

Abstract
A method to efficiently immobilize and partition large quantities of microbeads in an array format in microfabricated polydimethylsiloxane (PDMS) cassette for high-throughput in situ releasable solution-phase cell-based screening of one-bead-one-compound (OBOC) combinatorial libraries is described. Commercially available Jeffamine triamine T-403 (∼440 Da) was derivatized such that two of its amino groups were protected by Fmoc and the remaining amino group capped with succinic anhydride to generate a carboxyl group. This resulting tri-functional hydrophilic polymer was then sequentially coupled two times to the outer layer of topologically segregated bilayer TentaGel (TG) beads with solid phase peptide synthesis chemistry, resulting in beads with increased loading capacity, hydrophilicity and porosity at the outer layer. We have found that such bead configuration can facilitate ultra high-throughput in situ releasable solution-phase screening of OBOC libraries. An encoded releasable OBOC small molecule library was constructed on Jeffamine derivatized TG beads with library compounds tethered to the outer layer via a disulfide linker and coding tags in the interior of the beads. Compound-beads could be efficiently loaded (5-10 minutes) into a 5 cm diameter Petri dish containing a 10,000-well PDMS microbead cassette, such that over 90% of the microwells were each filled with only one compound-bead. Jurkat T-lymphoid cancer cells suspended in Matrigel® were then layered over the microbead cassette to immobilize the compound-beads. After 24 hours of incubation at 37°C, dithiothreitol was added to trigger the release of library compounds. Forty-eight hours later, MTT reporter assay was used to identify regions of reduced cell viability surrounding each positive bead. From a total of about 20,000 beads screened, 3 positive beads were detected and physically isolated for decoding. A strong consensus motif was identified for these three positive compounds. These compounds were re-synthesized and found to be cytotoxic (IC50 50-150 μM) against two T-lymphoma cell lines and less so against the MDA-MB 231 breast cancer cell line. This novel ultra high-throughput OBOC releasable method can potentially be adapted to many existing 96- or 384-well solution-phase cell-based or biochemical assays.
Keywords: OBOC library, combinatorial chemistry, high-throughput screening, in situ solution phase, releasable screening, cell-based screening, Jeffamine-triamine, microbead cassette
Introduction
In the one-bead-one-compound (OBOC) combinatorial library method, hundreds of thousands to millions of chemical compounds can be synthesized within days and screened against a variety of targets using on-bead binding or other functional assays.1 Initially developed to screen libraries consisting of short linear peptides,2 OBOC libraries have since been expanded to cyclic peptides,3 peptoids (N-substituted oligoglycine),4 peptidomimetic and small molecule libraries5 and has been successfully used in the identification of protein kinase substrates and inhibitors,5c, 6 protease substrates and inhibitors,3f, 7 G-coupled protein receptor inhibitors5a enhancers of the latrunculin B actin polymerization inhibitor,5d transcriptional activators,4a ligands against mRNA precursors,8 integrin-specific peptides against T-lymphoid leukemia cells,9 idiotype-specific peptides for murine lymphoma cells,10 molecular mimics to induce autoimmunity,11 MHC molecule anchor residues and antigenic epitopes for monoclonal antibodies,12 protein sequestering ligands for proteomic analysis13 ligands that bind and filter pathogenic prion proteins from blood14 and various ligands for the preparation of affinity column media.15 Methods have also been developed to prepare topologically segregated bilayer beads, permitting the synthesis of library molecules on the outer layer and the coding tag in the inner core.16 Such methods enable one to develop encoded small molecule, peptidomimetic or macrocyclic OBOC combinatorial libraries.
In addition to on-bead screening assays in which the library compounds remained tethered to the bead throughout the screen, library compounds can also be released from the OBOC libraries for in situ releasable solution-phase format.17 Such releasable schemes have been used with limited success to carry out what has been referred to as OBOC “lawn assays” in which beads are immobilized in soft agar or gelatin such that the beads are oriented randomly throughout the entire dish without any specific alignment18. Compounds are then released into the vicinity of each immobilized bead. There is no partition between the beads. As a consequence, the compounds surrounding the beads tend to be quite dilute. Unrestricted diffusion of released compounds also promotes increased heterogeneous spatial distributions of released compounds from multiple beads within the same region. To overcome these problems, we have recently developed a method such that large number (over 10,000) beads can be quickly and efficiently immobilized into a highly consistent and spatially distinct, uniform array format. To accomplish this, we used a photolithographic method to generate reusable molds from which the fabrication of hundreds of polydimethylsiloxane (PDMS) casts or microbead cassettes can be made. Each of these bead cassettes consist of thousands of arrayed “wells” optimized to anchor in place individual beads by making use of the innate swelling properties of the bead resin in different solvent environments. In addition, the individual wells that comprise the bead cassettes not only function to uniformly array tens of thousands of beads in a highly efficient manner, but also serve to restrict lateral diffusion of the released library compounds, thereby achieving greater concentrations of homogeneous compounds in specified bead vicinities. These advancements are major feats in the optimization of high-throughput in situ releasable solution-phase screening methodology.
TentaGel (TG) resin (Rapp Polymer Inc), with its size uniformity, rigidity, and compatibility with aqueous and organic conditions is most suitable for OBOC library construction and on-bead screening assays.6d However, it is not optimal for in situ solution phase releasable assays due to the tendency of the released hydrophobic and heterocyclic compounds to retain inside the polystyrene matrix of the bead resin.17c This can result in great variability between the quantity of compounds released from each bead, thereby complicating the screening process. To overcome this problem, we have developed a shell-core bead with a shell of hydrogel grafted onto the surface of the TG resin, such that the highly hydrophilic outer shell can be used for library synthesis and the inner core for chemical encoding.17c However, the grafting of a hydrogel shell onto TG resin is rather cumbersome and not easy to control. Here we report an alternative but more robust method of derivatizing the outlayer of TG beads with hydrophilic dendrimeric structures such that the loading capacity of the outer layer is high enough for OBOC in situ releasable solution-phase assays. To accomplish this, we derivatized commercially available Jeffamine triamine T-403 (∼440 Da) such that two of its amino groups were protected by 9H-fluoren-9-ylmethoxycarbonyl (Fmoc) and the remaining amino group capped with succinic anhydride to generate a carboxyl group. This tri-functional hydrophilic polymer ((Fmoc)2 Jeffamine-acid linker 1) was then sequentially coupled two times to the outer layer of topologically segregated bilayer TG beads via solid phase peptide synthesis chemistry, resulting in the formation a hydrophilic dendrimeric coating on the TG bead outer layer with increased surface loading capacity, hydrophilicity and porosity. Jeffamine triamine T-403 is a polyether amine characterized by repeating oxypropylene units and exists as a complex mixture of oligomers with an average molecular weight of 440 Da.19 Each Oligomer differs in weight by 58 Da. Jeffamine triamine is completely miscible with a wide range of organic solvents as well as water. To evaluate the utility of Jeffamine modified TG beads in OBOC releasable library screening, we studied the release of oligo-arginine-FITC from these beads and the uptake of the fluorescent cell-penetrating peptide into adjacent living cells. An encoded OBOC combinatorial small molecule library was also constructed on these beads; and when used in conjunction with the highly efficient microbead cassette as the cell-based screening platform, we were able to identify potent cytotoxic compounds against malignant T-lymphoid cells.
Results and Discussion
Immobilization of OBOC library beads
A major challenge of a robust OBOC in situ releasable solution-phase assay is to develop methods such that tens of thousands to hundreds of thousands of compound-beads can be efficiently immobilized in a solid substrate and individual beads partitioned from one another. To achieve this, a micro-bead cassette was developed using standard soft lithography techniques such that a reusable mold patterned over a silica wafer was constructed using a negative tone photoresist (Figure 1A and B). The developed mold was then used to fabricate a polydimethylsiloxane (PDMS) cast consisting of optimally sized wells for bead loading/immobilization (Figure 1C). The PDMS cast was bonded to the bottom of a standard polystyrene Petri dish with a thin layer of PDMS spin-coated inside the bottom of the dish. Alternatively, the PDMS casts could be permanently bonded to a secondary substrate (glass slide or polystyrene Petri dish) by oxygen plasma treatment of the contact surfaces. The resulting micro-bead cassettes were sterilized with 70% alcohol prior to cell-based assays.
Figure 1.
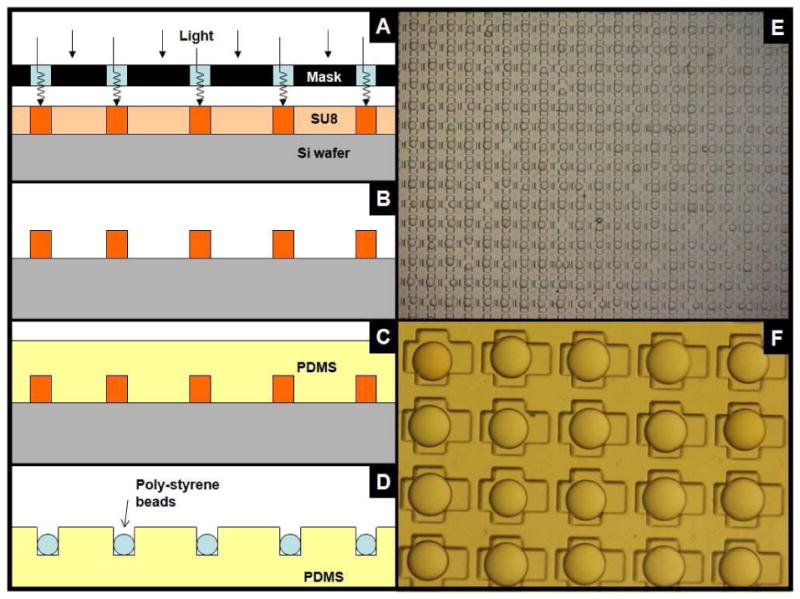
Immobilization of TG beads using photo-patterned microfabricated device. A) Si wafer substrate is coated with SU-8 2150 negative tone ultra thick photoresist and photo patterned. B) After developing, C) the device mold is coated with PDMS prepolymer D) to construct the reverse replica PDMS cast which can be loaded with polystyrene beads. E and F) PDMS cast consisting of arrayed “wells” after loading of 120 μm TG beads and swelled in 70% EtOH.
TG resin tends to swell in water and shrink in ethanol. We took advantage of this swelling property for loading of the library compound-beads into the micro-bead cassette. The patterned wells of the micro-bead cassette were first submerged in 100% ethanol. The compound-beads were swollen in 70% ethanol which served to sterilize the beads and also dramatically reduce bead clumping. The bead suspension was then added drop-wise over the surface of the 100% ethanol layer covering the wells. As the beads shrunk from the change in solvent environment, they became less buoyant, thereby causing them to sink and fall into the empty wells (Figure 1D-F). To reduce the propensity for trapping air inside the loaded wells, especially under aqueous conditions, a “cross-like” shape was adopted which featured an optimal 140 × 140 μm internal square to accommodate swollen 120 μm TG beads. The outer “cross” extensions can vary in shape and size depending on the needs of the screen. For example, multi-parametric high-content cell-based assays that require high resolution fluorescent microscopy and a visual field free from obstruction by the library bead will require a large extension area from the bead (Figure 1F). Once the beads are immobilized inside the micro-wells of the bead cassette, a variety of cell-based and biochemical assays can be performed.
Synthesis of (Fmoc)2Jeffamine-acid Linker
Lee et al. has reported the development of novel core-shell-type resins in which the functional groups were located on the outer shell layer composed of Jeffamine ED-600.20 The synthesis of the core-shell (CS) resins was achieved by cross-linking aminomethyl polystyrene resin with 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride, CNC) and the resulting resins were further treated with an excess of Jeffamine ED-600. As a result, the long chained Jeffamines were selectively coupled to the active chloride groups of the CNC on the outside of the bead resin. After coupling of FITC (5-fluorescein isothiocyanate) to the CS free amino group, confocal microscopy revealed that the CS resin-bound FITC existed only on the surface area, whereas the TentaGel and AM-PS resin-bound FITC was observed continuously throughout the bead volume. Furthermore, the initial loading onto the CS resins progressed more efficiently than that of other gel-type resins and the CS resin demonstrated significant compound release potential in a photo-cleavage reaction.20
In order to precisely control the grafting of Jeffamine onto TG, we have decided to use a step-wise dendrimer approach to decorate the outer layer of the TG resin, using (Fmoc)2-Jeffamine-acid as the building block and standard solid phase peptide chemistry as the coupling method. In the first step, Jeffamine triamine was selectively protected with succinyl moiety on treatment with succinic anhydride (1 eq) in dioxane for 30 minutes at 0°C. After removal of solvent, the resulting oily material was dissolved in water and extracted in ether to remove side products. To protect the two remaining amino groups with Fmoc, Fmoc-OSu (2 eq) in dioxane was added to reaction mixture at 0°C for 30 minutes and then stirred overnight at room temperature. After removal of solvent, the resulting oily material was dissolved in water and extracted in ether to remove side products. The remaining two amino groups were Fmoc protected with Fmoc, Fmoc-OSu (2 eq) in dioxane was added to reaction mixture at 0°C for 30 minutes and then stirred overnight at room temperature. After removal of solvent, the oily material was dissolved in water and extracted in ether to remove impurities. The aqueous layer was acidified and extracted with dichloromethane to get an oily material which was further purified by SiO2 column chromatography to generate pure (Fmoc)2-Jeffamine-acid linker (1) as colorless oil. HPLC chromatogram of linker 1 (Supporting Material) exhibited the presence of PPG oligomeric mixtures as reported for Jeffamine T-403.19 Matrix assisted laser ionization time-of-flight mass spectrometry (MALDI-TOF-MS) spectra of (Fmoc)2-Jeffamine-acid linker linker (1) (Supporting Material) showed that peaks of the oligomers were separated by 58 amu corresponding to polypropyleneoxide (PPO) unit, whereas the average molecular weight (MW) of (Fmoc)2-Jeffamine-acid linker linker (1) was determined to be 988.9.
To introduce a hydrophilic dendrimer to the outer layer of TG resin, (Fmoc)2-Jeffamine-acid linker (1) was coupled to the bead outer layer followed by Fmoc-deprotection. The process was repeated to yield a Jeffamine dendrimer layer of four-fold functional amplification, making the outer layer of the bead more porous, hydrophilic and high-loading. The outer layer of Jeffamine modified beads were then linked to previously reported disulphide linker17c (2, structure is shown at the bottom of Figure 3) for the development of an efficient OBOC in situ releasable solution-phase assay platform for ultra- high throughput screening.
Figure 3.

Synthesis of Jeffamine-derivatized TG beads displaying octa-arginine/FITC and LLP2A. Reagents: (a) TG resin was swollen in H2O, 48 h; Fmoc-OSu, DCM/Ether, DIEA; Boc2O, DIEA, DMF; (b) 20% Piperidine/DMF; Fmoc2-JA-OH (1), HBTU, HOBt, DIEA, NMP; this step was repeated once (c) 20% Piperidine/DMF; a mixture of Fmoc-OSu and Alloc-OSu (1:1), DIEA, DMF; (d) 20% Piperidine/DMF; Fmoc-[S-S]-OH (2), HOBt, DIC, DMF; (e) 20% Piperidine/DMF; Fmoc-K(Dde)-OH, HOBt, DIC, DMF; (f) 20% Piperidine/DMF; Fmoc-R(Pbf)-OH, HOBt, DIC, DMF (repeated 7 more times); (g) 20% Piperidine/DMF; Boc2O, DIEA, DMF; (h) Pd(PPh3)4, PhSiH3, DCM; Fmoc-Ach-OH, HOBt, DIC, DMF; (i) 20% piperidine/DMF; Fmoc-Aad(OtBu), HOBt, DIC, DMF; (j) 20% piperidine/DMF; HOBt, DIC, DMF; Fmoc-K(Alloc); (k) 20% piperidine/DMF; 2-{3-[(2-toluidinocarbonyl)amino]phenyl} acetic acid, HOBt, DIC, DMF; (l) Pd(PPh3)4, PhSiH3, DCM; (E)-3-(3-pyridinyl)-2-propenoic acid, HOBt, DIC, DMF; (m) 2% hydrazine/DMF; FITC, DIEA, NMP;TFA:Thioanisole:Phenol:H2O:TIS (84:5:5:5:1) (v/v).
Compound Release from Jeffamine-derivatized TG resin
To evaluate the efficiency of the derivatized beads to release the library compounds in solution phase assays, we prepared Jeffamine-derivated TG beads displaying FITC-octa-arginine (the surrogate library compound) and LLP2A ligand (cell-capturing ligands)9 on the outer layer (Figure 3). The FITC-octa-arginine conjugate was the surrogate library compound tethered to the outer layer via a disulfide linker.17c LLP2A is a high-affinity ligand against α4β1 integrin of malignant lymphoid cells and was used as a ligand to capture lymphoma cells to the bead surface. To prepare such beads, topologically segregated bilayer TG beads bearing 10 % Fmoc group on outer surface and 90% Boc inside were prepared followed by Fmoc deprotection and then (Fmoc)2 Jeffamine-acid linker (1) was linked to outer surface using HBTU/DIEA in NMP producing bifunctional beads.16 After Fmoc deprotection, (Fmoc)2 Jeffamine-acid linker (1) coupling was repeated. Fmoc groups on the outer surface were replaced with an equal combination of Alloc and Fmoc groups (Figure 3). After Fmoc-removal, disulfide linker (2),17c followed by octa-arginine, a cell penetrating peptide,21 was synthesized on the highly branched outer surface of the bead. In the next step, Alloc group was removed followed by synthesis of LLP2A,9 on the outer surface of the bead. A fluorescent dye, FITC, was covalently linked to octa-arginine after removal of Dde. Finally, side chain deprotection as well as removal of Boc group from inner amines was achieved by treatment with TFA cocktail for 12 hours twice to ensure complete removal of side chain protecting group from octa-arginine.
To demonstrate the release and cellular uptake of the octa-arginine-FITC conjugate, Molt-4 T-lymphoma cells were first captured on the bead surface with the LLP2A ligand. Compound release was initiated by the addition of 0.05% dithiothreitol (DTT) which reduces and cleaves the disulfide linker tethering the octa-arginine-FITC conjugate to the outer layer of the bead. Figure 4A and B show results 30 minutes after addition of DTT, with release of oligoarginine–FITC conjugate, with background light plus FITC filter (ìex 488, 555; ìem 520, 605), and FITC filter alone, respectively. Strong fluorescence signal emanating from individual cells suggests successful diffusion of the released compound from the bead into the cell. To characterize the diffusion of compound from the bead surface in gel matrix, the octa-arginine-FITC conjugated beads were immobilized with soft agar inside a Petri dish (Figure 4C) or inside a well of the PDMS micro-bead cassette (Figure 4D). Fluorescent images were obtained 10 minutes after compound release was initiated by the addition of 0.1% DTT in PBS over the agarose gel layer. In Figure 4C, a uniform distribution of fluorescence signal can be seen which gradually fades as the distance from the bead increases. This suggests a relatively uniform compound diffusion through the soft agar. Beads immobilized in PDMS microbead cassette wells demonstrated a strong build-up of fluorescence intensity inside the individual PDMS micro-well harboring the bead. These results indicate a significantly higher concentration of released compound is made possible by the “wall effect” of the microwells.
Figure 4.
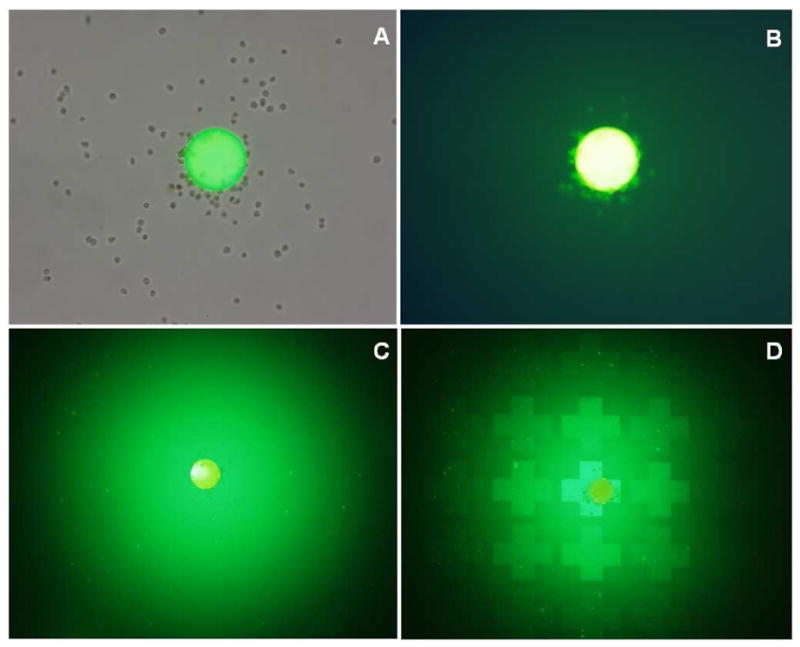
Release of octa-arginine-FITC from a bead displaying LLP2A, the lymphoma cell capturing ligand (see Figure 3). Compound release was triggered by addition of 0.1% DTT into the medium. (A, B) Molt-4 cells were first incubated and bound to the bead. Surface release of octa-arginine–FITC was facilitated by the addition of 0.1% DTT to the culture medium. (C, D) Molt4 cells were (C) suspended in 1% low melt agarose and coated over Petri dish and (D) seeded over the PDMS microbead cassette and coated with a thin layer of 1% low melt agarose. Fluorescent micrographs were obtained 10 minutes after the addition of DTT (488nm/520nm ex/em).
Small Molecule Releasable Library Synthesis
To evaluate the utility of Jeffamine-derivatized beads in high-throughput releasable solution-phase assay, a OBOC small molecule releasable library was generated (Figure 5) using our previously reported encoding methods16. Topologically segregated bilayer Tentagel beads bearing Jeffamine dendrimer with Fmoc-disulfide linker as the dendrimeric termini on the outer layer were first prepared to initiate the coding arm. Fmoc-Lys(Dde)-OH was then coupled to the inner core after Boc-deprotection of the interior coding arm. The Fmoc-group on both outer and inner layers was then removed, making ready for the construction of the OBOC dipeptide library using 30 different amino acids (Supporting Material) as the building blocks. After the synthesis of the dipeptide library, the bead was partitioned into outer layer and inner core and differentially protected with Fmoc and Boc, respectively. Upon treatment with 2% hydrazine, the Fmoc group at the exterior testing arm (N-terminus of the dipeptide) and Dde at the interior coding arm (lysine side chain) were removed concurrently and then acylated with 25 different carboxylic acids (Supporting Material). The 25 carboxylic acids were chosen because they could be readily coupled to amino group with high yield, but more importantly, such acylated lysine derivatives at the interior coding tag could be ambiguously decoded with microseqeucing.16a After treatment with TFA and thorough washing, the releasable small molecule OBOC library (R-XX, 22,500 diversity, wherein R = acyl group, and X = both L and D amino acids) was ready for screening.
Figure 5.

Synthesis of an encoded OBOC small molecule releasable library. Reagents: (a) (3), 20% Piperidine/DMF; Fmoc-[S-S]-OH (2), HOBt, DIC, DMF; (b) TFA/DCM (1:1); Fmoc-K(Dde)-OH, HOBt, DIC, DMF; (c) 20% Piperidine/DMF; Fmoc-X-OH, HOBt, DIC, DMF (repeated 1 more time); (d) 20% Piperidine/DMF; H2O, 48 h; Fmoc-OSu, DCM, Ether DIEA; Boc2O, DIEA, DMF; (e) 2% hydrazine/DMF; R-COOH, HOBt, DIC, DMF; (f) TFA:Thioanisole:Phenol:H2O:TIS (82.5:5:5:5:2.5) (v/v).
Screening for Cytotoxic Compounds
To screen for cytotoxic compounds against Jurkat T-lymphoma cells, two 25cm2 Petri dishes, each containing a 10,000 well bead cassette, were loaded with beads from the OBOC releasable small molecule library (Figure 5). Bead loading was carried out in ethanol and each cassette was loaded to over 90% bead capacity with one bead in each microwell. The loaded cassettes were then placed under vacuum to completely dry down the loaded beads. A suspension of Jurkat cells in Matrigel™ was gently poured over the bead cassettes to form 3-D cell cultures surrounding each immobilized bead. Cell culture media was added over the top of the “seeded” bead cassettes and cultures were incubated until cell colonies reached desired confluency, in this case 24 hours (Figure 6A). Compound release was initiated with the addition of fresh media containing 0.05% dithiothreitol (DTT) for a 30 minute period. After 2 hours, the cassettes were gently washed with sterile PBS to remove any residual DTT. Fresh media was then added over the 3-D gel matrix and the cassettes were incubated for a 48- hour treatment period. Cell viability was detected with the addition of fresh media containing 0.05% MTT reagent (tetrazolium bromide). After four hours of incubation, cassettes were inspected under a dissecting microscope for regions of decreased MTT reduction. Figure 6B-D shows three distinct regions of uniform decreased cell viability/metabolism as indicated by a reduction in MTT reduction. These regions were closely associated to proximities of individual beads and contained recognizable evidence of non-viable cells (Figure 6D). Accordingly, these positive beads were selected and the interior peptide coding tags were sequenced with Edman chemistry. The resulting sequences had striking consensus with 3-hydroxy-2-quinoxaline carboxylic acid and L-2-naphthylalanine as the N-terminal carboxylic acid and the adjacent amino acid building block, respectively (Figure 7). The probability for this to happen by chance alone is (1/750)3 or 1/421,875,000.
Figure 6.

In-situ release of surface bound compound from dipeptide library and resulting MTT cell viability assay. A) Jurkat cells were suspended in BD Matrigel™ and seeded over loaded bead cassette and cultured to desired confluency before compound release was initiated with the addition of 0.1% DTT. B-D) After 48 hr treatment period, cell viability was measured topologically throughout the cassette with the addition 0.05% MTT reagent (terazolium bromide). Beads with uniform regions of metabolically inactive cells were selected and sequenced.
Figure 7.

Positive compounds identified from initial cell killing screen (LTS1, LTS2 and LTS3) were synthesized on TG resin with disulfide cleavable linker and Rink cleavable resin resulting in sulfhydryl (-SH) and amide (-CONH2) forms of the three compounds, respectively.
Validation of cytotoxic compounds
Due to the fact that the initial screening process made use of a disulfide cleavable linker for on-demand release of library compounds, the resulting solution phase compounds contain a sulfhydryl group at the cleavage site. To compare the contribution of the sulfhydryl group to their biological activities, the three compounds were re-synthesized on both Rink resin and TG resin harboring a disulfide cleavable linker. The compounds were cleaved from Rink resin using trifluoroacetic acid (TFA) and from TG resin using DTT reducing agent, generating amide (-CONH2) and sulfhydryl (-SH) forms of the three compounds, respectively (Figure 7). The compounds were purified with C-18 RP HPLC and evaluated for their cytotoxic activities in 96-well-plate format. Figure 8 shows the results of quantitative cell viability assays using the both cleavage forms of two of the three synthesized compounds (LTS2 and LTS3) identified during the OBOC releasable screen against three cancer cell lines. Because the initial screen was carried out using Jurkat T lymphoma cell line, both Jurkat (Figure 8) and Molt-4 (data not shown) T lymphoma cancer cell lines were used for validation of cytotoxic effects of the identified compounds. In addition, an MDA-MB 231 breast cancer cell line was tested as a preliminary measure of cytotoxic specificity. Cells were cultured in 96 well plates at desired concentration (∼5000 cells/well). After 48 hour treatment period, Alamar Blue® cell viability indicator (AbD Serotec) was added to each well and the difference of cell viability relative to untreated cells was measured for each compound. Against both lymphoma lines, the amide form of LTS2 (LTS2-NH2) demonstrated the most pronounced cytotoxic activity (IC50 50 μM) while LTS3-NH2 elicited a cytotoxic response that was approximately three-fold weaker (IC50 150 μM) (Figure 8). The sulfhydryl forms of both compounds (LTS2-SH and LTS3-SH) were far less cytotoxic to both lymphoma lines (IC50 >200 μM). The reasons for these findings are not clear but these results may be due to compound solubility or stability as opposed to any direct effect of the sulfhydryl group on the activity of the compounds. All three compounds share marked hydrophobic properties. Indeed, at concentrations above 200 μM, noticeable signs of precipitate are evident when the compounds are transferred from a solvent (DMSO) to aqueous environment. In the case of the sulfhydryl forms of the compounds, it is also possible that the sulfhydryl group can become oxidized and subsequent cross-linking to form dimer may ensue with time whereas during the preliminary screen, the compound is released from the bead in its monomeric active state. The fact that, upon subsequent analysis, the sulfhydryl forms of the compounds seemed to lose effectiveness with time also indicates instability may be the source of the decreased effectiveness. Nevertheless, the observed cytotoxicity of LTS2-NH2 and LTS3-NH2 not only provide evidence to validate the results of the initial screen, but also suggests that the cleavable disulfide linker releasable mechanism used during the screen did not itself play a role in the activity of these compounds. Interestingly, the cytotoxic effects of these compounds against MDA-MB 231 breast cancer cell line were far less active in the concentration range tested. While the sulfhydryl forms of the compounds seemed to have no effect on cell viability, a mild cell killing effect was observed for LTS2-NH2 (IC50 ∼200 μM) and an even less substantial effect was observed for LTS3-NH2 (Figure 8). The other compound identified during the screen (LTS1) in which two of the three building block constituents were identical, demonstrated inconclusive results during the cytotoxicity analysis (data not shown). The reason for this is not clear but could be in part due to the insolubility of the purified product stemming from the increased hydrophobicity of its unique amino acid constituent, D-homocitruline. This may also explain why LTS2, containing L-histidine, the most hydrophilic unique amino acid constituent of the three identified compounds, is also the most active based on the results shown here. In the future, it may be possible to increase the activity of this motif by further diversifying its structure to increase the compound's solubility. Further analysis is also required to elucidate the specific mechanism of action of one or more of these compounds. Although we used disulfide linker as the on-demand cleavable linker in this study, we can easily adapt other cleavage linkers such as photo-cleavable linker17b, 18d and reverse diketopiperazine linker18e to this ultra-high throughput screening platform.
Figure 8.
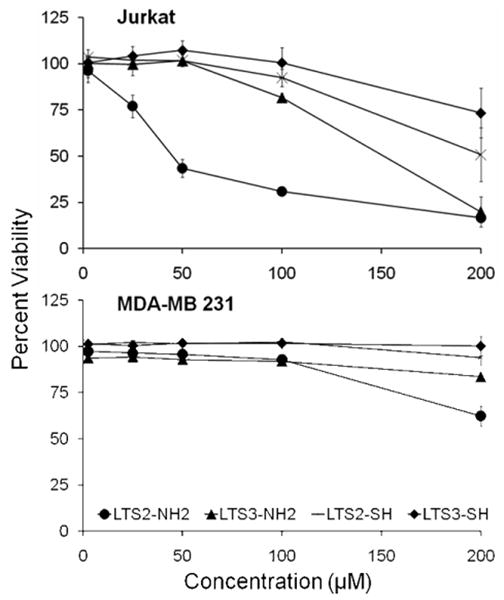
Quantitative cell viability assays plotted as (y) difference in percent reduction of alamarBlue® substrate vs. (x) compound concentration using two cancer cell lines.
Conclusions
From a primary screen of approximately 20,000 beads obtained from a library with 22,500 diversities for cytotoxicity against Jurkat T-lymphoma cells, three positive compounds with strong structural consensus were identified. These compounds were re-synthesized and demonstrated to be cytotoxic (IC50∼50μM) against two T-lymphoma cell lines but much less so against a breast cancer cell line. This limited library synthesis and cell-based screening experiment clearly demonstrates that our novel ultra-high-throughput, releasable solution-phase OBOC combinatorial platform is robust and could be adapted to many other existing 96- and 384-well cell-based or biochemical assays for drug discovery or chemical biology research. The main advantage of the OBOC combinatorial library method is that it is a practical, economical, and effective chemical library format that can be applied by a large number of academic investigators around the world. Although chemical decoding is needed in OBOC library method, it eliminates the need to have an expensive storage and archival system to track each and every compound in conventional chemical libraries (stored in 96-well plates under DMSO at -20°C), which is unaffordable by most academic institutions. Furthermore, the shipping and handling of OBOC libraries is easy, e.g. 100,000-500,000 discrete small molecule chemical compounds can be stored and shipped in a small 1 mL vial via Federal Express at room temperature. No expensive robotic systems are needed in the synthesis and screening of the OBOC libraries. Since the assay is highly miniaturized, the amount of expensive reagents needed for screening is minimal. In recent years, more investigators are using OBOC method successfully in their research and we expect this trend to continue.3c, 3g, h, 4, 5d, e, 8, 11, 13-14, 22
Experimental Section
General Considerations
SU-8 2150 was obtained from MicroChem USA. Fmoc-protected amino acids were purchased from Advanced ChemTech and Novabiochem and Chem-Impex International, Inc. (Wood Dale, IL). TentaGel S NH2 ∼ 92 μm, 0.27 μmol/g resin was purchased from Rapp Polymere Gmbh (Tubingen, Germany). HOBt (1 hydroxybenzotriazole) and Fmoc-OSu were purchased from GL Biochem (Shanghai, China). DIC, DIEA, cystamine · 2HCl, and FITC were purchased from Aldrich (Milwaukee, WI) and were used after drying at 25 °C in vacuum. All organic solvents and other chemical reagents were purchased from Fisher Scientific (Houston, TX) and Aldrich. For Fmoc deprotection, the resin was treated with 20% piperidine/DMF for 15 min and then washed thoroughly with DMF, MeOH, and DCM 3 times each. All infrared spectra were determined on a Genesis II Mattson FT-IR. UV-visible absorbances were obtained using Spectra Max M2 spectrometer of Molecular Devices (Sunnyvale, CA). Column chromatography was performed on silica gel using a mixture of MeOH and DCM and AcOH as the eluent. Analytical HPLC analyses (Vydac column; 4.6 mm × 250 mm; 5 μm; 300 Å; C18; 1.0 mL/ min; 25 min gradient from 100% aqueous H2O (0.1% TFA) to 100% CH3CN(0.1% TFA); 214, 220, 254, and 280 nm) were performed on a Beckman System Gold HPLC system (Fullerton, CA) or on Waters 2996 photodiode array detector, a Waters 2525 binary gradient module, and a Waters 2767 sample manager equipped with a 4.6 × 150 mm Waters Xterra MS C18 5.0 μm column employing a 20 min gradient from 100% aqueous H2O (0.1% TFA) to 100% CH3CN (0.1% TFA) at a flow rate of 1.0 mL/min. High-resolution fluorescence data and images were obtained using an Olympus fluorescence microscope (IX70).
Microfabrication of the Bead Cassette
Si wafer molds were constructed using ultra thick negative tone SU-8 photoresist patterned using standard photolithography techniques using L-Edit software and GCA Pattern generator to construct the photomask. The parameters used to obtain the 200 μm SU-8 2150 features on 4” Si wafer surface were as follows: CEE Spinner/hotplate to spread 500 rpm,15s 100 rpm/sec; spin 2500 rpm, 30 sec 300 rpm/sec; soft bake 65 °C, 6 min, 95 °C, 45 min; Karl Suss MA4 to expose 380 mJ/cm2; post exposure bake 65 °C, 5 min, 95 °C, 15 min; develop SU8 Developer 10 min light sonicating; hot bake 150 °C, 30 min.
The PDMS cast was fabricated using standard using softlithography techniques. For PDMS release, the wafer was pre coated with SL2 SigmaCote® and vacuum baked 100 °C, 30 min. The PDMS prepolymer was poured onto wafer, out-gassed and cured at 100 °C 120 min. After peeling off PDMS with transferred 200 μm well structures, the PDMS devices were cut to fit inside the bottom of 2 inch polystyrene Petri dishes. The devices were washed and sonicated in 100% EtOH and dried. To permanently bond the PDMS devices, the dishes were pre spin-coated with a thin layer of PDMS prepolymer which was partially cured at room temperature for 12 hours.
TG bead loading was performed by adding TG beads thoroughly swelled in 70% EtOH over the top of the PDMS wells submerged in 100% EtOH. Excess solvent and unloaded beads were carefully removed using a vacuum filter trap. Loaded “bead cassettes” were dried down overnight in desiccators to be used directly for cell based screening. *Optional transfer method to secondary substrate (data not shown) was performed by using a glass substrate spin-coated with PDMS prepolymer and partially cured for 20 hours. Coated substrate was then placed over the loaded PDMS wells and minimal force was used to transfer the dried down beads from the wells to the partially cured layer of PDMS which cured completely for an additional 48 hrs at room temperature to render the “bead chip”.
Synthesis of Jeffamine Linker
Jeffamine triamine T-403 (15 mmol, 6.6 mL) was dissolved in 1,4-dioxane (25 mL). The mixture was cooled in an ice-water bath with a vigorous stirring. A solution of anhydride (13 mmol, 1.3 g) in 1,4-dioxane (50mL) was added to the flask over 30-40 min., and the resulting solution was stirred for 15 min. at room temperature. Upon the addition of anhydride a white sticky material was formed. The solvent was removed under vacuo. An aqueous Na2CO3 solution (13 mmol, 1.37g) was then added into the flask, and the resulting solution was treated with ether (200mL) to remove any unreacted material and side products. The bottom aqueous layer was then collected into 1L round bottom flask, followed by the addition of Na2CO3 (30 mmol, 3.18g). The resulting solution was placed in an ice-water bath with a vigorous stirring. After the complete dissolution of Na2CO3, Fmoc-OSu (30mmol, 10.11g) in 1,4-dioxane was added drop wise and the resulting mixture was stirred for 30 min and then stirred at room temperature overnight. A white precipitate was formed which was removed by filtration and the filtrate was then concentrated to remove the solvent. The aqueous solution was treated with ether to remove the side products and unreacted material. The aqueous layer was acidified with 1M HCl to pH 1-2, and then extracted with dichloromethane. The organic layer was dried over MgSO4 and filtered. The filtrate was dried in vacuo to yield product which was further purified by SiO2 column chromatography using CH2Cl2:MeOH:AcOH (9.5:.4:.1, v/v/v) as eluent to yield purified desired product as colorless oil (1) (yield 65 %); MALDI-TOF-MS: m/z = 872, 930, 988, 1024, 1105 (average 988). These peaks are 58 (PPO) apart.
Synthesis of Disulfide Cleavable Linker
A suspension of cystamine dihydrochloride 1 (22.2 mmol, 5g) DIEA (44.4 mmol, 7.7 mL) and Methanol (25mL) was cooled in an ice-water bath with a vigorous stirring. A solution of succinic anhydride (18 mmol, 1.8g) in 1,4-dioxane (50 mL) was added to the flask over 30-40 min., and the resulting solution was stirred for 15 min. at room temperature. The solvent was removed under vacuo. An aqueous Na2CO3 solution (25 mmlo, 2.6g) was then added into the flask, and the resulting solution was treated with ether (200 mL) to remove any unreacted material and side products. The bottom aqueous layer was then collected into 1L round bottom flask, followed by the addition of Na2CO3 (36 mmol, 3.8g). The resulting solution was placed in an ice-water bath with a vigorous stirring. After the complete dissolution of Na2CO3, Fmoc-OSu (22.2 mmol, 7.4 g) in 1,4-dioxane was added drop wise and the resulting mixture was stirred for 30 min and then stirred at room temperature overnight. A white precipitate was formed which was removed by filtration and the filtrate was then concentrated to remove the solvent. The aqueous solution was treated with ether (200 mL) to remove the side products and unreacted material. The aqueous layer was acidified with 1M HCl to pH 1-2, and then extracted with dichloromethane. The organic layer was dried over MgSO4 and filtered. The solution was concentrated to a small volume and diluted with Et2O to give a white solid. The crystallized product was collected, washed with Et2O, and dried in vacuum. A white solid powder (2) was obtained with a yield of 47.6%. ESI-MS m/z 475.1 (M+).
General Procedure for Preparation of Two Layer TG Beads
[Inner (90% Boc-Protected), Outer-Layer (10% Fmoc-Protected)]. For preparation of topologically segregated bifunctional TG beads (3) with 10 % Fmoc outside and 90 % Boc inside, TG beads (1.0 g, 0.27 mmol) were swollen in water for 48 h. The water was drained, and a solution of Fmoc-OSu (9.09 mg, 27.0 μmol) in 50 mL of DCM/Et2O mixture (55:45, v/v) was added to the resin, followed by addition of DIEA (9.41 μL, 54.0 μmol). The resulting mixture was shaken vigorously for 30 min. The resin was washed with DCM (5 mL × 3) and DMF (5 mL × 6). A solution of (Boc)2O (212.09 mg, 0.97 mmol) in 10 mL of DCM was then added to the beads, followed by the addition of DIEA (338.8 μL, 1.94 mmol). The mixture was shaken until the ninhydrin test was negative. The obtained outside-Fmoc inside- Boc-bifunctional beads (3) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) each and dried in vacuum.
Preparation of Jeffamine Derivatized TentaGel Beads (6)
Bifunctional beads (3) (1 g, 10 % Fmoc-outside (27.0 μmol, 90% Boc-inside (0.243 mmol) were swollen in DMF overnight, followed by Fmoc deprotection. A mixture of Jeffamine linker (1) (80.02 mg, 81 μmol), HOBt (10.93 mg, 81 μmol), HBTU (30.69, 81 μmol) DIEA (28.246 μL, 162 μmol), and 5 mL NMP was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The resin (4) (1 g, Fmoc-outside (108.0 μmol, Boc-inside (0.243 mmol) was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DCM (5 mL × 3), three times each and then followed by Fmoc deprotection. This step was repeated once. A mixture of Alloc-OSu (10.74 μL, 54 μmol) and Fmoc-OSu (18.198 μL, 54 μmol), DIEA (627.56 μL, 216 μmol) and DMF (5mL) was added to resin beads and the reaction mixture was agitated for 30 minutes. The obtained outside-Fmoc and Alloc inside- Boc-trifunctional beads (5) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) each, followed by Fmoc deprotection. A mixture of Fmoc-disulfide linker (2) (77.11 mg, 162 μmol), HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), and 5 mL DMF was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The resin was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DCM (5 mL × 3), followed by Fmoc deprotection. A mixture of Fmoc-K (Dde)-OH (86.184 mg, 162 μmol) HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), and 5 mL DMF was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The resulting beads (6) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) each and dried in vacuum.
Synthesis of Oligoarginine-Compound on Outer Surface of TG Beads (8)
Beads (6) (1 g, Fmoc-outside (54 μmol), Alloc-outside (54 μmol) Boc-inside (0.243 mmol) were swollen in DMF overnight, followed by Fmoc deprotection. A mixture of Fmoc-Arg (Pbf)-OH (104.97 mg, 162 μmol) HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), and 5 mL DMF was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. This step was repeated 7 more times which was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DCM (5 mL × 3), followed by Fmoc deprotection. A mixture of (Boc)2O (35.31μL, 162 μmol), DIEA (54.48 μL, 324 μmol) was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The beads (8) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) each and dried in vacuum.
Synthesis of LLP2A on Outer Surface of Resin Beads 8
Beads (8) were swollen in DMF overnight. A mixture of Pd(PPh3)4 (15.59 mg, 0.25 equiv), PhSiH3 (134.31 μL, 20 equiv) and CH2Cl2 (1 mL) were added to each reaction column to remove Alloc protecting groups. After 30 min, the resin was washed with CH2Cl2 (3×10 mL) and DMF (3×10 mL). This step was repeated and then resin was swollen in DMF. A mixture of Fmoc-AcH-OH (58.80 mg, 162 μmol) HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), and 5 mL DMF was added to the resin beads. The reaction mixture was agitated until the chloranil test was negative. The obtained beads (9) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) followed by Fmoc deprotection and treated with a mixture of Fmoc-Aad(OtBu)-OH (71.11mg, 162 μmol) HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), in 5 mL DMF. The reaction mixture was agitated until the ninhydrin test was negative. Beads (10) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) followed by Fmoc deprotection and treated with a mixture of Fmoc-K (Alloc)-OH (71.11mg, 162 μmol) HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), in 5 mL DMF. The reaction mixture was agitated until the ninhydrin test was negative. The beads (11) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) followed by Fmoc deprotection and treated with a mixture of 2-{3-[(2-toluidinocarbonyl)amino]phenyl} acetic acid (46.0 mg, 162 μmol) HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), in 5 mL DMF. The reaction mixture was agitated until the ninhydrin test was negative. Beads (12) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3). A mixture of Pd(PPh3)4 (15.59 mg, 0.25 equiv), PhSiH3 (134.31 μL, 20 equiv) and CH2Cl2 (1 mL) were added to each reaction column to remove Alloc protecting groups. After 30 min, the resin was washed with CH2Cl2 (3×10 mL) and DMF (3×10 mL). This step was repeated and then resin was swollen in DMF and treated with a mixture of (E)-3-(3-pyridinyl)-2-propenoic acid (24.13mg, 162 μmol) HOBt (21.87 mg, 162 μmol), DIC (25.20 μL, 162 μmol), in 5 mL DMF. The reaction mixture was agitated until the ninhydrin test was negative. Beads (13) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) and dried in vacuo.
Attachment of FITC on Outer Surface of Resin Beads 13 and Side Chain Deprotection
Resin beads (13) were swollen in DMF overnight and treated with a 2 % NH2NH2/DMF for 15 minutes to remove Dde protecting group. This step was repeated twice to ensure complete removal of Dde group. A mixture of FITC (63.01 mg, 162 μmol), DIEA (56.48 μL, 324 μmol), and 5 mL NMP was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. Side-chain deprotection was achieved by repeated treatment with a solution of TFA/thioanisole/phenol/water/TIS (84:5:5:5:1, v/v/w/ v/v) for 12 hours. After neutralization with 10% DIEA/DMF twice, the resin (14) was washed sequentially with DMF (10 mL × 3), methanol (10 mL × 3), DCM (10 mL × 3), DMF (10 mL × 3), DMF/water (10 mL × 3), water (10 mL × 3), and finally PBS (10 mL × 3).
Oligoarginine-FITC Release
Bead cassettes were loaded as needed with oligoarginine–FITC beads (14) (Figure 2) as described previously. A suspension of Molt4 cells was added over the surface of the bead cassette and cells were settled into PDMS wells. Alternatively, cells were prebound to the oligoarginine–FITC beads and settled on the bottom of petri dish. 1% low melt agarose:PBS mixture was boiled, cooled to 37 °C, and poured over the top of the bead cassette and allowed to gel before adding media + 0.1% DTT over the top. Fluorescent micrographs were obtained 10 minutes post release (488nm/520nm ex/em).
Figure 2.

Synthesis of (Fmoc)2-Jeffamine-acid linker. (a) Succinic anhydride (1 eq.), Dioxane, 0 °C; (b) Fmoc-OSu, Dioxane/H2O, Na2CO3.
Synthesis of a OBOC Small Molecule Library
The library synthesis was carried out according to literature procedures.16 Bi-functionalized resin (3) (1 g, Fmoc-outside, Boc-inside was swollen in DMF overnight followed by Fmoc deprotection. A mixture of Fmoc2-Jeffamine linker (1) (80.02 mg, 81 μmol), HOBt (10.93 mg, 81 μmol), HBTU (30.69, 81 μmol) DIEA (28.246 μL, 162 μmol), and 5 mL NMP was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The resin (4) was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DCM (5 mL × 3), followed by Fmoc deprotection. This step was repeated once. A mixture of Fmoc-disulphide linker (2) (154.2 mg, 324 μmol), HOBt (43.74 mg, 324 μmol), DIC (50.4 μL, 324 μmol), and 5 mL DMF was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The resin (15) was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DCM (5 mL × 3), followed by Boc deprotection by shaking with TFA:DCM (1:1) for 30 min. The resin was washed with DCM (5 mL × 3) and DMF (5 mL × 6) and 2 % DIEA/DMF and DMF (5 mL × 6). A mixture of Fmoc-K (Dde)-OH (172.36 mg, 324 μmol) HOBt (43.74 mg, 324 μmol), DIC (50.4 μL, 324 μmol), and 5 mL DMF was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The beads (16) were washed with DCM (5 mL × 3), DMF (5 mL × 3), and MeOH (5 mL × 3), DMF (5 mL × 3), followed by Fmoc deprotection. The beads were split into 30 columns. Thirty Fmoc-amino acids (3 eq, each) preactivated by HOBt/DIC/ were added to the corresponding columns (i.e., one amino acid in each column. The coupling reaction was carried out at room temperature for 12 h. After the coupling was completed, all beads in 30 columns were combined, mixed and the Fmoc deprotected and the whole procedure repeated. The resulting beads (17) were washed thoroughly with DMF, MeOH, and DMF. After randomization of all 30 portions of beads, Fmoc group was removed and beads were thoroughly washed with DMF (10 mL × 3), methanol (10 mL × 3), and DCM (10 mL × 3), dried and then swollen in water for 48 h. The water was drained, and a solution of Fmoc-OSu (36.39 mg, 108 μmol) in 50 mL of DCM/Et2O mixture (55:45, v/v) was added to the resin, followed by addition of DIEA (37.64 μL, 216 μmol). The resulting mixture was shaken vigorously for 30 min. The resin was washed with DCM (5 mL × 3) and DMF (5 mL × 6). A solution of (Boc)2O (212.09 mg, 0.97 mmol) in 10 mL of DCM was then added to the beads, followed by the addition of DIEA (338.8 μL, 1.94 mmol). The mixture was shaken until the ninhydrin test was negative. The resulting outside-Fmoc inside-Boc bifunctional beads (18) were washed with DCM (5 mL × 3), DMF (5 mL × 3), DCM (5 mL × 3), and MeOH (5 mL × 3) each and dried in vacuum. Fmoc in the testing arm and Dde in the coding arm were removed in a single step by 2% hydrazine/DMF. The beads were divided into 25 portions, and 25 different carboxylic acids (3 equiv each) were preactivated by HOBt/DIC and added to the corresponding columns. This final acylation was carried out at room temperature for 12 h and repeated once. After combination of all 25 portions of beads, the beads were washed with DMF (10 mL × 3), methanol (10 mL × 3), and diethyl ether (10 mL × 3). The beads were then dried under vacuum for 1 h. Side-chain deprotection was achieved with a solution of TFA/phenol/ water/TIS (88:5:5:2, v/w/v/v) for 30 min. After neutralization with 10% DIEA/DMF twice, the resin (19) was washed sequentially with DMF (10 mL × 3), methanol (10 mL × 3), DCM (10 mL × 3), DMF (10 mL × 3), DMF/water (10 mL × 3), water (10 mL × 3), and finally PBS (10 mL × 3). The thirty Fmoc amino acids used as the first and second building blocks and twenty-five carboxylic acids for acylation (third building block) are available in the supporting material.
OBOC Releasable Screen Against Small Molecule Releasable Library
Jurkat cells were grown to approximately 1 to 5 × 105 cells/ml in 75 cm culture flask containing RPMI media. 20 ml cell suspension was pelleted and placed and chilled on ice. Using chilled pipette tips, 1 ml thawed (4 °C) BD Matrigel™ Matrix (BD Biosciences) was used to carefully resuspend cell pellet. Matrigel™ suspension was then added over chilled bead cassette containing releasable small molecule library (Figure 5). Suspension was spread across cassette by manually tilting as needed. “Seeded” cassettes were then placed at 37 °C for 30 min to solidify Matrigel™ layer. 5 ml cell media was added over the top and cells were grown to desired confluency. To initiate compound release, fresh media containing 0.1% DTT was added for 30 min 37 °C. Media + DTT was then aspirated and seeded cassette was placed, uncapped, inside a secondary larger Petri dish containing sterile deionized H2O to prevent drying out and incubated 2 hr 37 °C to allow adequate compound diffusion. Fresh media was then added to the cassette and incubated for 48 hr at 37 °C. Media was aspirated and fresh media containing 0.05% MTT reagent (terazolium bromide) was added. After 6 hr incubation at 37 °C, cassette was washed in sterile PBS and observed topologically for regions of decreased cell metabolism as colorimetrically indicated by inhibition of MTT reduction. Such regions that closely correlated with individual beads were used to select beads for micro sequencing and lead compound identification.
Synthesis of LTS-1-3 amide from Rink resin
Rink amide resin beads (0.2 g, 0.1 mmol) were swollen in DMF for 4 h followed by Fmoc deprotection with 20% piperidine in DMF for ten minutes (5 mL × 2). A mixture of Fmoc-amino acid (0.3 mmol), HOBt (0.37 mmol, 50 mg) and DIC (0.47 mmol, 60 μl) in 3 mL of DMF was added to the resin beads. Fmoc-D-HoCit (0.3 mmol, 125 mg), Fmoc-L-His(Boc) (0.3 mmol, 185 mg), or Fmoc-L-Thr(OtBu) (0.3 mmol, 105 mg) were used for LTS1-3, respectively. The reaction mixture was agitated until the ninhydrin test was negative. The resin was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DMF (5 mL × 3), followed by Fmoc deprotection. A mixture of Fmoc-L-Nal-2 (0.3 mmol, 125 mg) HOBt (0.37 mmol, 50 mg), DIC (0.47 mmol, 60 μl) in 3 mL of DMF was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The resin was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DMF (5 mL × 3), followed by Fmoc deprotection. A mixture of 3-hydroxy-2-quinoxaline carboxylic acid (0.3 mmol, 60 mg) HOBt (0.37 mmol, 50 mg), DIC (0.47 mmol, 60 μl) in 3 mL biotech grade DMF was added to the resin beads. The reaction mixture was agitated until the ninhydrin test was negative. The resin was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DCM (5 mL × 3) and dried down completely followed by side chain deprotection and compound cleavage from resin with TFA:TIS:H2O (95 : 2.5 : 2.5) (3 mL) for 4 hrs with agitation. The TFA/compound solution was drained from the resin and collected along with additional TFA wash (1 mL) of the bead resin. Compounds was dried down under N2 gas and precipitated with 4 mL ether followed by 3 subsequent ether washes.
Synthesis LTS-1-3 with SH from TG resin
The synthesis of LTS-1-3 with SH group is similar to LTS-1-3 except a disulfide linker is first coupled to the amino group of TG resin. In brief, TG resin beads (0.2 g, 0.056 mmol) were swollen in DMF for 2 h. A mixture of Fmoc-disulfide linker 2 (0.07 mmol, 33 mg), HOBt (0.25 mmol, 33 mg), DIC (0.43 mmol, 40 μl) in 3 mL of DMF was added to the resin beads. The reaction mixture was agitated for 12 hours until the ninhydrin test was negative. The resin was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DMF (5 mL × 3), followed by Fmoc deprotection and construction of LTS-1-3 as described above. The resin was washed with DMF (5 mL × 3), MeOH (5 mL × 3), and DCM (5 mL × 3) and dried down completely followed by side chain deprotection with TFA:TIS: H2O (95 : 2.5 : 2.5) for 4 hrs with agitation. The resin was washed with DMF (5 mL × 3), neutralized with 2% DIEA in DMF (5 mL × 2), washed with DMF (5 mL × 3) and MeOH (5 mL × 3) and stored in MeOH for subsequent cleavage of compound from bead resin. Resin (100 mg, 0.028 mmol) was transferred to 1 ml column and washed with H2O (1 mL × 5). 1 mL 1%DTT in H2O (0.01g, 0.065 mmol) was added to the resin for 30 minutes with agitation. Eluant was collected and 1 mL 2% DTT in H2O (0.02g, 0.13 mmol) was added to the resin for 30 minutes with agitation. Eluant was collected and resin was washed with MeOH (1 mL ×1), DCM (1 mL ×2), and DMF(1 mL ×1) collecting eluant each time for subsequent purification and analysis.
Cell Viability Assays
Cells were cultured in 96 well plates at desired concentration (∼5000 cells/well). A serial dilution of each compound in DMSO was constructed. 0.25 μl of each dilution was added to 100 μl media + cells resulting in a 2.5 μM to 200 μM final concentration range for each compound. Negative controls received 0.25 ul (0.25%) DMSO only. After 48 h treatment period, 10% Alamar Blue® cell viability indicator (AbD Serotec) was added to each well and the difference of cell viability relative to untreated cells (D or percent difference) was measured for each compound according to suggested manufacturer protocol using Eq.1, wherein A1= absorbance of test wells at 570nm, A2= absorbance of test wells at 600nm, O1= molar extinction coefficient (E) of oxidized alamarBlue® at 570nm (80,586), O2 = E of oxidized alamarBlue® at 600nm (117,216), P1= absorbance of positive control wells (cells plus alamarBlue® and 0.25% DMSO but no test compound) at 570nm, and P2 = absorbance of positive control wells at 600nm.
| Eq. 1 |
Supplementary Material
Acknowledgments
This work was supported by NCI U19 CA113298 and the J. William Fulbright Scholarship Program. Special thanks to David Dyer, Frank Yaghmaie, and Rick Perron at the UC Davis Northern California Nanotechnology Center for their advice and support and to Mary Saunders for help with synthesis of disulfide cleavable linker.
Footnotes
Supporting Information Available. Fig. S1. Thirty Fmoc-amino acids used as building blocks for encoded small molecule library, Fig S2. Chemical structure of 25 acylating reagents, Figure S3. HPLC Chromatogram of Fmoc-Jeffamine Linker [(Fmoc)2-JA-OH], Figure S4. MALDI-TOF-MS spectra of Jeffamine Linker (Fmoc)2-JA-OH. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354(6348):82–4. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lam KS, Lebl M, Krchnak V, Lake DF, Smith J, Wade S, Ferguson R, Ackerman-Berrier, Wertman K. Application of Selectide Technology in Identifying (i) a Mimotope for a Discontinuous Epitope, and (ii) D-amino Acid Ligands. In: Hodges RS, editor. Peptides: Chemistry, Structure and Biology, (Proc 12th Amer Peptide Symp) 1994. pp. 1003–1004. [Google Scholar]; (b) Lam KS, Lebl M, Wade S, Stierandova A, Khattri PS, Collins N, Hruby VJ. Streptavidin-peptide interaction as a model system for molecular recognition. Pept : Chem, Struct Biol, Proc Am Pept Symp 13th. 1994:1005–1006. [Google Scholar]
- 3.(a) Aina OH, Marik J, Liu R, Lau DH, Lam KS. Identification of novel targeting peptides for human ovarian cancer cells using “one-bead one-compound” combinatorial libraries. Mol Cancer Ther. 2005;4:806–813. doi: 10.1158/1535-7163.MCT-05-0029. [DOI] [PubMed] [Google Scholar]; (b) Aina OH, Sroka TC, Chen M, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66:184–199. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]; (c) Benito JM, Meldal M. Bicyclic organo-peptides as selective carbohydrate receptors: design, solid-phase synthesis, and on-bead binding capability. QSAR Comb Sci. 2004;23:117–129. [Google Scholar]; (d) Lau DH, Guo L, Liu R, Song A, Shao C, Lam KS. Identifying peptide ligands for cell surface receptors using cell-growth-on-bead assay and one-bead one-compound combinatorial library. Biotechnol Lett. 2002;24:497–500. [Google Scholar]; (e) McBride JD, Freeman HNM, Leatherbarrow RJ. Selection of human elastase inhibitors from a conformationally constrained combinatorial peptide library. Eur J Biochem. 1999;266:403–412. doi: 10.1046/j.1432-1327.1999.00867.x. [DOI] [PubMed] [Google Scholar]; (f) McBride JD, Freeman N, Domingo GJ, Leatherbarrow RJ. Selection of chymotrypsin inhibitors from a conformationally-constrained combinatorial peptide library. J Mol Biol. 1996;259:819–827. doi: 10.1006/jmbi.1996.0360. [DOI] [PubMed] [Google Scholar]; (g) Mikawa M, Wang H, Guo L. Novel peptide ligands for integrin a4b1 overexpressed in cancer cells. Mol Cancer Ther. 2004;3:1329–1334. [PubMed] [Google Scholar]; (h) Wittmann V, Seeberger S. Combinatorial solid-phase synthesis of multivalent cyclic neoglycopeptides. Angew Chem, Int Ed. 2000;39:4348–4352. doi: 10.1002/1521-3773(20001201)39:23<4348::AID-ANIE4348>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 4.(a) Liu B, Alluri PG, Yu P, Kodadek T. A Potent Transactivation Domain Mimic with Activity in Living Cells. J Am Chem Soc. 2005;127:8254–8255. doi: 10.1021/ja0515295. [DOI] [PubMed] [Google Scholar]; (b) Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. Isolation of protein ligands from large peptoid libraries. J Am Chem Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]; (c) Reddy MM, Bachhawat-Sikder K, Kodadek T. Transformation of Low-Affinity Lead Compounds into High-Affinity Protein Capture Agents. Chem Biol. 2004;11:1127–1137. doi: 10.1016/j.chembiol.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 5.(a) Appell KC, Chung TDY, Ohlmeyer MJH, Sigal NH, Baldwin JJ, Chelsky D. Biological screening of a large combinatorial library. J Biomol Screening. 1996;1:27–31. [Google Scholar]; (b) Baldwin JJ, Burbaum JJ, Henderson I, Ohlmeyer MHJ. Synthesis of a Small Molecule Combinatorial Library Encoded with Molecular Tags. J Am Chem Soc. 1995;117:5588–5589. [Google Scholar]; (c) Lam KS, Liu R, Miyamoto S, Lehman AL, Tuscano JM. Applications of one-bead one-compound combinatorial libraries and chemical microarrays in signal transduction research. Acc Chem Res. 2003;36:370–377. doi: 10.1021/ar0201299. [DOI] [PubMed] [Google Scholar]; (d) Lo MMC, Neumann CS, Nagayama S, Perlstein EO, Schreiber SL. A Library of Spirooxindoles Based on a Stereoselective Three-Component Coupling Reaction. J Am Chem Soc. 2004;126:16077–16086. doi: 10.1021/ja045089d. [DOI] [PubMed] [Google Scholar]; (e) Stavenger RA, Schreiber SL. Asymmetric catalysis in diversity-oriented organic synthesis: Enantioselective synthesis of 4320 encoded and spatially segregated dihydropyrancarboxamides. Angew Chem, Int Ed. 2001;40:3417–3421. doi: 10.1002/1521-3773(20010917)40:18<3417::aid-anie3417>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 6.(a) Al-Obeidi FA, Lam KS. Development of inhibitors for protein tyrosine kinases. Oncogene. 2000;19(49):5690–701. doi: 10.1038/sj.onc.1203926. [DOI] [PubMed] [Google Scholar]; (b) Kamath JR, Liu R, Enstrom AM, Lou Q, Lam KS. Development and characterization of potent and specific peptide inhibitors of p60c-src protein tyrosine kinase using pseudosubstrate-based inhibitor design approach. J Pept Res. 2003;62(6):260–8. doi: 10.1046/j.1399-3011.2003.00094.x. [DOI] [PubMed] [Google Scholar]; (c) Ramdas L, Bunnin BA, Plunkett MJ, Sun G, Ellman J, Gallick G, Budde RJ. Benzodiazepine compounds as inhibitors of the src protein tyrosine kinase: screening of a combinatorial library of 1,4-benzodiazepines. Arch Biochem Biophys. 1999;368(2):394–400. doi: 10.1006/abbi.1999.1313. [DOI] [PubMed] [Google Scholar]; (d) Lam KS, Lebl M, Krchnak V. The “One-Bead-One-Compound” Combinatorial Library Method. Chem Rev. 1997;97(2):411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 7.(a) Meldal M. Multiple column synthesis of quenched solid-phase bound fluorogenic substrates for characterization of endoprotease specificity. Methods (San Diego, CA, U S) 1994;6:417–424. [Google Scholar]; (b) Meldal M, Svendsen I. Direct visualization of enzyme inhibitors using a portion mixing inhibitor library containing a quenched fluorogenic peptide substrate. Part 1. Inhibitors for subtilisin Carlsberg. J Chem Soc, Perkin Trans. 1995:1591–1596. [Google Scholar]; (c) Meldal M, Svendsen I, Breddam K, Auzanneau FI. Portion-mixing peptide libraries of quenched fluorogenic substrates for complete subsite mapping of endoprotease specificity. Proc Natl Acad Sci USA. 1994;9:3314–3318. doi: 10.1073/pnas.91.8.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Abato P, Conroy JL, Seto CT. Combinatorial library of serine and cysteine protease inhibitors that interact with both the S and S′ binding sites. J Med Chem. 1999;42(19):4001–9. doi: 10.1021/jm990272g. [DOI] [PubMed] [Google Scholar]; (e) Kick EK, Roe DC, Skillman AG, Liu G, Ewing TJ, Sun Y, Kuntz ID, Ellman JA. Structure-based design and combinatorial chemistry yield low nanomolar inhibitors of cathepsin D. Chem Biol. 1997;4(4):297–307. doi: 10.1016/s1074-5521(97)90073-9. [DOI] [PubMed] [Google Scholar]
- 8.Chirayil S, Chirayil R, Luebke KJ. Discovering ligands for a microRNA precursor with peptoid microarrays. Nucleic Acids Res. 2009;37(16):5486–97. doi: 10.1093/nar/gkp549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng L, Liu R, Marik J, Wang X, Takada Y, Lam KS. Combinatorial chemistry identifies high-affinity peptidomimetics against alpha4beta1 integrin for in vivo tumor imaging. Nat Chem Biol. 2006;2(7):381–9. doi: 10.1038/nchembio798. [DOI] [PubMed] [Google Scholar]
- 10.Lam KS, Lou Q, Zhao ZG, Smith J, Chen ML, Pleshko E, Salmon SE. Idiotype specific peptides bind to the surface immunoglobulins of two murine B-cell lymphoma lines, inducing signal transduction. Biomed Pept Proteins Nucleic Acids. 1995;1(3):205–10. [PubMed] [Google Scholar]
- 11.Maverakis E, Menezes JS, Ametani A, Han M, Stevens DB, He Y, Wang Y, Ono Y, Miyamura Y, Lam KS, Ward ES, Sercarz EE. Molecular mimics can induce a nonautoaggressive repertoire that preempts induction of autoimmunity. Proc Natl Acad Sci U S A. 2010;107(6):2550–5. doi: 10.1073/pnas.0914508107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu R, Enstrom AM, Lam KS. Combinatorial peptide library methods for immunobiology research. Exp Hematol. 2003;31(1):11–30. doi: 10.1016/s0301-472x(02)01008-1. [DOI] [PubMed] [Google Scholar]
- 13.(a) Righetti PG, Boschetti E, Lomas L, Citterio A. Protein Equalizer Technology : the quest for a “democratic proteome”. Proteomics. 2006;6(14):3980–92. doi: 10.1002/pmic.200500904. [DOI] [PubMed] [Google Scholar]; (b) Guerrier L, Thulasiraman V, Castagna A, Fortis F, Lin S, Lomas L, Righetti PG, Boschetti E. Reducing protein concentration range of biological samples using solid-phase ligand libraries. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;833(1):33–40. doi: 10.1016/j.jchromb.2005.12.048. [DOI] [PubMed] [Google Scholar]
- 14.Gregori L, Gurgel PV, Lathrop JT, Edwardson P, Lambert BC, Carbonell RG, Burton SJ, Hammond DJ, Rohwer RG. Reduction in infectivity of endogenous transmissible spongiform encephalopathies present in blood by adsorption to selective affinity resins. Lancet. 2006;368(9554):2226–30. doi: 10.1016/S0140-6736(06)69897-8. [DOI] [PubMed] [Google Scholar]
- 15.(a) Tozzi C, Anfossi L, Baggiani C, Giovannoli C, Giraudi G. A combinatorial approach to obtain affinity media with binding properties towards the aflatoxins. Anal Bioanal Chem. 2003;375:994–999. doi: 10.1007/s00216-003-1754-z. [DOI] [PubMed] [Google Scholar]; (b) Amatschek K, Necina R, Hahn R. Affinity chromatography of human blood coagulation factor VIII on monoliths with peptides from a combinatorial library. J High Resol Chromatogr. 2000;23:47–58. [Google Scholar]; (c) Kaufman DB, Hentsch ME, Baumbach GA. Affinity purification of fibrinogen using a ligand from a peptide library. Biotechnol Bioeng. 2002;77:278–289. doi: 10.1002/bit.10120. [DOI] [PubMed] [Google Scholar]
- 16.(a) Liu R, Marik J, Lam KS. A Novel Peptide-based Encoding System for “One-bead One-compound” Peptidomimetic and Small Molecule Combinatorial Libraries. J Am Chem Soc. 2002;124:7678–7680. doi: 10.1021/ja026421t. [DOI] [PubMed] [Google Scholar]; (b) Liu R, Wang X, Song A, Bao T, Lam KS. Development and Applications of Topologically Segregated Bilayer Beads in One-bead One-compound Combinatorial Libraries. QSAR Comb Sci. 2005;24(10):1127–1140. doi: 10.1111/j.1399-3011.2005.00192.x. [DOI] [PubMed] [Google Scholar]
- 17.(a) Salmon SE, Lam KS, Lebl M, Kandola A, Khattri PS, Wade S, Patek M, Kocis P, Krchnak V, Thorpe D, et al. Discovery of biologically active peptides in random libraries: solution-phase testing after staged orthogonal release from resin beads. Proc Natl Acad Sci U S A. 1993;90(24):11708–12. doi: 10.1073/pnas.90.24.11708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lebl M, Patek M, Kocis P, Krchnak V, Hruby VJ, Salmon SE, Lam KS. Multiple release of equimolar amounts of peptides from a polymeric carrier using orthogonal linkage-cleavage chemistry. Int J Pept Protein Res. 1993;41(2):201–3. doi: 10.1111/j.1399-3011.1993.tb00132.x. [DOI] [PubMed] [Google Scholar]; (c) Baek HG, Liu R, Lam KS. Development of hydrogel TentaGel shell-core beads for ultrahigh throughput solution-phase screening of encoded OBOC combinatorial small molecule libraries. J Comb Chem. 2009;11(1):91–102. doi: 10.1021/cc800092y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Evans B, Pipe A, Clark L, Banks M. Identification of a potent and selective oxytocin antagonist, from screening a fully encoded differential release combinatorial chemical library. Bioorg Med Chem Lett. 2001;11(10):1297–300. doi: 10.1016/s0960-894x(01)00201-3. [DOI] [PubMed] [Google Scholar]; (e) Hiemstra HS, Benckhuijsen WE, Amons R, Rapp W, Drijfhout JW. A new hybrid resin for stepwise screening of peptide libraries combined with single bead Edman sequencing. J Pept Sci. 1998;4(4):282–8. doi: 10.1002/(sici)1099-1387(199806)4:4<282::aid-psc145>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 18.(a) Silen JL, Lu AT, Solas DW, Gore MA, MacLean D, Shah NH, Coffin JM, Bhinderwala NS, Wang Y, Tsutsui KT, Look GC, Campbell DA, Hale RL, Navre M, DeLuca-Flaherty CR. Screening for novel antimicrobials from encoded combinatorial libraries by using a two-dimensional agar format. Antimicrob Agents Chemother. 1998;42(6):1447–53. doi: 10.1128/aac.42.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jayawickreme CK, Sauls H, Bolio N, Ruan J, Moyer M, Burkhart W, Marron B, Rimele T, Shaffer J. Use of a cell-based, lawn format assay to rapidly screen a 442,368 bead-based peptide library. J Pharmacol Toxicol Methods. 1999;42(4):189–97. doi: 10.1016/s1056-8719(00)00083-6. [DOI] [PubMed] [Google Scholar]; (c) Quillan JM, Jayawickreme CK, Lerner MR. Combinatorial diffusion assay used to identify topically active melanocyte-stimulating hormone receptor antagonists. Proc Natl Acad Sci U S A. 1995;92(7):2894–8. doi: 10.1073/pnas.92.7.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Iuga AO, Reddy VB, Lerner EA. Identification of novel hexapeptide agonists at the Xenopus laevis melanophore melanocortin receptor. Peptides. 2005;26(11):2124–8. doi: 10.1016/j.peptides.2005.03.001. [DOI] [PubMed] [Google Scholar]; (e) Salmon SE, Liu-Stevens RH, Zhao Y, Lebl M, Krchnak V, Wertman K, Sepetov N, Lam KS. High-volume cellular screening for anticancer agents with combinatorial chemical libraries: a new methodology. Mol Divers. 1996;2(1-2):57–63. doi: 10.1007/BF01718701. [DOI] [PubMed] [Google Scholar]
- 19.(a) Rissler K. Separation of acetylated polypropylene glycol di- and triamines by gradient reversed-phase high-performance liquid chromatography and evaporative light scattering detection. Journal of Chromatography A. 1994;667(1-2):167–174. [Google Scholar]; (b) Rissler K. High-performance liquid chromatography and detection of polyethers and their mono(carboxy)alkyl and -arylalkyl substituted derivatives. Journal of Chromatography A. 1996;742(1-2):1–54. [Google Scholar]
- 20.Kim H, Cho JK, Chung WJ, Lee YS. Core-shell-type resins for solid-phase peptide synthesis: comparison with gel-type resins in solid-phase photolytic cleavage reaction. Org Lett. 2004;6(19):3273–6. doi: 10.1021/ol048815q. [DOI] [PubMed] [Google Scholar]
- 21.Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, Li ST, Kobayashi N, Matsumoto S, Tanaka K, Tanaka N, Matsui H. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat Med. 2004;10(3):305–9. doi: 10.1038/nm994. [DOI] [PubMed] [Google Scholar]
- 22.(a) Garaud M, Pei D. Substrate profiling of protein tyrosine phosphatase PTP1B by screening a combinatorial peptide library. J Am Chem Soc. 2007;129(17):5366–7. doi: 10.1021/ja071275i. [DOI] [PubMed] [Google Scholar]; (b) Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J Am Chem Soc. 2006;128(39):13000–9. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.