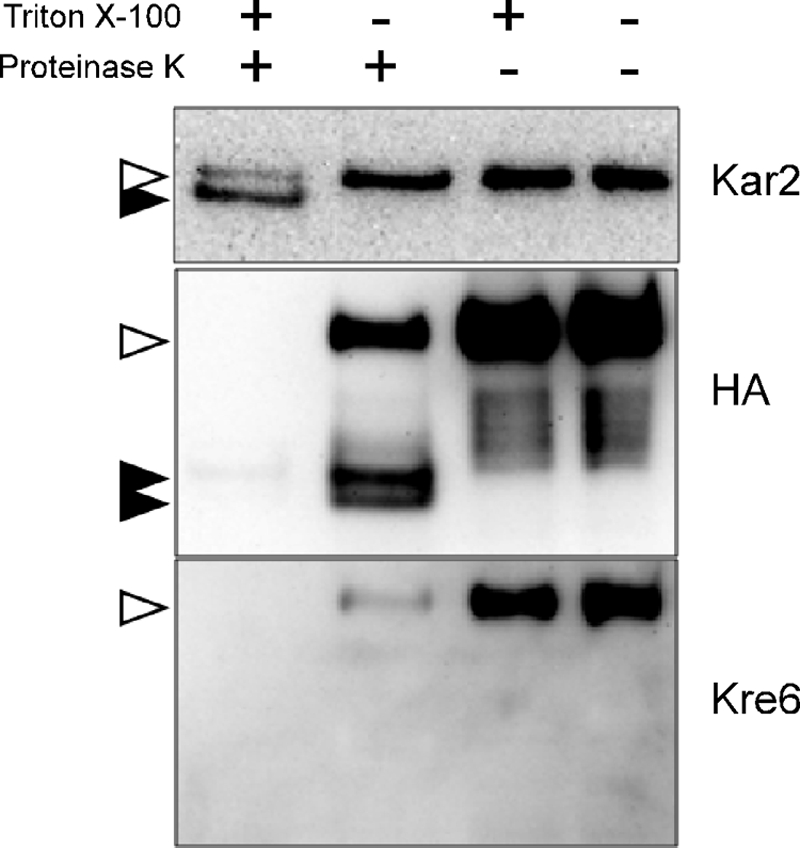
Topology analysis of Kre6–3HA protein by protease protection assay. Kre6 was tagged at the C terminus with 3HA epitope by homologous recombination on the chromosome. The protoplast was prepared by lyticase digestion and gently lysed in the presence of 200 mm sorbitol. After digestion with proteinase K with or without Triton X-100, Kre6–3HA was detected by Western blotting with either anti-HA mAb or anti-Kre6 antiserum. The ER lumenal protein Kar2 was used as a control, which becomes digestion-resistant smaller polypeptide after treatment with proteinase K and Triton X-100. Open and filled arrowheads indicate bands of undigested and digested proteins, respectively.
Fractionation of Kre6-containing Compartments by Sucrose Density Gradient Centrifugation
To further obtain cell biological information about membranes that contain Kre6 and accumulate at the bud tip, we fractionated the wild-type cell lysate by centrifugation in 18–60% stepwise sucrose density gradients. After SDS-PAGE and Western blotting of 12 fractions (Fig. 4A), Kre6 was found broadly in the fractions of higher density than fraction 5. Kre6 distribution apparently has two peaks at fractions 5–8 and fractions 10–12, and the latter peak seems larger (Fig. 4A). In this condition, fractions 5–8 usually contain small vesicles such as the Golgi and endosomes. Fractions 10–12 usually contain ER- and PM-derived membranes, and we actually detected the main peaks of the marker proteins of the ER (Scs2) and PM (Gas1) by Western blotting with their specific antisera. In Fig. 4A, althoughKre6 had a small peak in fractions 5–7, the ER marker Scs2 was absent, and the PM-marker Gas1 was at a low level in these fractions. This peak of fractions 5–7 suggests that a part of Kre6 is in some membrane that is distinct from the ER or PM.
When EDTA is added to the lysate and sucrose solutions to chelate the divalent cations, the density of ER decreases by releasing the peripheral-binding proteins such as ribosomes. Therefore, the ER and PM form separate peaks in the sucrose density gradient in the presence of EDTA (16). The ER marker Scs2 had a broad peak in fractions 6–9, and only a little signal was found in fractions 10–12 (Fig. 4B). The PM-marker Gas1 was more broadly distributed in most of the fractions, but a significant amount was also found in the bottom fractions of 11 and 12. Under this condition, the wild-type Kre6 also had two peaks; a larger but broad peak at fractions 6–9 and a smaller but significant peak at fractions 10–12. This small peak suggests that a part of Kre6 is present in the membranes that are distinct from the ER, which could be a part of the PM.
The result of cell fractionation in different conditions suggests the major portion of Kre6 is present in the ER. It is compatible with the previous report (9). However, it was also suggested that the localization of Kre6 is not restricted in the ER. Kre6 was found in fractions 5–8 in the absence of EDTA and in fractions 10–12 in the presence of EDTA, which suggests a part of Kre6 may be present in small vesicles and the PM, respectively.
Fractionation of Kre6-containing Compartments of Budding Cell-enriched Synchronized Culture
Using the same anti-Kre6 serum, sucrose density gradient fractionation suggests that a major portion of Kre6 is in the ER, but immunofluorescence microscopic observation indicated the most brilliant signal accumulates in the growing bud. A possible reason for why visibly different results were obtained by these experiments may be because we observe individual cells at various cell cycle stages by microscopy, but fractionation combines these different cells altogether. Kre6 may localize in some specific membranes distinct from the ER in small buds for a short term, and they are only detectable by microscopic observation. To test this possibility, we synchronized the cell cycle of the culture and fractionated the lysate enriched in the cells with small buds.
We synchronized the culture by the cell cycle block and release using α-factor. The cells, more than half of which had daughter cells smaller than 20% of the size of the mother cell, were collected (supplemental Fig. 2). The lysate was prepared from these cells and subjected to a sucrose density gradient fractionation of two different protocols as described above. As shown in Figs. 4, C and D, the results were essentially the same as the results obtained from the random cell culture. The positions and relative heights of the peaks of Kre6, Scs2, and Gas1 are totally reproduced. This suggests that the distribution of Kre6 among the membranes separable in the sucrose density gradient is hardly affected by the cell cycle stages. If some parts of the membrane may gather at a specific area, the same parts are always present at any of the cell cycle stages.
The Accumulation of Kre6 at the Daughter Cells Precedes to the ER Inheritance
We investigated the localization of Kre6 in cells of various cell cycle stages. In sucrose density gradient fractionation, a major portion of Kre6 was always found in the same fractions that have the ER-marker, Scs2. We compared the fluorescence signals of anti-Kre6 staining and the GFP-tagged signals of the ER-resident ceramide synthase subunit Lip1 (Fig. 5). GFP-Lip1 is found in the mother cell with the typical ER pattern of double rings; that is, the nuclear envelope ring and peripheral ring underneath the PM. Kre6 showed very different images of bright foggy fluorescence mainly in the daughter cells as analyzed in Fig. 1. Even when no bud was detectable, large cells had a bright fluorescence at an area of one end of the cell, which is likely to represent the incipient bud. When the size of daughter cells was very small, GFP-Lip1 fluorescence was mostly in the mother cells, and it was hardly detected in the daughter cell. The GFP-Lip1 was detectable after the size of the daughter cell reached about 20% that of the mother cell. These observations are consistent with the reports on the ER inheritance showing that the small daughter cells do not have the cortical ER (25–27). The bright Kre6 signals gather at the sites of polarized growth before the ER enters the daughter. The signal seems concentrated at the periphery of the daughter cells. This should be present in the membranes that form peaks distinct from the ER when the cells were disrupted and fractionated in the sucrose density gradient. It is very curious why the majority of Kre6 in the ER was not found as double ring signals in the mother cells. It is currently unclear, but a weak fluorescence signal of Kre6 was found in the mother cell.
FIGURE 5.

Immunofluorescent images of Kre6 in comparison with the ER-marker Lip1. The fixed cells with the chromosomal YPT1promoter-GFP-LIP1 gene were observed by immunofluorescent staining with anti-Kre6 serum and compared with fluorescent images of GFP-Lip1 of the same cells. D/M indicates the ratio of areas of daughter cells to mother cells.
We previously showed that an ER-resident essential membrane protein Keg1 binds to Kre6 by immunoprecipitation (9). It will be important to see whether a part of Keg1 is also delivered into the incipient buds with Kre6 or not. GFP-Keg1 was produced by a CEN plasmid in our previous report, but we constructed more stable chromosomal integration of the YPT1promoter-GFP-KEG1 units at the chromosomal ura3-52 locus and examined the localization of GFP-Keg1 at various expression levels (supplemental Fig. 3A). The images of fluorescence of GFP-Keg1 were typical ER profiles, and no accumulation in the buds was found at either the highest or lowest expression level (supplemental Fig. 3B). We also examined the localization of GFP-Keg1 in sucrose density gradient fractionation of both random and synchronized cultures, but GFP-Keg1 was detected in the ER and never found in the PM at the bottom (supplemental Fig. 3C). These results suggest that although Kre6 binds to Keg1 in the ER, the subpopulation of Kre6 accumulated in the buds is free from Keg1 that is localized exclusively in the ER.
The Localization of Kre6 at the Site of Polarized Growth Is Similar to the Localization of the Secretory Vesicle/PM Marker Snc1
We next compared the localization of Kre6 and GFP-tagged Snc1. Snc1 is a v-SNARE (vesicle soluble N-ethylmaleimide-sensitive factor attachment protein receptor) of SVs that was transferred from the Golgi to the PM. It recycles between the PM, endosomes, and Golgi and is found at the PM of small buds (14, 23, 29). The fluorescence of GFP-Snc1 generally coincided with the immunostaining of Kre6 (Fig. 6A). By comparing the serial sections of confocal fluorescence microscopy data, the patterns at the periphery of the cells are mostly identical, but the Kre6 signal, which was also found inside of the cells, was much stronger than the GFP-Snc1 signal.
Immunoelectron Microscopic Observation of Kre6-containing Membranes
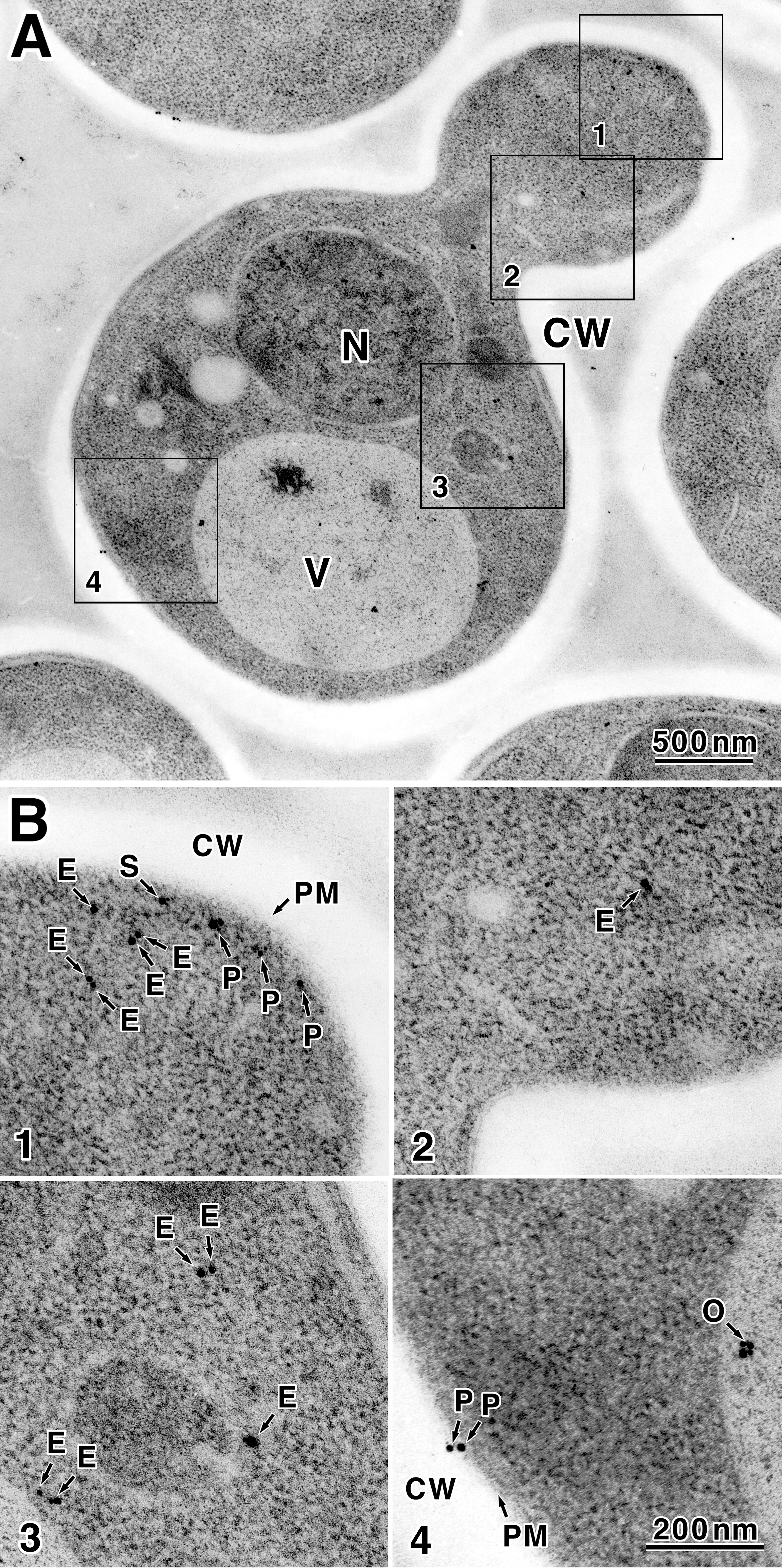
To determine the cellular localization of Kre6 with higher magnification, we conducted immunoelectron microscopic observation as described (22). Anti-Kre6 serum could not be used because a nonspecific reaction was found even in the area without yeast cells. As a nonspecific reaction was not found in the case of anti-HA mAb, we analyzed the thin sections of KTY284 cells with anti-HA mAb. In total, 2807 colloidal gold particles in 63 cells in good sections were examined in detail. The colloidal gold particles bound to anti-HA mAb were found in all regions of the cell except the transparent cell wall (Fig. 7A). We could not find the difference between the mother and daughter regions of the budding cells. The localization of colloidal gold particles was classified in four classes (ER, SV, PM, and Others) as described below. The particles were counted, the percentage of each class in a cell was calculated, and the average of the percentage of the particle class of 63 cells was calculated.
FIGURE 7.

Localization of Kre6–3HA decided by immunoelectron microscopic observation. A, S. cerevisiae KTY284 (chromosomal KRE6–3HA) cells were subjected to high pressure freezing and freeze substitution. Kre6–3HA was detected with anti-HA mAb and 1-nm ultra-small gold particle-conjugated goat anti-mouse IgG. The gold particles were enhanced by silver deposition as described under “Experimental Procedures.” N, nucleus; V, vacuole; CW, cell wall. B, four regions indicated by squares are enlarged, and gold particles are indicated with arrows. Classified localization is: E, ER; S, SV; P, PM; O, others.
Most of colloidal gold particles were dispersed in cytoplasm (1838 particles in total, class ER, average of 63 cells is 66 ± 13%, Fig. 7B, 1–3, labeled E). Most colloidal gold particles were found at or near the small areas where ribosomes were excluded. As Kre6 is a type II membrane protein, it should be in some membrane. The cytoplasmic region without ribosome is likely to be the ER or Golgi compartments, but the membranes were not stained in the immunoelectron microscopic specimen and are hard to identify. As the cell fractionation data indicated the major portion of Kre6 behaved as the ER marker, those cytoplasmic Kre6-3HA are likely to be in the ER membrane.
Next, 721 colloidal gold particles in total (class Others, average 24 ± 12%) were found within the nucleus or vacuole. It is difficult for a type II membrane protein Kre6 to localize in these regions. Because the nucleus and vacuole usually tend to bind colloidal gold nonspecifically by the basic nature of their constituent proteins, we conclude these are nonspecific signals (Fig. 7B4, labeled O).
In the 58 of 63 cells examined (92%), some colloidal gold was found at or very close to the PM. If Kre6-3HA resides in the PM, the epitope 3HA is at the C terminus of Kre6 in the outside of the cell. Considering the thickness of the PM and length of mAb molecule and antigen (30), the colloidal gold detected within 35 nm of the center of the PM may be bound to Kre6-3HA in the PM. Such particles were 248 in total. When some membranous regions like secretory vesicles were found beneath the PM, it was impossible to decide to which of the membranes the colloidal gold particle bound. Such particles were 135 in total (class SV, average 5.6 ± 6.2%, Fig. 7B1, labeled S). The colloidal gold particles bound to the Kre6-3HA in the PM were 113 in total (class PM, average 4.5 ± 5.5%, Fig. 7B, 1 and 3, labeled P). It may be possible that Kre6 in the SV will be collected in fractions 5–7 in the absence of EDTA by sucrose density gradient fractionation, and Kre6 in the PM may be collected in fractions 10–12 in the presence of EDTA.
Localization of the Truncated Kre6 Proteins without Parts of Its N-terminal Cytoplasmic Domain
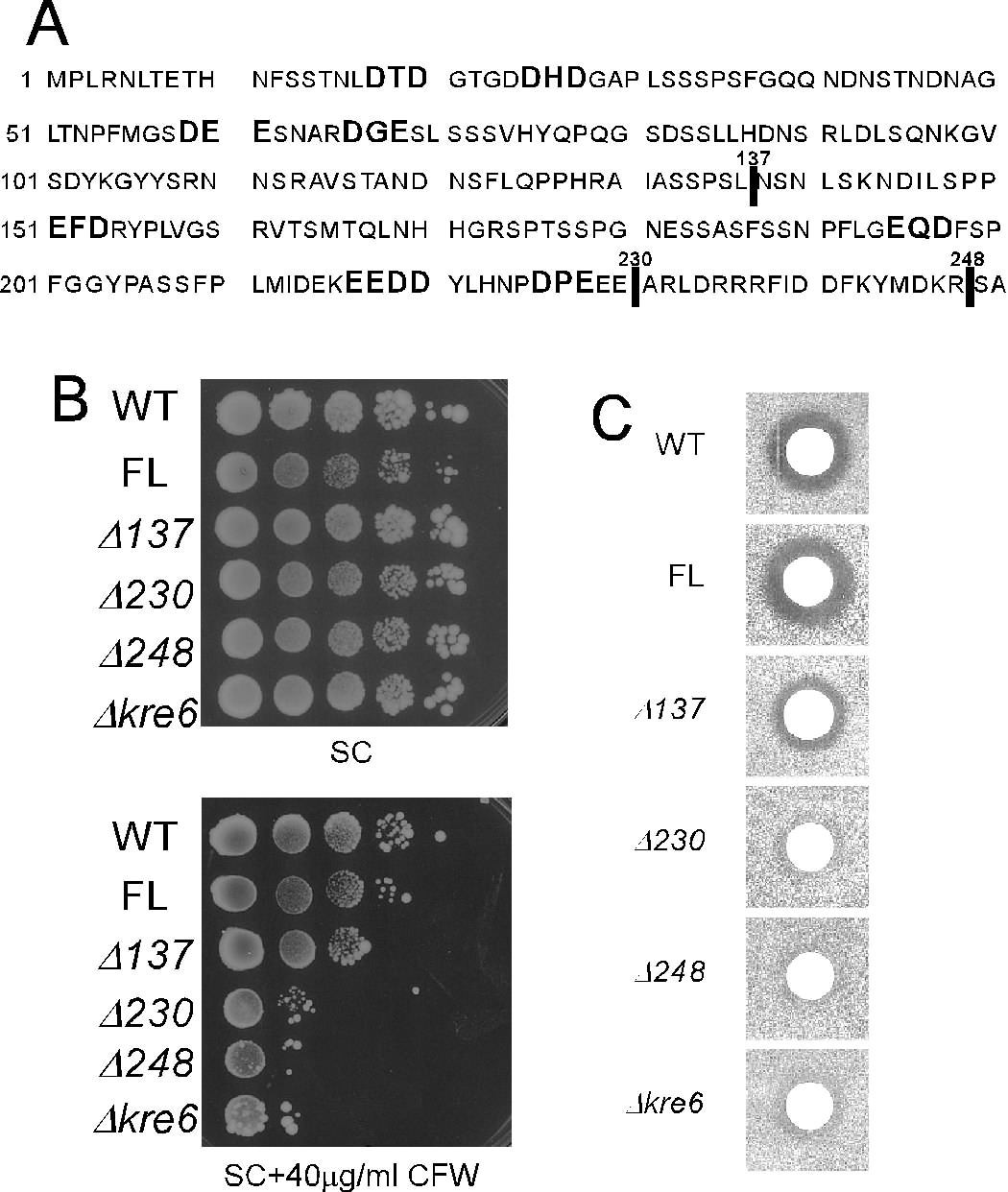
To determine whether the localization of Kre6 in the polarized membranes and its biological function in the β-1,6-glucan synthesis has any essential correlation, we sought to test some variant forms of Kre6 that are not delivered out of the ER. Li et al. (3) reported that a strain with the truncated Kre6 losing its N-terminal 137 amino acids showed a partial decrease both in the K1 killer toxin sensitivity and in the β-1,6-glucan content, and deletion of its whole cytoplasmic domain of 248 amino acids resulted in almost complete loss of Kre6 activity that was not rescued by replacement with the corresponding domain of the Kre6-paralog Skn1. They also reported that the loss of the cytoplasmic domain results in decreased stability and aberrant localization of Kre6, although their conclusions may have some problems because the Kre6 derivatives were produced by multicopy plasmids. We reproduced the same deletion constructs (Kre6Δ137 and Kre6Δ248) but with the 3HA tag and integrated them at the chromosomal ura3-52 locus. Another deletion construct (Kre6Δ230) that has the smallest cytoplasmic domain without possible ER-exit determinants (DXE and similar di-acidic sequences (31)) was also constructed as shown in Fig. 8A.
FIGURE 8.

Deletions including possible ER exit motifs in the cytoplasmic domain and the biological activity of Kre6. A, the amino acid sequence of the cytoplasmic domain of Kre6 is shown with indication of di-acidic sequences. Vertical lines indicate the ends of deletions. B, Calcofluor White sensitivity of the wild-type (WT) and Δkre6 mutant with chromosomal integration of various KRE6–3HA derivatives (full-length (FL) or three truncated versions) at the ura3-52 locus are shown. Cells were grown in SC medium until A600 nm = 1, diluted 2-fold, and then serially diluted 4 times by 10-fold. Ten μl of these samples were spotted on SC plates with or without 40 μg/ml Calcofluor White (CFW) and incubated at 30 °C for 3 d. C, K1 killer toxin sensitivity of the strains were used in B. The cells were grown in YPD at 30 °C until A600 nm = 1.0 and plated on a low-pH YPD plate. Five μl of a fresh overnight culture of the K1 killer producer was spotted, and the plates were incubated at 25 °C for 2 days to show the growth inhibitory zone.
Western blots indicated that 146-kDa (full-length Kre6), 82-kDa (Kre6Δ137), 73-kDa (Kre6Δ230), and 68-kDa (Kre6Δ248) proteins were produced with small amounts of possible degradation products of similar pattern (supplemental Fig. 4A). By sucrose density gradient fractionation (supplemental Figs. 4, B and C, without and with EDTA, respectively), a portion of the full-length Kre6 and Kre6Δ137 was found in the PM (fractions 11 and 12). In contrast, the Kre6Δ230 and Kre6Δ248 were not found in these fractions.
Immunofluorescence images revealed that most of the Kre6-3HA derivatives represented a number of dots dispersed in the whole cytoplasm (supplemental Fig. 4D) in contrast to the brilliant fluorescent signals in the buds of the cells of the full-length Kre6. By extensive examination, we could found a small number of cells with stronger fluorescence at their buds in the Kre6Δ137 cells, but we could not find them in the Kre6Δ230 and Kre6Δ248 cells. The nature of aberrant dots is not clear at present, but sucrose density fractionation data suggest that they are likely to be in the ER rather than some smaller vesicular structures like the Golgi or SV.
Exit from the ER Is Required for the Function of Kre6 Protein in β-1,6-Glucan Synthesis
To examine the activity of the Kre6 derivatives with altered localization from the full-length protein, we tested the Calcofluor White sensitivity by serial dilution. As shown in Fig. 8B, although the Kre6Δ137 cells had similar sensitivity to the wild-type cells, the Kre6Δ230 and Kre6Δ248 cells were hypersensitive to Calcofluor White as the Δkre6 disruptant cells, which suggests the deletions caused a severe functional defect. Next, we examined the ability of β-1,6-glucan synthesis by K1 killer toxin sensitivity. Fig. 8C shows the Kre6Δ137 cells had a smaller clear zone compared with the full-length Kre6 cells. The Kre6Δ230 and Kre6Δ248 cells were indistinguishable from the Δkre6 disruptant cells, which indicates the cytoplasmic 138–229th amino acid region is very important for the activity of Kre6. Although these variants have the complete lumenal domain with homology to glycosidase and are fairly stable in the cell, they did not complement the severe defect of Δkre6 disruptant in the β-1,6-glucan synthesis. The activity of β-1,6-glucan synthesis and polarized accumulation at the buds of the Kre6 derivatives correspond well with each other, which suggests that transport of Kre6 from the ER to PM is required for its function.
DISCUSSION
The process of the cell wall β-1,6-glucan synthesis is one of the most mysterious problems in yeast cell biology. It is still uncertain where it is actually synthesized and which enzyme directly catalyzes the reaction. Kre6 is believed to be a key protein in β-1,6-glucan synthesis because it has an amino acid motif of glycoside hydrolase and its loss results in a severe defect. The localization of Kre6 is also debated. Because the signal of GFP-tagged Kre6 was hardly detectable using a low copy vector, Li et al. (3) used a multicopy plasmid. Their conclusion was that Kre6 is in the early Golgi as identified by Och1. The analysis using overexpression and a tagged protein generally has a risk of artifacts. We produced Kre6-6myc using CEN plasmid in Δkre6 cell and found that it partially recovers the Δkre6 defects and is localized in the ER. The co-immunoprecipitation of Kre6 and ER-resident Keg1 supported the presence of Kre6 in the ER (9).
In this study we raised anti-Kre6 antibody and succeeded in the detection of the intrinsic Kre6 in the wild-type cell. The immunofluorescent image of Kre6 by anti-Kre6 staining was almost identical to that of chromosomal Kre6-3HA stained by anti-HA. However, the pattern was quite distinct from the pattern of Golgi (Fig. 1B, Och1) or ER (Fig. 5, Lip1) but similar to the pattern of SV or PM with accumulation at the site of polarized growth (Fig. 6, Snc1). Sucrose density fractionation in two different conditions indicated some parts of Kre6 are in particular membranes distinct from the ER (Fig. 4), which is consistent with immunofluorescence and immunoelectron microscopic observations. However, the majority of Kre6 is likely to be present in the ER as suggested by the peak size in the sucrose density gradient fractionation and abundant colloidal gold in the cytoplasmic regions of immunoelectron microscopy data. It is curious that the Kre6 in the ER was not clearly observed by immunofluorescence. It may be possible that although the total amount of Kre6 in the ER is larger than in the SV or PM, the concentration is low, and so the signal is weaker than the other Kre6 in the concentrated regions. Otherwise, although we are uncertain if it may be possible, the intrinsic Kre6 forms some complex in the ER, and the access of antibody is somehow blocked. It may be worth noting that the ER membrane protein Keg1 binds to Kre6 but does not exit from the ER, and therefore, Kre6 in the SV or PM should be free from Keg1. When the total amount of Kre6 increased a little by using a CEN plasmid, the free or unprotected Kre6 in the ER became detectable as double-ring immunofluorescence images.
The catalytic subunit of chitin synthase, Chs3, mainly works either at bud neck and PM. Most of Chs3 is present in the secretory organelles in the cell at 25 °C but is transported to the PM when the chitin synthesis is up-regulated by heat shock (32). It is considered to be one of the biological tactics of the cell to rapidly respond to the environment by producing excess enzymes for cell wall synthesis and retaining them in the organelle until they become necessary. The localization of Kre6 may resemble the localization of Chs3. The ER may be the major reservoir of Kre6, and it may transport Kre6 to the sites of polarized growth for function by yet unknown mechanism. The proper trafficking of Chs3 is controlled by many transport factors, one of which is Chs7 in the ER (1). As an analogous story, Keg1 that binds to Kre6 may possibly control the transport of Kre6 from the ER.
Logically speaking, all secretory proteins are transported to the resident compartment via the ER, they all have time to reside in the ER before getting to their final destination or most suitable localization. If the protein is trapped by the ER quality control, the time will become longer. A subunit of the protein complex is generally unable to leave the ER unless it is integrated in the proper complex. If Kre6 has its active/mature form and precursor/immature form, it may be possible that the immature Kre6 remains in the ER and mature Kre6 is transported to the site of polarized growth.
Some of the genes related to β-1,6-glucan synthesis are considered to play a role in the quality control in the ER; CNE1 that is related to calnexin, KRE5 that encodes UDP-glucose:glycoproteins glucosyltransferase, CWH41 and ROT2 that encode glucosidases I and II, respectively. It is a mystery why these genes related to the ER quality control can have influence in the synthesis of β-1,6-glucan in the outside of the PM (1, 6). The loss of Rot1 in the ER results in a severe growth defect and reduction of the cell wall β-1,6-glucan content. Takeuchi et al. (12) showed that Rot1 functions as the molecular chaperon of Kre6 and Kre5 and is engaged in the folding and stabilization of these proteins. They suggested that the reduction of the β-1,6-glucan content in the Δrot1 cells may be a secondary effect through the defect of Kre6 and Kre5 (12). We confirmed that a large amount of Kre6 is present in the ER in this study. This is consistent with the idea that the folding/maturation of Kre6 in the ER becomes an important step of β-1,6-glucan synthesis.
It has been reported that the mutants of END3 and SLA1 related to endocytosis at the PM or LAS17 and VRP1 related to cell polarity and actin patch show an abnormal increase of the cell wall β-1,6-glucan (7). It is also reported that the Δend3 or Δsla1 disruptant cells show mislocalization of the polarized proteins at the PM and the abnormal multiple-layer synthesis of the cell wall (33, 34). These observations indicate that the β-1,6-glucan synthesis can be largely affected by genes of endocytosis and cell polarity. Probably, the cell wall-synthesizing enzymes are delivered in the vesicles by the transport system to the PM and work to synthesize the wall polymers there until there is enough of the product, then they are taken up from the PM by endocytosis and transported to next places to work. β-1,6-Glucan synthesis will also be conducted in concert manner with the syntheses of other constituents. This paper reports for the first time that a significant part of Kre6 is present in the PM, and it is concentrated at the sites of polarized growth. The interaction of the cytoplasmic domain of Kre6 with Sla1 and Las17 has been reported (3), and this domain is required for trafficking or retention of Kre6 to the PM. It is important to examine if the effect of the PM endocytosis genes on β-1,6-glucan content is related to the presence of Kre6 in the PM.
The cell wall chitin and β-1,3-glucan are synthesized by the enzyme localized in the PM by polymerizing the unit sugars from UDP-sugar in the cytosol. These enzymes have a subunit with multiple transmembrane domains that is required to translocate the products after polymerization. Kre6 with a single transmembrane domain is quite unlikely to work as do the synthetases of chitin and β-1,3-glucan. When Kre6 is in the PM, the lumenal domain of this type II membrane protein with homology to glycoside hydrolase is outside of the PM. Although β-1,6-glucan is only detected in the outside of the PM by antiserum, it may be possible that small chains of β-1,6-glucan are synthesized from UDP-glucose in the cell and then somehow transported outside and joined by transglycosylation there. It remains to be examined if Kre6 has such an enzyme activity to elongate short β-1,6-glucan chains. Recently, it is reported that small molecular β-1,6-glucan chains were actually detected in the cytosol of S. cerevisiae (35). It may be better to consider the de novo β-1,6-glucan synthesis in the cell and its elongation to the final length outside of the cell.
At the very least, this study links the most important separated parts, the gene product related to β-1,6-glucan in the cell and β-1,6-glucan on the outside of the PM. Further studies on Kre6, especially on its cellular trafficking, possible enzyme activity, and interaction with other proteins, will elucidate the complete process of β-1,6-glucan synthesis in S. cerevisiae.
Supplementary Material
Acknowledgments
We thank Dr. Satoshi Kagiwada and Dr. Masao Tokunaga for antibodies, Dr. Hugh R. B. Pelham and Dr. Keisuke Sato for GFP-tagged gene constructs, and Dr. Keisuke Sato for critical reading of the manuscript.
This work was supported by a grant-in-aid for Scientific Research from the Japan Society for the Promotion of Science (to Y. N. and K. Y.) and a grant from the Noda Institute of Scientific Research (to Y. N.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4.
- PM
- plasma membrane
- ER
- endoplasmic reticulum
- SV
- secretory vesicles.
REFERENCES
- 1. Lesage G., Bussey H. (2006) Microbiol. Mol. Biol. Rev. 70, 317–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Montijn R. C., Vink E., Müller W. H., Verkleij A. J., Van Den Ende H., Henrissat B., Klis F. M. (1999) J. Bacteriol. 181, 7414–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li H., Pagé N., Bussey H. (2002) Yeast 19, 1097–1112 [DOI] [PubMed] [Google Scholar]
- 4. Vink E., Rodriguez-Suarez R. J., Gérard-Vincent M., Ribas J. C., de Nobel H., van den Ende H., Durán A., Klis F. M., Bussey H. (2004) Yeast 21, 1121–1131 [DOI] [PubMed] [Google Scholar]
- 5. Aimanianda V., Clavaud C., Simenel C., Fontaine T., Delepierre M., Latgé J. P. (2009) J. Biol. Chem. 284, 13401–13412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shahinian S., Bussey H. (2000) Mol. Microbiol. 35, 477–489 [DOI] [PubMed] [Google Scholar]
- 7. Pagé N., Gérard-Vincent M., Ménard P., Beaulieu M., Azuma M., Dijkgraaf G. J., Li H., Marcoux J., Nguyen T., Dowse T., Sdicu A. M., Bussey H. (2003) Genetics 163, 875–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levinson J. N., Shahinian S., Sdicu A. M., Tessier D. C., Bussey H. (2002) Yeast 19, 1243–1259 [DOI] [PubMed] [Google Scholar]
- 9. Nakamata K., Kurita T., Bhuiyan M. S., Sato K., Noda Y., Yoda K. (2007) J. Biol. Chem. 282, 34315–34324 [DOI] [PubMed] [Google Scholar]
- 10. Roemer T., Bussey H. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 11295–11299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roemer T., Paravicini G., Payton M. A., Bussey H. (1994) J. Cell Biol. 127, 567–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takeuchi M., Kimata Y., Kohno K. (2008) Mol. Biol. Cell 19, 3514–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sato K., Noda Y., Yoda K. (2009) Mol. Biol. Cell 20, 4444–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lewis M. J., Nichols B. J., Prescianotto-Baschong C., Riezman H., Pelham H. R. B. (2000) Mol. Biol. Cell 11, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sato K., Noda Y., Yoda K. (2007) Mol. Biol. Cell 18, 3472–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Powers J., Barlowe C. (1998) J. Cell Biol. 142, 1209–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amberg D. C., Burke D., Strathern J. N. (2005) Methods in Yeast Genetics, pp. 165–167, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 18. Imai K., Noda Y., Adachi H., Yoda K. (2005) J. Biol. Chem. 280, 8275–8284 [DOI] [PubMed] [Google Scholar]
- 19. Konomi M., Kamasawa N., Takagi T., Osumi M. (2000) Plant Morphol. 12, 20–31 [Google Scholar]
- 20. Humbel B. M., Konomi M., Takagi T., Kamasawa N., Ishijima S. A., Osumi M. (2001) Yeast 18, 433–444 [DOI] [PubMed] [Google Scholar]
- 21. Takagi T., Ishijima S. A., Ochi H., Osumi M. (2003) J. Electron Microsc. (Tokyo) 52, 161–174 [DOI] [PubMed] [Google Scholar]
- 22. Konomi M., Fujimoto K., Toda T., Osumi M. (2003) Yeast. 20, 427–438 [DOI] [PubMed] [Google Scholar]
- 23. Valdez-Taubas J., Pelham H. R. B. (2003) Curr. Biol. 13, 1636–1640 [DOI] [PubMed] [Google Scholar]
- 24. Roemer T., Delaney S., Bussey H. (1993) Mol. Cell. Biol. 13, 4039–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fehrenbacher K. L., Davis D., Wu M., Boldogh I., Pon L. A. (2002) Mol. Biol. Cell 13, 854–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Estrada P., Kim J., Coleman J., Walker L., Dunn B., Takizawa P., Novick P., Ferro-Novick S. (2003) J. Cell Biol. 163, 1255–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Du Y., Walker L., Novick P., Ferro-Novick S. (2006) EMBO J. 25, 4413–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arai S., Noda Y., Kainuma S., Wada I., Yoda K. (2008) Curr. Biol. 18, 987–991 [DOI] [PubMed] [Google Scholar]
- 29. Gurunathan S., Chapman-Shimshoni D., Trajkovic S., Gerst J. E. (2000) Mol. Biol. Cell 11, 3629–3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fujimoto K. (1997) Histochem. Cell Biol. 107, 87–96 [DOI] [PubMed] [Google Scholar]
- 31. Epping E. A., Moye-Rowley W. S. (2002) J. Biol. Chem. 277, 34860–34869 [DOI] [PubMed] [Google Scholar]
- 32. Valdivia R. H., Schekman R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10287–10292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ayscough K. R., Eby J. J., Lila T., Dewar H., Kozminski K. G., Drubin D. G. (1999) Mol. Biol. Cell 10, 1061–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tang H. Y., Xu J., Cai M. (2000) Mol. Cell. Biol. 20, 12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hosomi A., Tanabe K., Hirayama H., Kim I., Rao H., Suzuki T. (2010) J. Biol. Chem. 285, 24324–24334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}