

Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) are linked to familial as well as sporadic forms of Parkinson's disease (PD), a neurodegenerative disease characterized by dysfunction and degeneration of dopaminergic and other types of neurons. The molecular and cellular mechanisms underlying LRRK2 action remain poorly defined. Here, we show that LRRK2 controls synaptic morphogenesis at the Drosophila neuromuscular junction. Loss of Drosophila LRRK2 results in synaptic overgrowth, whereas overexpression of Drosophila LRRK or human LRRK2 has opposite effects. Alteration of LRRK2 activity also affects neurotransmission. LRRK2 exerts its effects on synaptic morphology by interacting with distinct downstream effectors at the presynaptic and postsynaptic compartments. At the postsynapse, LRRK2 interacts with the previously characterized substrate 4E-BP, an inhibitor of protein synthesis. At the presynapse, LRRK2 phosphorylates and negatively regulates the microtubule (MT)-binding protein Futsch. These results implicate synaptic dysfunction caused by deregulated protein synthesis and aberrant MT dynamics in LRRK2 pathogenesis and offer a new paradigm for understanding and ultimately treating PD.
Introduction
Parkinson's disease (PD) is one of the most common neurodegenerative diseases and is characterized by locomotor abnormalities as a result of the dysfunction and eventual loss of dopaminergic (DA) neurons (Dawson and Dawson, 2003). Most PD cases are sporadic with no known cause. Recent advances in PD genetics have led to the identification of familial PD (FPD) genes. It is anticipated that understanding the disease mechanisms of the FPD cases will provide insights into PD pathogenesis in general. Despite intensive studies of the FPD gene products at the biochemical and cell biological levels, our understanding of their physiological function and the molecular and cellular pathways underlying disease pathogenesis is still fragmentary. Of all FPD genes, leucine-rich repeat kinase 2 (LRRK2) is the most frequently mutated. LRRK2 encodes a large serine/threonine kinase with multiple other domains (Paisán-Ruíz et al., 2004; Zimprich et al., 2004). Some pathogenic mutations in LRRK2, such as the I2020T and G2019S substitutions in the kinase domain and R1441C substitution in the ROC domain, appear to augment kinase activity (West et al., 2005; Gloeckner et al., 2006). In Drosophila and mouse models, pathogenic human (hLRRK2) or Drosophila (dLRRK) LRRK2 induce parkinsonian phenotypes in an age-dependent manner (Imai et al., 2008; Liu et al., 2008; Li et al., 2009). A number of LRRK2-interacting proteins and substrates have been identified through in vitro studies (Jaleel et al., 2007; Imai et al., 2008; Shin et al., 2008; Gillardon, 2009a,b), which implicate diverse biological functions for LRRK2 in translational control, vesicular trafficking, and cytoskeletal regulation. The physiological relevance of these interacting proteins and substrates remain to be established.
Actin and microtubule (MT) cytoskeleton dynamics play a crucial role in the formation of the nervous system, regulating such fundamental processes as axonal guidance and synaptogenesis. Dynamic modulation of synaptic structure and function is fundamental to neural network formation during development and is the molecular basis of learning and memory. Synaptic dysfunction is tightly linked to the pathogenesis of major neurodegenerative diseases such as Alzheimer's disease (Selkoe, 2002), and its role in PD is beginning to be appreciated (Calabresi et al., 2007). In Drosophila, the MT-associated protein 1B (MAP1B) homolog Futsch is required for axonal and dendritic growth during embryogenesis and for synaptic morphogenesis during larval neuromuscular junction (NMJ) development (Hummel et al., 2000; Roos et al., 2000). In this study, we show that dLRRK phosphorylates and negatively regulates Futsch function at the presynapse. We also show that the previously characterized dLRRK substrate 4E-BP functionally interacts with LRRK2 at the postsynapse. Our results implicate defects in presynaptic MT cytoskeleton dynamics and postsynaptic protein synthesis in LRRK2 pathogenesis.
Materials and Methods
Fly strains.
The dLRRKe03680, thor1, UAS–dLRRK–WT, UAS–dLRRK–I1915T, UAS–dLRRK–3KD, UAS–d4E-BP–WT, UAS–d4E-BP–T37/46A, UAS–PAR-1, and UAS–mito–GFP were described previously (Zhang et al., 2007; Imai et al., 2008; Yang et al., 2008). UAS–Fos–DN was provided by Dr. Aaron DiAntonio (Washington University, St. Louis, MO) (Collins et al., 2006), and UAS–hLRRK2–WT–Flag and UAS–hLRRK–G2019S–Flag were provided by Dr. Wanli Smith (Johns Hopkins University, Baltimore, MD) (Liu et al., 2008). Myosin heavy chain–GAL4 (Mhc–GAL4) driver was provided by Dr. Troy Littleton (Massachusetts Institute of Technology, Boston, MA). The dLRRKdf, EP1419, futschN94, futschK68, elav–GAL4, and Da–GAL4 lines were obtained from Bloomington Drosophila Stock Center. For pharmacological approach, either rapamycin or DMSO was added to fly food at 0.5 μm concentration, and larvae were raised on drug-containing food at 25°C until dissection at the third-instar stage.
Immunohistochemistry.
For immunohistochemistry, third-instar larvae were selected, dissected in PBS, and fixed in 4% formaldehyde in PBS for ∼15 min. Aged flies were anesthetized with CO2, with wings and legs removed, dissected in PBS, and fixed in 4% formaldehyde in PBS for 1 h. The primary antibodies used were as follows: anti-cysteine string protein (CSP) (1:20; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), anti-Dlg (4F3) (1:50; Developmental Studies Hybridoma Bank), anti-Futsch (22C10) (1:50; Developmental Studies Hybridoma Bank); anti-tyrosinated Tubulin (tyr–Tub) (1:40, MAB1864; Millipore Bioscience Research Reagents), and anti-green fluorescent protein (GFP) (1:1000; Abcam). Primary antibody incubation was performed at 4°C overnight. All secondary antibodies (Invitrogen) and FITC-conjugated anti-HRP (Jackson ImmunoResearch) were used at 1:200 and incubated for ∼1.5–2 h at room temperature. Laval preparations were mounted in Vectashield (Vector Laboratories). Confocal images were collected from a Carl Zeiss LSM Meta 510 microscope with 40× or 100× inverted NX oil lens. Carl Zeiss LSM Meta 510 software was used to capture, process, and analyze images. For analysis of the neuromuscular junction, wandering third-instar larvae were selected from the culture medium, dissected, and immunostained as described above. To determine the total number of boutons, dissected body-wall muscle samples were stained with anti-HRP. Type Ib boutons were determined based on Dlg staining or bouton size (normally type Ib boutons are between 3 and 6 μm in diameter). Bouton numbers were quantified by analyzing confocal sections of whole-mount stained muscle samples by focusing on muscle 6/7 of abdominal segment 3 (A3 segment) and then normalizing counted bouton numbers by muscle size for all the genotypes. Muscle surface was visualized by phalloidin staining, and surface area was measured using NIH Image J software. NMJ lengths were also normalized with surface area of muscles 6 and 7 in A3 segment. The normalized bouton number or NMJ length in control genotypes in each experiment was set at 100%, and all the other genotypes were standardized relative to the control genotype. All crosses for genetic interaction were performed at 25°C.
Immunoprecipitation.
For coimmunoprecipitation (co-IP) experiments, 50 third-instar larvae expressing UAS–hLRRK2–Flag transgene driven by Da–GAL4 or elav–GAL4 were dissected, homogenized in IP buffer (50 mm Tris-HCl pH 8.0, 1% Triton X-100, 150 mm NaCl, 2 mm Na3VO4, 10 mm NaF, 10% glycerol, protease inhibitors), and then centrifuged at 12,000 × g for 25 min at 4°C. Supernatants were precleared by incubation with protein G agarose (Pierce) for 1 h at 4°C. The samples were incubated with anti-FLAG M2 agarose affinity resin (Sigma), or, in the case of 22C10 immunoprecipitation (IP), samples were first incubated with 22C10 antibody overnight at 4°C and then incubated with protein G agarose for 4 h at 4°C. Beads were washed six times with IP buffer and boiled in SDS sample buffer, or, in the case of Flag IP, beads were eluted with 3XFlag peptide and the elute was mixed with an equal volume of 2× SDS sample buffer. Samples were subjected to gel electrophoresis and Western blotting.
In vitro phosphorylation assay.
The Futsch substrate was affinity purified from adult fly heads by IP with monoclonal antibody (mAb) 22C10. The hLRRK2 proteins used as kinase sources were affinity purified from HEK293 cells transfected with the corresponding hLRRK2 constructs. The in vitro kinase assay was performed essentially as described (Imai et al., 2008).
Electrophysiology.
Electrophysiological recordings of spontaneous miniature excitatory junctional currents (mEJCs) and evoked EJCs were performed according to previously described protocols (Zhang et al., 2007). Quantal content was calculated as mean EJC amplitude divided by mean mEJC amplitude.
Results
dLRRK is required for normal synaptic morphogenesis at the Drosophila NMJ
To probe the expression pattern of dLRRK in Drosophila, we performed immunohistochemical analysis using an anti-dLRRK antibody, the specificity of which has been characterized previously (Imai et al., 2008). In wild-type (WT) third-instar larvae, dLRRK is present ubiquitously, with its immunosignals observed in both the cytoplasm of body-wall muscles and the presynaptic and postsynaptic compartment of the NMJ (supplemental Fig. S1A–F, available at www.jneurosci.org as supplemental material). These signals are specific to dLRRK, because they were dramatically reduced in the NMJ and muscle cytoplasm of dLRRKe03680 homozygous mutant (Imai et al., 2008; Wang et al., 2008) (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material).
To assess the physiological function of dLRRK at the NMJ, we analyzed NMJ nerve terminal morphology in dLRRK mutants. To exclude possible contribution of background mutations on NMJ morphogenesis, a transheterozygote between dLRRKe03680 and a chromosomal deficiency that uncovers dLRRK (dLRRKe03680/dLRRKDf) was used. We focused our analysis on the morphology of NMJs located on muscle 6/7 of A3. To exclude possible effects of muscle size on bouton number, we normalized bouton number with muscle size. dLRRK mutants displayed NMJ overgrowth and irregularly shaped boutons (Fig. 1D). Quantification of the total number of type I boutons showed that dLRRK mutant had a 41% increase compared with control (Fig. 1A). We also separately counted type Ib (big) and type Is (small) boutons. Type Ib bouton number was not significantly changed in dLRRK mutants, whereas type Is or irregularly shaped small-sized boutons were significantly increased (Fig. 1A). No significant change in muscle size was observed in dLRRK mutant. Average bouton size in dLRRK mutant was noticeably smaller (control, 7.9 ± 1.0 μm2, n = 16; dLRRKe03680/dLRRKDf, 4.2 ± 1.0 μm2, n = 13; p < 0.0001). Furthermore, NMJ length and branch number in dLRRK mutant after normalization with muscle size were increased by 56 and 38%, respectively (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). dLRRKe03680 homozygous mutants showed similar phenotypes, although the bouton number increase was slightly less than that seen in dLRRKe03680/dLRRKDf transheterozygotes (Fig. 1A).
Figure 1.
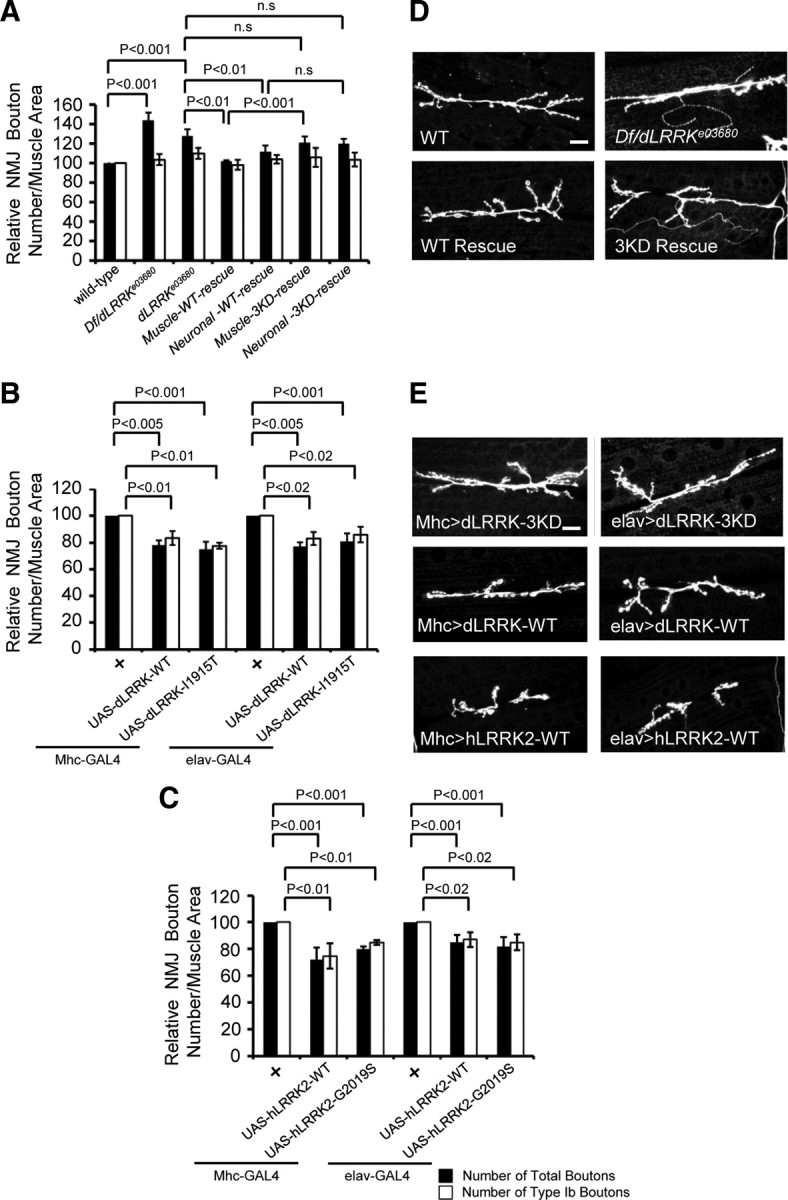
Effects of LOF and GOF of dLRRK on synaptic differentiation. A, Quantification of the total number of boutons (black bar) and the number of type 1b boutons (white bar) per muscle area on muscle 6/7 of A3 in dLRRK mutant and rescued animals: WT, n = 19; dLRRKe03680, n = 54; dLRRKDf/dLRRKe03680, n = 33; muscle WT-rescue (UAS–dLRRK–WT/+; Mhc–GAL4, dLRRKe03680/dLRRKe03680), n = 30; neuronal WT-rescue (elav–GAL4/+; UAS–dLRRK–WT/+; dLRRKe03680/dLRRKe03680), n = 26; muscle 3KD-rescue (Mhc–GAL4, dLRRKe03680/dLRRKe03680, UAS–dLRRK–3KD), n = 23; neuronal 3KD-rescue (elav–GAL4/+; dLRRKe03680/dLRRKe03680, UAS–dLRRK–3KD), n = 20). Data represent values relative to control. Note that the rescuing effect of postsynaptic dLRRK is significantly different from dLRRK–3KD when directly compared. B, C, Quantification of the total number of boutons (black bar) and the number of type 1b boutons (white bar) on muscle 6/7 of A3, after normalization with muscle area, in dLRRK overexpression animals of the indicated genotypes: Mhc–GAL4/+, n = 23; Mhc–GAL4>UAS–dLRRK–WT/UAS–dLRRK–WT, n = 36; Mhc–GAL4>UAS–dLRRK–I1915T/UAS–dLRRK-I1915T, n = 35; elav–GAL4/+, n = 31; elav–GAL4>UAS–dLRRK–WT/UAS–dLRRK–WT, n = 30; elav–GAL4>UAS–dLRRK–I1915T/UAS–dLRRK-I1915T, n = 40 (B), Mhc–GAL4/+, n = 25; Mhc–GAL4>hLRRK–WT, n = 23; Mhc–GAL4>hLRRK–G2019S, n = 23; elav–GAL4/+, n = 24; elav–GAL4>hLRRK–WT, n = 23; elav–GAL4>hLRRK–G2019S, n = 21 (C). Data represent values relative to control. D, E, HRP immunostaining showing the effects of dLRRK LOF and dLRRK or hLRRK2 GOF on synaptic morphology. NMJs of wild-type, dLRRK mutant, and dLRRK mutant with neuronal or muscle rescue (D) or animals overexpressing dLRRK–WT, dLRRK–3KD, or hLRRK2 specifically in muscle or neurons (E) are shown. Genotypes shown are as follows: dLRRK–3KD muscle overexpression (genotype, UAS-dLRRK–3KD; Mhc–GAL4), dLRRK–WT muscle overexpression (genotype, UAS–dLRRK–WT/+; UAS–dLRRK–WT/Mhc–GAL4), hLRRK2 muscle overexpression (genotype, Mhc–GAL4/UAS–hLRRK2–WT–Flag), dLRRK–3KD neuronal overexpression (genotype, elav–GAL4; UAS–dLRRK–3KD), dLRRK–WT neuronal overexpression (genotype, elav–GAL4/+; UAS–dLRRK–WT/+; UAS–dLRRK–WT/+), and hLRRK2 neuronal overexpression (genotype, elav–GAL4/+; UAS–hLRRK2–WT–Flag/+). Scale bars, 20 μm.
Given that dLRRK protein is ubiquitously present at the NMJ at both the presynaptic and postsynaptic compartments, we next asked whether it is required for normal synaptic growth by acting in the presynaptic neurons or postsynaptic muscle cells or both. For this purpose, we examined bouton number and size in dLRRKe03680 mutants that express a UAS–wild-type dLRRK (dLRRK–WT) transgene driven by the postsynaptic Mhc–GAL4 or presynaptic elav–GAL4 driver. As a control, we used an upstream activated sequence (UAS) transgene expressing a kinase-dead form of dLRRK (dLRRK–3KD) (Greggio et al., 2006; Imai et al., 2008). Muscle expression of UAS–dLRRK–WT essentially rescued the synaptic morphology phenotype of dLRRKe03680 mutant, and dLRRK–WT rescue was significantly better than dLRRK–3KD rescue (Fig. 1A,D). Neuronal expression of UAS–dLRRK–WT showed a partial but still significant rescue of synaptic morphology in dLRRKe03680 mutant (Fig. 1A). Average bouton size was also restored by either muscle or neuronal expression of dLRRK–WT (dLRRKe03680/dLRRKe03680, 4.7 ± 1.2 μm2, n = 10; dLRRK–WT muscle rescue, 7.3 ± 0.8 μm2, n = 15; dLRRK–WT neuronal rescue, 7.6 ± 1.1 μm2, n = 11; p < 0.0001). In contrast, muscle or neuronal expression of dLRRK–3KD failed to rescue under similar conditions (Fig. 1A,D). These results demonstrate that dLRRK is functioning both presynaptically and postsynaptically to control synaptic morphogenesis at the NMJ, with postsynaptic dLRRK playing a seemingly more prominent role than presynaptic dLRRK. Furthermore, the kinase activity of dLRRK appears to be essential for normal NMJ development.
Having observed dLRRK loss-of-function (LOF) effect on NMJ differentiation, we next examined its gain-of-function (GOF) effect. Either presynaptic or postsynaptic overexpression of dLRRK–WT resulted in an estimated 20% decrease in the total number of type I boutons and NMJ branch number on muscle 6/7 of A3 (Fig. 1B,E) (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material), with no observable effect on muscle size. NMJ length was normal (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). The type I bouton number in animals either presynaptically or postsynaptically overexpressing dLRRK–3KD was not significantly different from that in nontransgenic control animals (Fig. 1E). Overexpression of the pathogenic dLRRK–I1915T resulted in similar NMJ length, branch number, and bouton loss phenotypes as dLRRK–WT overexpression (Fig. 1B) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). We next overexpressed the WT and pathogenic G2019S mutant forms of human LRRK2. Overexpression of hLRRK2–WT or hLRRK2–G2019S showed a similar degree (∼20%) of reduction of the total number of type I boutons, on muscle 6/7 from A3 (Fig. 1C,E). In addition, NMJ length and branch number were decreased by 25 and 30%, respectively (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Separate quantification of type Ib versus type Is boutons on muscle 6/7 revealed a similar degree (∼20%) of decrease in the number of each type of boutons in dLRRK–WT, dLRRK–I1915T, hLRRK2–WT, or hLRRK–G2019S transgenic animals (Fig. 1B,C). It is worth noting that, although dLRRK GOF has no effect on NMJ length, hLRRK2 GOF leads to a reduction of NMJ length. This is consistent with previous findings that the hLRRK2 proteins tend to induce stronger phenotypes than the dLRRK proteins (Imai et al., 2008; Liu et al., 2008).
To determine whether LRRK2 affects normal synaptic assembly, we examined the NMJs of dLRRK mutant and dLRRK overexpression animals with various presynaptic and postsynaptic markers. No significant change in the localization or expression levels of the presynaptic vesicle marker CSP or postsynaptic scaffolding protein Dlg was detected by immunohistochemistry or Western blot analysis (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
We also analyzed the NMJs of adult animals by quantifying the total number of NMJ boutons on the dorsal muscle of A3 segment in young (3-d-old) and aged (30-d-old) dLRRK mutant and dLRRK overexpression flies. There was a similar pattern of bouton number change in adult animals as in larvae for each genotype, and no significant difference was observed between young and old animals (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material), suggesting that the NMJ morphology defects could be primarily attributed to a developmental role of dLRRK.
d4E-BP interacts with dLRRK at the postsynaptic compartment
We next sought to identify the downstream mediators of LRRK2 function at the synapse. Because the kinase activity of LRRK2 is essential for NMJ morphogenesis, we focused on LRRK2 substrates. Previous studies revealed that dLRRK phosphorylates d4E-BP at the T37/46 sites and that the d4E-BP pathway of translational control may mediate dLRRK-induced DA neuron degeneration (Imai et al., 2008). Given the known role of translational control in regulating synaptogenesis at the Drosophila NMJ (Sigrist et al., 2000; Menon et al., 2004), we tested whether d4E-BP may mediate the effect of dLRRK on synaptic morphogenesis. We first tested the effects of overexpressing d4E-BP–WT or d4E-BP–T37/46A (d4E-BP-TA), a nonphosphorylatable form of d4E-BP with the LRRK2 target sites mutated. Postsynaptic overexpression of d4E-BP–WT or d4E-BP–TA led to a decrease in the total number of type I boutons (both type Ib and type Is) on muscle 6/7 of A3 (Fig. 2B,C,J). In contrast, their presynaptic overexpression had no detectable effect (Fig. 2J). These results suggest that d4E-BP plays a more specific role at the postsynaptic compartment. We next examined the effect of coexpressing d4E-BP–WT or d4E-BP–TA on dLRRK overexpression phenotypes. The reduction of NMJ boutons caused by postsynaptic dLRRK overexpression was fully rescued by the coexpression of d4E-BP–WT. In contrast, d4E-BP–TA coexpression showed no rescue (Fig. 2D–F,K). We also examined the effect of d4E-BP RNA interference (RNAi) in dLRRK overexpression background. The bouton loss phenotype caused by postsynaptic dLRRK overexpression was not affected by the coexpression of a d4E-BP RNAi transgene (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). The differential effect of d4E-BP–WT and d4E-BP–TA in dLRRK overexpression background is a bit surprising, considering that d4E-BP–TA is supposedly a more active form of d4E-BP in repressing translation. It is possible that the ability of d4E-BP to undergo reversible phosphorylation/dephosphorylation is required for the rescue. As a control, we found that d4E-BP–WT failed to rescue the bouton-loss phenotype caused by postsynaptic overexpression of PAR-1 kinase at the NMJ (Zhang et al., 2007), indicating that the genetic interaction between dLRRK and d4E-BP is specific (supplemental Fig. S4C, available at www.jneurosci.org as supplemental material). Consistent with d4E-BP playing a more important role at the postsynaptic compartment, coexpression of neither d4E-BP–WT nor d4E-BP–TA was able to rescue the NMJ phenotype caused by presynaptic dLRRK overexpression (Fig. 2K). We further examined the genetic interaction between dLRRK and d4E-BP in NMJ development using LOF mutants. The synaptic overgrowth phenotype in dLRRK mutant was rescued by the simultaneous loss of d4E-BP (also known as thor) in dLRRK thor double mutant, although d4E-BP mutation in a wild-type background had no discernable effect on synaptic differentiation (Fig. 2G–I,K). These data support a functional interaction between dLRRK and d4E-BP at the postsynapse.
Figure 2.

Genetic interactions between LRRK2 and d4E-BP at the postsynapse. A–I, NMJ terminals of the indicated genotypes revealed by HRP immunostaining. Mhc–GAL4/+ (A), postsynaptic overexpression of d4E-BP–WT (B), and postsynaptic overexpression of d4E-BP–T37/46A (marked as d4E-BP–TA) (C). D–F, The reduction of synaptic bouton number caused by postsynaptic overexpression of dLRRK is rescued by d4E-BP-WT (D) but not d4E-BP-TA (E) coexpression (F). G–I, LOF analysis of the genetic interaction between dLRRK and 4E-BP. Scale bar, 20 μm. J, Quantification of the total number of boutons (black bar) and the number of type 1b boutons (white bar) in animals overexpressing wild-type or a nonphosphorylatable form of d4E-BP presynaptically or postsynaptically. K, Quantification of data showing genetic interaction between dLRRK and d4E-BP under GOF or LOF conditions. The numbers inside the black bars (n) in this figure and subsequent figures represent the total number of boutons and the number of type 1b boutons quantified for each genotype.
Rapamycin, an inhibitor of target of rapamycin (TOR) signaling (Brown et al., 1994), is a useful reagent for a pharmacological approach to induce 4E-BP activation by attenuating its phosphorylation in vitro or in vivo. To investigate whether rapamycin treatment may have similar effect as postsynaptic overexpression of d4E-BP in either control or hLRRK2 overexpression animals, we fed Mhc–GAL4>hLRRK2 and control Mhc–GAL4/+ flies with food containing 0.5 μm rapamycin or DMSO alone (vehicle control). The total number of NMJ boutons or the number of type Ib boutons was reduced in control third-instar larvae treated with rapamycin compared with DMSO-only treatment (Fig. 3A,B,E). Significantly, the NMJ morphology phenotype caused by postsynaptic hLRRK2 overexpression was rescued by rapamycin treatment (Fig. 3C–E). Although rapamycin is known to induce autophagy in a number of organisms, including Drosophila (Ravikumar et al., 2004; Hennig et al., 2006), it is unlikely that its effect on synaptogenesis shown here is attributable to its induction of autophagy, because autophagy induction by presynaptic or postsynaptic overexpression of Atg1 had no significant effect on synapse number (supplemental Fig. S4D, available at www.jneurosci.org as supplemental material). It appears that the effect of rapamycin on protein translation may be playing a more dominant role in determining synaptic growth. Together, these results provide strong evidence for a functional interaction between d4E-BP-mediated protein translation and dLRRK in NMJ development.
Figure 3.

Pharmacological rescue of hLRRK2 overexpression phenotype by rapamycin. A–D, Representative anti-HRP immunostaining of NMJ terminals of the indicated genotypes with or without rapamycin treatment. A, Mhc–GAL4/+ treated with DMSO (vehicle control); B, Mhc–GAL4/+ treated with rapamycin; C, Mhc–GAL4>hLRRK2–WT animals treated with DMSO; and D, Mhc–GAL4>hLRRK2–WT animals treated with rapamycin. Scale bar, 20 μm. E, Quantification of the pharmacological effect of rapamycin on the total number of boutons (black bar) and the number of type 1b boutons (white bar) on muscle 6/7 of A3, after normalization with bouton area.
LRRK2 and Futsch interact at the presynaptic compartment
Our observation that presynaptic dLRRK overexpression causes a reduction of bouton number in a 4E-BP-independent manner suggests that other factor(s) may mediate the presynaptic effect of dLRRK2. Futsch, a Drosophila homolog of neuronally enriched MAP1B (Hummel et al., 2000; Roos et al., 2000), is a candidate for such a factor. In a proteomic approach to identifying hLRRK2-associated proteins in mammalian HEK293T cells, we found MAP1B in the hLRRK2 immunocomplex (Y. Imai and B. Lu, unpublished observation). Interestingly, the synaptic phenotype induced by neuronal overexpression of dLRRK or hLRRK2 is similar to that observed in futsch mutants (Roos et al., 2000). Thus, the total number of boutons was decreased, whereas NMJ bouton size was increased in hLRRK2 presynaptic overexpression animals, as in futsch mutants (supplemental Fig. S5A–C, available at www.jneurosci.org as supplemental material). To test for a functional link between Futsch and LRRK2 in NMJ development, we first asked whether LRRK2 and Futsch physically associate in a protein complex in vivo. In co-IP experiments using extracts prepared from body-wall muscle and central brain complexes of Da–GAL4>UAS–hLRRK2–Flag transgenic larvae, the 22C10 monoclonal antibody against endogenous Futsch was able to specifically pull down Flag-tagged hLRRK2 (Fig. 4A). Reciprocally, the anti-Flag antibody against hLRRK2–Flag was able to pull down endogenous Futsch (Fig. 4B). To test whether the specific interaction between dLRRK and Futsch occurs in endogenous proteins, co-IP was performed on wild-type fly head extracts with anti-Futsch antibodies or normal IgG. The 22C10 monoclonal antibody against endogenous Futsch was able to specifically pull down endogenous dLRRK (supplemental Fig. S5D, available at www.jneurosci.org as supplemental material). We next examined whether dLRRK colocalizes with Futsch at the NMJ. Immunohistochemical analysis showed that dLRRK and Futsch colocalized in the presynaptic region (supplemental Fig. S5E, available at www.jneurosci.org as supplemental material). These results support a direct physical interaction between hLRRK2 and Futsch in larval NMJ.
Figure 4.

LRRK2 interacts with and phosphorylates Futsch. A–D, hLRRK2 physically interacts with Futsch. A, Extracts from Da–GAL4>hLRRK2–Flag or control animals were immunoprecipitated with mAb 22C10 against Futsch, and the presence of hLRRK2–Flag in the immunocomplex was detected by Western blot with anti-hLRRK2. B, IP of extracts as described in A was performed to pull down hLRRK2–Flag with anti-Flag. The presence of Futsch was detected by Western blot with mAb 22C10. C, Genetic interaction between dLRRK and futsch. D, Futsch coexpression rescues synaptic defects caused by presynaptic overexpression of hLRRK2. E, hLRRK2 phosphorylates Futsch in vitro. In vitro kinase assays were performed with substrate only or substrate plus WT, G2019S, or KD forms of hLRRK2. Top, Autoradiography; bottom, Western blots as control for protein loading. Blots were probed with anti-Futsch and anti-hLRRK2.
We further tested the genetic relationship between dLRRK and futsch in synapse development, using muscle 6/7 of A3 as a model. Reduction of Futsch activity by removing one copy of futsch gene in dLRRK mutant background rescued the synaptic overgrowth phenotype, although futsch heterozygosity in a wild-type background had no effect on synaptic differentiation (Fig. 4C) (supplemental Fig. S6A, available at www.jneurosci.org as supplemental material). This result is consistent with the notion that dLRRK negatively regulates the activity of Futsch to control synaptic differentiation. This is further supported by the ability of Futsch coexpression to rescue hLRRK2 overexpression-induced synaptic morphology defects, as measured by the total number of type I boutons or the number of type Ib boutons (Fig. 4D) (supplemental Fig. S6A, available at www.jneurosci.org as supplemental material). Distinct from the effect of 4E-BP, the rescue of hLRRK2 overexpression phenotype by Futsch coexpression only occurred at the presynaptic side (Fig. 4D). We further verified the specificity of dLRRK/Futsch genetic interaction at the presynapse. As reported previously, presynaptic overexpression of a dominant-negative (DN) form of Fos resulted in reduced bouton number (Sanyal et al., 2002; Collins et al., 2006). This phenotype was not affected by Futsch coexpression (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material). These results strongly support that Futsch plays a specific role at the presynaptic compartment to mediate dLRRK function.
LRRK2 phosphorylates Futsch
The clear genetic interaction between LRRK2 and futsch and the physical interaction between these proteins at the NMJ raised the interesting possibility that LRRK2 might regulate Futsch function through direct phosphorylation. To test this possibility, we performed in vitro kinase assays, using hLRRK2–WT, hLRRK2–G2019S, or hLRRK2–3KD proteins affinity purified from transfected HEK293T cells as the kinase source. As substrate, we used Futsch protein affinity purified from wild-type fly heads using the 22C10 antibody. Phosphorylation events were detected by autoradiography after in vitro kinase reaction. The pathogenic hLRRK2–G2019S protein exhibited higher kinase activity than hLRRK2–WT, as shown by the increased level of autophosphorylation, whereas hLRRK2–3KD showed no kinase activity (Fig. 4E). Correlating with the specific kinase activity, hLRRK2–G2019S showed strongest phosphorylation of Futsch. Moderate Futsch phosphorylation was observed with hLRRK2–WT, whereas hLRRK2–3KD showed no detectable Futsch phosphorylation (Fig. 4E). The hLRRK2–3KD result ruled out the possibility that some contaminating, hLRRK2-interacting kinase might be responsible for the observed Futsch phosphorylation. No phosphorylation signal at the Futsch position was observed when Futsch substrate was omitted or when 22C10 immunoprecipitate prepared from futschN94 mutant was used as the substrate. These results demonstrated specific phosphorylation of Futsch by hLRRK2. Futsch is known to be phosphorylated by Shaggy at sites recognized by the phospho-specific antibody pAb–BUGS (Gögel et al., 2006). In dLRRK mutant or in hLRRK2 overexpression animals, Futsch phosphorylation as detected by pAb–BUGS was not changed (supplemental Fig. S6C, available at www.jneurosci.org as supplemental material), suggesting that LRRK2 and Shaggy may phosphorylate Futsch at different sites.
dLRRK regulates presynaptic cytoskeletal organization and organelle trafficking
Futsch has been shown to regulate synapse growth at the Drosophila NMJ through modification of the MT cytoskeleton. It colocalizes with MTs at cytoskeletal loop structures that traverse the lateral margins of select synaptic boutons (Roos et al., 2000). Ectopic formation of such loop structures correlates with NMJ overgrowth. We wondered whether the increase of boutons seen in dLRRK mutant might be related to change of MT cytoskeleton dynamics mediated by Futsch.
We found that dLRRK mutant NMJs show a significant increase of Futsch-positive MT loops (2.6 ± 0.4 for control, 4.3 ± 1.2 for dLRRK mutant, after normalization with muscle size; p < 0.01) (Fig. 5A–C), suggesting that LRRK2 controls the organization of MT cytoskeleton and may normally inhibit the formation of MT loop structures. To test whether postsynaptic LRRK2 activity could influence presynaptic MT loop structure, we examined Mhc>hLRRK2 animals. No change of MT loop structures was observed in these animals. We also examined thor; dLRRK double mutant. The number of MT loop structures in the double mutant (4.8 ± 0.5, after normalization with muscle size; p < 0.01) (Fig. 5C) was similar to that of dLRRK mutant alone (4.3 ± 1.2), suggesting that loss of the aforementioned postsynaptic function of d4E-BP had no significant effect on cytoskeleton dynamics mediated by Futsch at the presynaptic compartment (Fig. 5C).
Figure 5.

Regulation of MT cytoskeleton dynamics by LRRK2. A–C, Analysis of MT dynamics in dLRRK mutant. Arrows mark MT loops detected with anti-Futsch antibody 22C10. The bar graph (C) shows statistical analysis of the number of MT loops in the indicated genotypes. Scale bar: A, B, 10 μm. D–G, Immunostaining for tyr–Tub in terminal boutons of muscle 6/7 in wild-type (D), dLRRK mutant (E), elav–GAL4>UAS–hLRRK2 (F), and elav–GAL4>UAS–hLRRK2+EP1419 (G) larval NMJs. Third-instar larvae were double stained with anti-HRP (Dii, Eii, Fii, Gii) and anti-tyr–Tub (Di–Gii). Scale bar: D–G, 5 μm. H, Bar graphs showing statistical analysis of the MT phenotypes on muscle 6/7 of A3 in the indicated genotypes (wild-type, n = 13; dLRRKe03680/dLRRKe03680, n = 12; elav–GAL4>UAS–hLRRK2–WT–flag, n = 15; elav–GAL4>UAS–hLRRK2–WT–flag+EP1419, n = 11). The abnormal phenotypes of MT in terminal boutons were classified as fragmented or tangled. I, hLRRK2 associates with Futsch and Tubulin in a complex. Extracts from control and elav–GAL4>UAS–hLRRK2–Flag animals were immunoprecipitated with anti-Flag antibody. Blots were probed with anti-hLRRK2, anti-Futsch, and anti-Tubulin.
To further examine the role of dLRRK in regulating presynaptic MT dynamics, we analyzed MT structures in NMJ boutons of dLRRK mutant and hLRRK2 overexpression animals. MT in terminal boutons was immunostained with anti-tyr–Tub antibody, which recognizes newly synthesized tubulin and thus specifically labels dynamic MTs (Ruiz-Canada et al., 2004). In wild-type boutons, anti-tyr–Tub labeled continuous tubular MT structures (Fig. 5D,H). In contrast, in animals overexpressing hLRRK2 presynaptically, tyr–Tub-positive MTs inside synaptic boutons appeared more fragmented (Fig. 5F,H). An opposite phenotype was observed in dLRRK mutant boutons, in which the tyr–Tub-positive MTs appeared more elaborate and tangled (Fig. 5E,H). The abnormal MT phenotype in hLRRK2 overexpression background was rescued by the coexpression of Futsch (Fig. 5G,H), supporting that Futsch is a major mediator of LRRK2 effect on MT dynamics. To further test the idea that hLRRK2 controls MT dynamics via Futsch, we performed co-IP studies to probe the existence of a multiprotein complex containing hLRRK2, Futsch, and Tubulin. The anti-Flag antibody could pull down both Tubulin and Futsch from elav–GAL4>hLRRK2–WT–Flag transgenic larval extracts, suggesting that hLRRK2 may simultaneously bind to Futsch and Tubulin (Fig. 5I).
In addition to maintaining proper synapse morphology, MT cytoskeleton is also important for the transport of synaptic vesicles and organelles such as mitochondria, which are important for providing the energy source for neurotransmission and for buffering excess Ca2+ at synapses. We next tested whether the synaptic distribution of mitochondria is affected in dLRRK LOF or hLRRK2 GOF conditions. We first tested the effects of Futsch LOF and GOF on synaptic mitochondrial distribution. Whereas Futsch GOF had no significant effect on synaptic mitochondria number, its LOF led to a reduction of synaptic mitochondria (Fig. 6E). Neuronal overexpression of hLRRK2 led to a decrease of mitochondria number in terminal boutons, whereas dLRRK LOF had the opposite effect (Fig. 6A–C,E). Coexpression of Futsch in hLRRK2 GOF background essentially rescued the mitochondria distribution phenotype (Fig. 6D,E), consistent with Futsch-mediated MT structural changes being responsible for the organelle distribution phenotype induced by LRRK2 overactivation.
Figure 6.
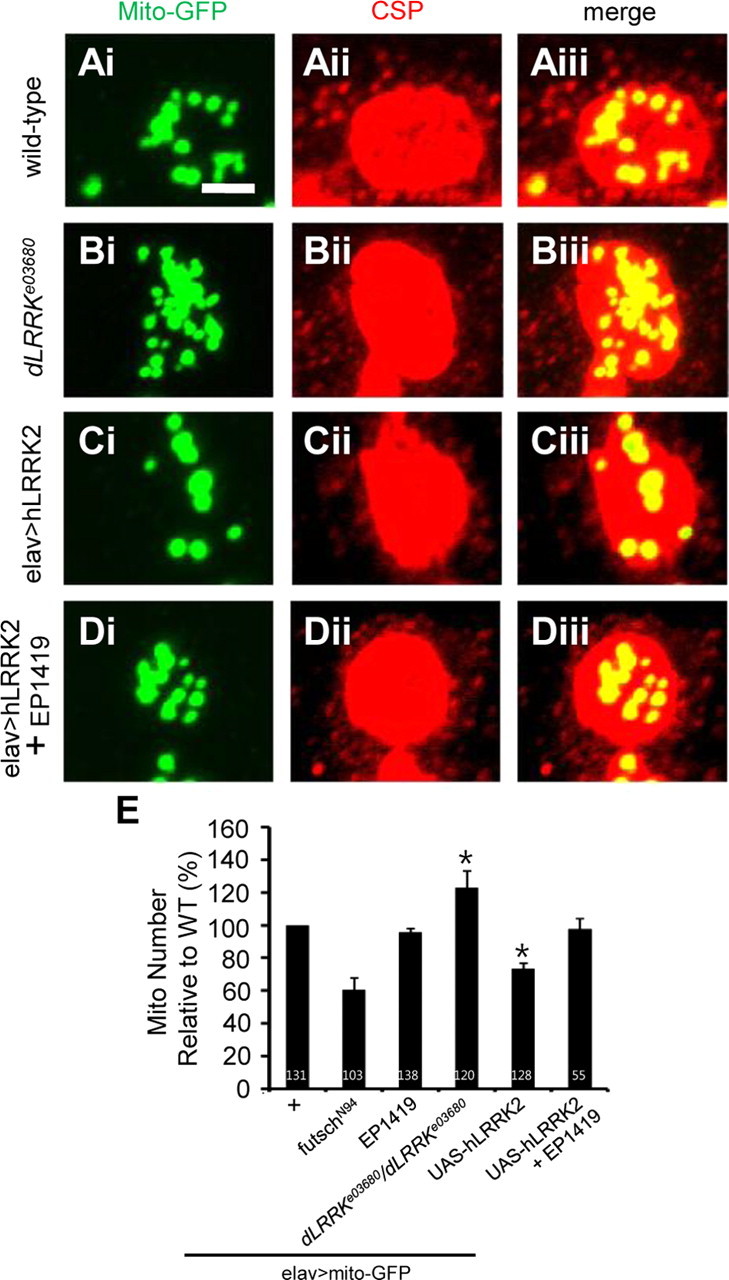
LRRK2 affects the distribution of mitochondria in the presynaptic region. A–D, Terminal boutons of wild-type (A), dLRRK mutant (B), elav–GAL4>UAS–hLRRK2–WT (C), and elav–GAL4>UAS–hLRRK2–WT+EP1419 (D) animals, all expressing a UAS–mito–GFP transgene, were double stained with anti-GFP (Ai, Bi, Ci, Di) and anti-CSP (Aii, Bii, Cii, Dii). Merged images are shown in Aiii, Biii, Ciii, and Diii. Scale bar: A–D, 2 μm. E, Quantification of the distribution of mitochondria relative to the elav–GAL4/+; UAS–mito–GFP/+ control (n = 131) in the indicated genotypes: futschN94; UAS–mito–GFP/+; elav–GAL4/+ (n = 103), elav–GAL4/EP1419; UAS–mito–GFP/+ (n = 138), elav–GAL4/+; UAS–mito–GFP/+; dLRRKe03680/ dLRRKe03680 (n = 120), elav–GAL4/+; UAS–mito–GFP/UAS–hLRRK2 (n = 128), and elav–GAL4/EP1419; UAS–mito–GFP/UAS–hLRRK2 (n = 55). Mitochondria distribution is quantified as the number of mitochondria normalized by bouton area. *p < 0.001.
Alterations of LRRK2 activity affect synaptic transmission
To address whether changes in synaptic morphology caused by alterations of LRRK2 activity affect synaptic physiology, we first performed electrophysiological analyses of evoked EJCs and spontaneous mEJCs on muscle 6 of wild-type (w−) control and dLRRK mutant third-instar larvae. Whereas no significant difference in mEJC amplitude was observed in dLRRK mutant compared with wild-type control (Fig. 7B), mEJC frequency was significantly increased in dLRRK homozygous mutant. dLRRK heterozygous animals showed a moderate increase of mEJC frequency (Fig. 7A). In terms of EJC amplitude, dLRRK mutant animals showed a significant reduction (Fig. 7C). Quantal content, a representation of synaptic junction efficacy, was also significantly reduced in dLRRK mutant (Fig. 7D). Muscle expression of a dLRRK transgene was unable to rescue the alternations of mEJC frequency, EJC amplitude, or quantal content in dLRRK mutant (Fig. 7A,C). Despite neuronally expressed dLRRK being able to partially rescued EJC amplitude in dLRRK mutant background, it did not significantly rescue quantal content (Fig. 7D). We further examined the electrophysiological effects when hLRRK2 proteins were overexpressed at the presynaptic or postsynaptic compartments. We observed that elav–GAL4-driven presynaptic overexpression of hLRRK2–G2019S led to a significant increase of mEJC frequency (Fig. 7E). Conversely, quantal content was decreased in elav–GAL4>hLRRK2–G2019S but not significantly changed in elav–GAL4>hLRRK2–WT animals when compared with elav–GAL4/+ control (Fig. 7H). The differential effects by hLRRK2–G2019S and hLRRK2–WT were unlikely attributable to different levels of transgene expression, because hLRRK2–G2019S was actually expressed at a lower level than hLRRK2–WT (supplemental Fig. S6E, available at www.jneurosci.org as supplemental material). Surprisingly, despite the obvious effects of postsynaptic overexpression of hLRRK2–WT or hLRRK2–G2019S on synaptic morphology, no obvious effect on synaptic transmission was observed when mEJC frequency, mEJC amplitude, EJC amplitude, and quantal content were measured (Fig. 7I–L). Thus, there is no specific pattern of electrophysiological phenotypes in the loss-of-function mutant and overexpression backgrounds that will allow us to draw a conclusion about the exact roles of LRRK2, suggesting that the mechanism of LRRK2 regulation of synaptic transmission is complex, with its precise level being important for synaptic efficacy. Our data do indicate that the pathogenic hLRRK2–G2019 appears to be more potent than hLRRK2–WT in disturbing neurotransmission at the presynaptic side.
Figure 7.
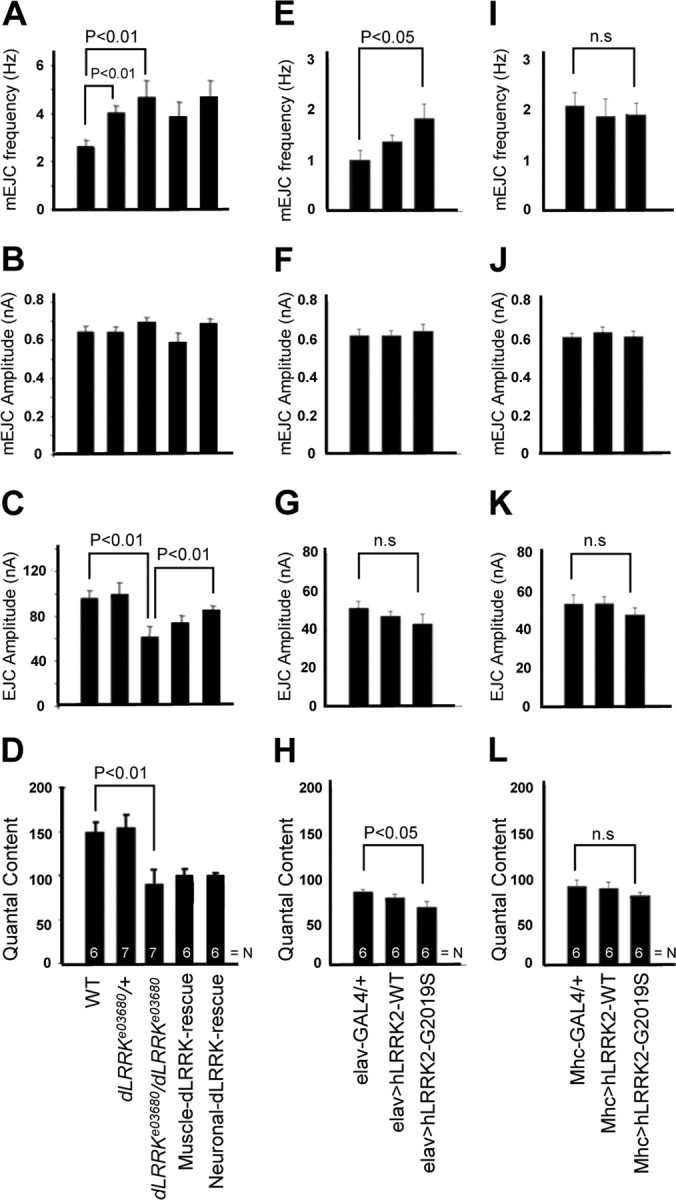
Electrophysiological phenotypes in animals with altered LRRK2 activities. Bar graphs showing quantitative analyses of mEJC frequency, mEJC amplitude, EJC amplitude, and quantal content in wild-type, dLRRKe03680/+, dLRRKe03680/dLRRKe03680, muscle dLRRK–WT-rescue, neuronal dLRRK–WT-rescue, elav–GAL4/+, elav–GAL4>UAS–hLRRK2–WT, elav–GAL4>UAS–hLRRK2–G2019S, Mhc–GAL4/+, Mhc–GAL4>UAS–hLRRK2–WT, and Mhc–GAL4>UAS–hLRRK2–G2019S animals are presented. A, E, I, mEJC frequency was increased in both dLRRKe03680/+ and dLRRKe03680/dLRRKe03680 relative to wild-type control (p < 0.01). There was no rescue of this dLRRK mutant phenotype by neuronal or muscle expression of the dLRRK transgene. mEJC frequency was increased in elav–GAL4>hLRRK2–G2019S (E) (p < 0.05) but not Mhc–GAL4>UAS–hLRRK2–G2019S (I) compared with elav–GAL4/+ and Mhc–GAL4/+ control, respectively. B, F, J, There was no significant change in the amplitude of mEJCs for the various genotypes compared with the corresponding controls. C, D, G, H, K, L, The EJC amplitude (C) and quantal content (D) were decreased in dLRRKe03680 mutant compared with wild-type control (p < 0.01). For EJC amplitude, there was partial rescue by neuronal but not muscle expression of the dLRRK transgene. However, for quantal content, neither neuronal nor muscle expression of the dLRRK transgene showed rescue. Although the EJC amplitude (G) was not significantly changed, quantal content (H) was decreased in elav–GAL4>UAS–hLRRK2–G2019S animals (p < 0.05). EJC amplitude (K) and quantal content (L) were not significantly changed in Mhc–GAL4>UAS–hLRRK2–G2019S animals. The total number of boutons quantified for each genotype is indicated by N inside the bars.
Discussion
Genetic mutations in LRRK2 are frequently found in familial and sporadic PD cases. Understanding the physiological function of LRRK2 will therefore offer insights into PD pathogenesis in general. Here we reveal a new physiological function of LRRK2 and offer molecular mechanisms underlying such function. The key findings from our study are that LRRK2 plays an important role in regulating synaptic morphogenesis and that it does so through distinct substrate proteins at the presynaptic and postsynaptic compartments. Our results also show that the precise level of LRRK2 activity is important for synaptic morphogenesis and neurotransmission, but the regulation of these two synaptic processes likely involve different mechanisms and players. Given the similarity of Drosophila NMJ synapse to mammalian excitatory glutamatergic synapses, it is possible that the findings reported here are relevant to mammalian systems.
Synaptic loss is a major neurobiological substrate of cognitive dysfunction in a number of neurological diseases (Wishart et al., 2006). Extensive studies in patients and animal models have documented that synaptic failure is one of the earliest events in the pathogenesis of Alzheimer's disease (Selkoe, 2002). Interestingly, neurotransmission defects have been repeatedly observed in rodent FPD models, including the LRRK2 model (Tong et al., 2009), although no obvious signs of neurodegeneration accompany the electrophysiological defects. These results suggest that synaptic dysfunction is a primary effect of FPD gene mutations and that synaptic failure is intimately involved in PD pathogenesis. The molecular mechanisms underlying these synaptic transmission defects, however, remain elusive. Our study of the LOF and GOF models of LRRK2 in Drosophila provides mechanistic insights into the possible cause of synaptic dysfunction in LRRK2-associated PD. We find that LRRK2 regulates synaptic morphogenesis at the presynaptic and postsynaptic compartments through distinct substrates.
In the presynaptic side, LRRK2 forms a complex with tubulin and the MT-binding protein Futsch. Furthermore, we found that LRRK2 phosphorylates Futsch and negatively regulates the presynaptic function of Futsch in controlling MT dynamics. MT cytoskeleton is critical for the generation and maintenance of neuronal axons and dendrites, transport of synaptic vesicles and organelles along the processes, and the initiation and maintenance of synaptic transmission. Disrupted MT dynamics in neuronal synapses has been implicated as an underlying cause for several neurological diseases, including hereditary spastic paraplegia and fragile X syndrome (Zhang et al., 2001; Sherwood et al., 2004; Trotta et al., 2004). LRRK2-associated PD may share this feature with the aforementioned diseases. Disrupted MT dynamics could be responsible for the presynaptic effects observed in LRRK2 LOF and GOF mutants, such as aberrant mitochondria distribution. The synaptic vesicle transport phenotypes seen in Caenorhabditis elegans LRK-1 mutant could also be attributable to altered MT dynamics (Sakaguchi-Nakashima et al., 2007). These could all contribute to synaptic dysfunction. Futsch/MAP1B proteins are large multidomain proteins that are phosphorylated by multiple kinases, including Sgg/GSK-3β, PAR-1/MARK, and Cdk5, which also phosphorylate tau and are implicated in tau pathology (Lee et al., 2001). Tau-related pathology has been observed in LRRK2 transgenic animals (Li et al., 2009). It would be interesting to test for possible interplay between LRRK2 and these other kinases in regulating MT-binding proteins and MT dynamics. In mammalian hippocampal neurons, overexpression of pathogenic hLRRK2 led to reduced neurite length and branching, whereas deficiency of LRRK2 had opposite effects (MacLeod et al., 2006). Whether MT dynamics regulated by Futsch/MAP1B is contributing to this LRRK2 effect in mammals will await additional investigation.
We also show that LRRK2 interacts with 4E-BP at the postsynapse. 4E-BP acts as a negative regulator of the translational initiation factor eIF4E through direct binding and sequestration (Haghighat et al., 1995). Phosphorylation of 4E-BP by LRRK2 weakens 4E-BP binding to eIF4E (Imai et al., 2008), therefore releasing the inhibitory effect of 4E-BP on eIF4E. Previous studies have demonstrated an important postsynaptic role for eIF4E-mediated protein synthesis in activity-dependent synaptic growth at the Drosophila NMJ (Sigrist et al., 2000). Our genetic interaction studies demonstrate a functional role for 4E-BP in mediating the synaptic effects of LRRK2. However, the exact roles of 4E-BP and LRRK2 in this process appear complex. For example, (1) despite the prominent effects of 4E-BP overexpression on NMJ development, its loss of function has no obvious effect. One would expect a gain of eIF4E function in the absence of 4E-BP and therefore a synaptic-overgrowth phenotype. (2) 4E-BP activity is predicted to be high in dLRRK mutant and low in LRRK2 overexpression condition, but we observed synapse phenotypes opposite of what is predicted based on the presumed roles of eIF4E and 4E-BP on Drosophila NMJ morphogenesis. One possible explanation of these seemingly disparate results is that phospho-4E-BP, the product of LRRK2-mediated phosphorylation of 4E-BP, instead of being inactive and inert, may actually perform some new synaptic function at the NMJ. In this scenario, loss of 4E-BP function in d4E-BP mutant would not show the same phenotype as LRRK2 overexpression, which produces more phospho-4E-BP. Our recent studies in Drosophila dopaminergic neurons suggest a functional role for phospho-4E-BP in vivo (Gehrke et al., 2010). Alternately, effectors other than 4E-BP may also mediate the effects of LRRK2 on NMJ development and neurotransmission. Although 4E-BP overexpression might have masked the effects of these other effectors, in dLRRK mutant background, the functional roles of these other effectors might manifest themselves. A similar situation was observed in pumillo mutant, in which a synapse-loss phenotype was observed despite the upregulation of eIF4E activity in this mutant attributable to the derepression of translational inhibition of eIF4E, which would have resulted in a predicted synapse-overgrowth phenotype (Menon et al., 2004). Involvement of other effectors in mediating the effects of LRRK2 on NMJ development and neurotransmission, and possibly different effectors for NMJ development versus neurotransmission, could also explain the complex electrophysiological phenotypes of dLRRK mutant and LRRK2 overexpression animals, as well as the uncoupling of the effects on synaptic differentiation and neurotransmission by the various genetic manipulations. Future studies will test these possibilities as well as the relevance of the NMJ studies to dopaminergic neuron synapses.
In addition to LRRK2, the TOR pathway also regulates 4E-BP function through phosphorylation (Miron et al., 2003). This pathway primarily regulates cell and organism growth through diverse outputs, including protein synthesis, cytoskeletal change, autophagy, and cell survival. We found that treatment of flies with rapamycin, an inhibitor of TOR, has the same effects as 4E-BP overexpression in wild type as well as LRRK2 overexpression backgrounds. Although rapamycin has been extensively studied in the context of autophagy induction and neurodegeneration models (Ravikumar et al., 2004), its effect on Drosophila NMJ development is unlikely attributable to autophagy, because our results show that presynaptic or postsynaptic induction of autophagy through Atg1 overexpression had no obvious effect on synapse number. The similar effects of rapamycin and 4E-BP overexpression on NMJ development support that rapamycin acts via the 4E-BP translational control pathway to impact NMJ development. A recent report showed that either 4E-BP overexpression or 4E-BP activation by rapamycin could suppress the muscle and dopaminergic neurodegeneration phenotypes seen in Drosophila Pink1 and Parkin models of PD (Tain et al., 2009). These results suggest that deregulation of protein synthesis could be generally involved in PD pathogenesis and that rapamycin or its analogs could be developed into effective PD therapeutics.
Footnotes
This work was supported by the McKnight Foundation Brain Disorders Award, Alzheimer's Association Grant IIRG0626723, National Institutes of Health Grants R01AR054926 and R01MH080378, and Taiwan Department of Health Clinical Trial and Research Center of Excellence Grant DOH99-TD-B-111-004. We are grateful to Dr. Gordon-Weeks for the gift of the anti-BUGS antibody and Drs. A. DiAntonio, T. Littleton, D. A. Kimbrell, W. Smith, and W. Saxton and the Vienna Drosophila Research Center and Bloomington Stock Centers for fly stocks. We thank members of the Lu laboratory for discussions and help.
References
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Galletti F, Saggese E, Ghiglieri V, Picconi B. Neuronal networks and synaptic plasticity in Parkinson's disease: beyond motor deficits. Parkinsonism Relat Disord. 2007;13(Suppl 3):S259–S262. doi: 10.1016/S1353-8020(08)70013-0. [DOI] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability: a point of convergence in parkinsonian neurodegeneration? J Neurochem. 2009a;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- Gillardon F. Interaction of elongation factor 1-alpha with leucine-rich repeat kinase 2 impairs kinase activity and microtubule bundling in vitro. Neuroscience. 2009b;163:533–539. doi: 10.1016/j.neuroscience.2009.06.051. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Gögel S, Wakefield S, Tear G, Klämbt C, Gordon-Weeks PR. The Drosophila microtubule associated protein Futsch is phosphorylated by Shaggy/Zeste-white 3 at an homologous GSK3beta phosphorylation site in MAP1B. Mol Cell Neurosci. 2006;33:188–199. doi: 10.1016/j.mcn.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995;14:5701–5709. doi: 10.1002/j.1460-2075.1995.tb00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig KM, Colombani J, Neufeld TP. TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J Cell Biol. 2006;173:963–974. doi: 10.1083/jcb.200511140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel T, Krukkert K, Roos J, Davis G, Klämbt C. Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron. 2000;26:357–370. doi: 10.1016/s0896-6273(00)81169-1. [DOI] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Menon KP, Sanyal S, Habara Y, Sanchez R, Wharton RP, Ramaswami M, Zinn K. The translational repressor Pumilio regulates presynaptic morphology and controls postsynaptic accumulation of translation factor eIF-4E. Neuron. 2004;44:663–676. doi: 10.1016/j.neuron.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Miron M, Lasko P, Sonenberg N. Signaling from Akt to FRAP/TOR targets both 4E-BP and S6K in Drosophila melanogaster. Mol Cell Biol. 2003;23:9117–9126. doi: 10.1128/MCB.23.24.9117-9126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Roos J, Hummel T, Ng N, Klämbt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/s0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Ruiz-Canada C, Ashley J, Moeckel-Cole S, Drier E, Yin J, Budnik V. New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron. 2004;42:567–580. doi: 10.1016/s0896-6273(04)00255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Sandstrom DJ, Hoeffer CA, Ramaswami M. AP-1 functions upstream of CREB to control synaptic plasticity in Drosophila. Nature. 2002;416:870–874. doi: 10.1038/416870a. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Sherwood NT, Sun Q, Xue M, Zhang B, Zinn K. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004;2:e429. doi: 10.1371/journal.pbio.0020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Sigrist SJ, Thiel PR, Reiff DF, Lachance PE, Lasko P, Schuster CM. Postsynaptic translation affects the efficacy and morphology of neuromuscular junctions. Nature. 2000;405:1062–1065. doi: 10.1038/35016598. [DOI] [PubMed] [Google Scholar]
- Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta N, Orso G, Rossetto MG, Daga A, Broadie K. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr Biol. 2004;14:1135–1147. doi: 10.1016/j.cub.2004.06.058. [DOI] [PubMed] [Google Scholar]
- Wang D, Tang B, Zhao G, Pan Q, Xia K, Bodmer R, Zhang Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol Neurodegener. 2008;3:3. doi: 10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–739. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Guo H, Kwan H, Wang JW, Kosek J, Lu B. PAR-1 kinase phosphorylates Dlg and regulates its postsynaptic targeting at the Drosophila neuromuscular junction. Neuron. 2007;53:201–215. doi: 10.1016/j.neuron.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]