

Abstract
Macroautophagy is a highly conserved mechanism of lysosomal mediated protein degradation that plays a key role in maintaining cellular homeostasis by recycling amino acids, reducing the amount of damaged proteins, and regulating protein levels in response to extracellular signals. We have found that macroautophagy is induced following effector T cell activation. Engagement of the T cell receptor and CD28 results in enhanced LC3 processing, increased numbers of LC3-containing vesicles and increased LC3 flux, indicating active autophagosome formation and clearance. The autophagosomes formed in stimulated T cells actively fuse with lysosomes to degrade their cargo. Using a conditional knockout mouse model where Atg7, a critical gene for macroautophagy, is specifically deleted in T cells, we have found that macroautophagy-deficient effector T helper cells have defective IL-2 and INFγ production and reduced proliferation following stimulation, with no significant increase in apoptosis. We have found that ATP generation is decreased when autophagy is blocked, and defects in activation-induced cytokine production are restored when an exogenous energy source is added to macroautophagy-deficient T cells. Furthermore, we present evidence showing that the nature of the cargo inside autophagic vesicles found in resting T cells differs from the cargo of autophagosomes in activated T-cells, where mitochondria and other organelles are selectively excluded. These results suggest that macroautophagy is an actively regulated process in T cells that can be induced in response to T cell receptor engagement to accommodate the bioenergetic requirements of activated T cells.
Introduction
Protein turnover is necessary not only to reduce the accumulation of damaged proteins in the cell and to recycle amino acids for new protein synthesis, but it also allows for the modification of protein levels in response to extracellular signals (1-4). A major pathway involved in the degradation of long-lived proteins is macroautophagy, a catabolic process that delivers cytoplasmic material to lysosomes. Degradation of proteins by autophagy plays a key role in maintenance of correct cell homeostasis, which requires a careful balance between protein synthesis and protein degradation (5).
Macroautophagy is a form of autophagy responsible for the degradation of cytosolic proteins and whole organelles. This process is critical to maintain cell function, as its failure leads to intracellular accumulation of damaged proteins, defective regulation of many cellular processes and altered responses to stress, which appear to underlie the basis for different human diseases (4, 6). Macroautophagy involves sequestering of cargo into a de novo formed double-membrane vesicle called the autophagosome (7). Eventually, the autophagosome fuses with lysosomes and breakdown of the cargo occurs. Two ubiquitin-like conjugation systems regulated by Atg7 are involved in the biogenesis of the autophagosome: Atg8 (LC3)-phosphatidyl-ethanolamine and Atg12-Atg5 (8, 9). Macroautophagy is regulated by the PI3K class III, Vps34 that forms a complex with Beclin1 and stimulates autophagosome nucleation (7). Knocking down the proteins involved in the conjugation processes (e.g. Atg5 or Atg7) or inhibiting PI3K class III causes inhibition of macroautophagy.
Although the role of macroautophagy in several tissues and systems has been characterized, it is still unclear what role this process may play in the regulation of the adaptive immune system. Presentation of intracellular antigens on MHC class II molecules by dendritic cells has been shown to be mediated by macroautophagy, which is also active in thymic epithelial cells, where it plays a key role in the regulation of thymocyte selection and therefore in shaping the T cell repertoire(10). Macroautophagy also regulates B cell survival and development (11).
It is only recently that a possible role for macroautophagy in the regulation of T cell homeostasis has been proposed. Indeed, macroautophagy has been shown to control apoptosis and proliferation of peripheral T cells, and also to regulate growth-factor withdrawal-induced cell death in CD4+ T cells (12, 13). Macroautophagy is also activated in CD4+ T cells when the CXCR4 receptor is engaged by the HIV-1 Env protein, which leads to cell death (14). Recently, macroautophagy has been proposed to be a major regulatory process of mitochondrial turnover during T cell development (15). However, the functional role of macroautophagy during T cell activation is still not fully understood.
Following antigen recognition, T cells need to rapidly go from a resting to an activated state in order to respond to that antigen and proliferate. T cell activation imposes a vast bioenergetics challenge in order to sustain a rapid transition that involves the activation of a new transcriptional program (16). This process results in a change in the cell proteome that is needed for activation and survival. Macroautophagy has been shown in other systems to play a critical role in maintaining amino acid and energy homeostasis (3, 17). Here we show that macroautophagy is upregulated upon activation in CD4+ T cells. Furthermore, using macroautophagy-deficient T cells, we show that macroautophagy activation is required to maintain cell proliferation and cytokine production. Finally, we describe a novel role for macroautophagy during T cell activation in which selective cargo sequestration allows this catabolic process regulate energy metabolism during T cell activation.
Material and Methods
Mice
Six to eight-week-old C57BL/6 mice were purchased from Jackson Laboratories and were maintained in pathogen-free conditions. To generate mice with a T cell specific knockout of macroautophagy, Atg7F/F mice (provided by M. Komatsu and K. Tanaka, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) were crossed with mice expressing Cre recombinase under the control of an Lck promoter purchased from Jackson Laboratories. Atg7F/F mice were also crossed to B6.Cg-Tg(CAG-Cre/Esr1)5Amc/J mice (Cre-ER mice), which express a tamoxifen-inducible Cre-mediated recombination system (The Jackson Laboratory). Studies were performed in Atg7F/F-Lck-Cre or Atg7F/F-Cre-ER mice and littermate controls lacking the Cre transgene (Atg7F/F mice). In vitro deletion of Atg7 was performed by incubation of T cells with 2 μM of 4-hydroxytamoxifen (Sigma-Aldrich) for 48 hrs before macroautophagy was assessed. Deletion of Atg7F/F was assessed by PCR and confirmed by immunoblot or real time quantitative PCR. All animal work was approved and performed according to the guidelines set by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee.
Cell culture
Primary CD4+ T cells were isolated from lymph nodes and spleen of mice using anti-CD4-coupled magnetic beads (Invitrogen). Isolated T cells were stimulated with 0.5 μg/ml plate-bound anti-CD3 and 0.5 μg/ml anti-CD28 (BD Biosciences) and differentiated for 7 days with IL-12 (10 ng/ml) (Cell Sciences), anti-IL-4 (10 μg/ml) and 10 U/ml of recombinant human IL-2 (NCI BRB Preclinical Repository). Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, nonessential amino acids (Cambrex), essential vitamins (Cambrex), and 50 μM 2-mercaptoethanol. Where indicated cells were treated with 10 mM 3-methyladenine or 20 mM ammonium chloride and 100 μM leupeptin (Sigma-Aldrich) for 1-4 hrs prior to T cell activation to inhibit macroautophagy or lysosomal degradation, respectively.
ELISA
Th1 cells (2.5–5 × 104) were stimulated with 0.5 μg/ml plate-bound anti-CD3 and anti-CD28 in 96-well plates for 16 h. Supernatants were collected and IL-2 and IFNγ levels were measured in a sandwich ELISA following the manufacture’s recommendations (BD Biosciences).
Measurement of intracellular protein degradation
Activated T cells were incubated with [3H]Leucine (2 μCi/ml) for 48 h at 37°C, and then extensively washed and maintained during the chase in medium containing an excess of unlabeled Leucine (2.8 mM), to prevent reutilization of the radiolabeled Leucine. Aliquots of the medium taken at different times were precipitated with TCA, and proteolysis was calculated as the percentage of initial total acid precipitable radioactivity (protein) transformed to acid soluble (peptides and amino acids) at each time point (18). Total radioactivity incorporated into cellular proteins was determined in duplicate samples as the amount of acid precipitable radioactivity in labeled cells immediately after washing. In other studies, cells were labeled with 0.2 mCi/ml of 35S-Methionine/Cystein for 48 h and then extensively washed and maintained during the chase in medium containing an excess of unlabeled Methionine/Cysteine to prevent reutilization of the radiolabeled amino acids. Cytosolic extracts were prepared (supernatants of cell homogenates after a 100,000 g centrifugation for 1 h) at different time points, run on a SDS-gel and analyzed by autoradiography.
Measurement of ATP levels
ATP levels were assessed using a bioluminescence assay kit (Roche) following the manufacturer’s instructions.
Lactate measurements
Th1 cells were activated with plate bound anti-CD3 and anti-CD28 in the presence or absence of inhibitors of lysosomal activity. Lactate release into the media was determined at different time points using a lactate assay kit (MBL) following the manufacturer’s recommendations.
Measurement of TG utilization
T cells were cultured with 14C-Oleate-BSA complex for 4 h. Cells were then extensively washed and stimulated with plate-bound anti-CD3 and anti-CD28 in the presence or absence of NH4Cl and leupeptin. At different time points, cellular lipids were extracted with hexane/isopropanol (3:2), dried and redisolved in chloroform. Samples were resolved by thin-layer chromatography. Phosphorimager images were obtained and quantified with a Storm Imaging System (GE Healthcare).
Real time PCR
cDNA was synthesized from total RNA samples and gene expression was analyzed using SYBR Green in a Smart Cycler II thermocycler (Cepheid). Expression of each gene was normalized to levels of actin. In order to assess levels of Atg7 and Il2 mRNA the following primers were used: Atg7, 5’-CAGTTTCCAGTCCGTTGAAGTCCT-3’ and 5’-GGGTCCATACATCCACTGAGGTTC-3’. Il2, 5’-GGCATGTTCTGGATTTGACTC-3’ and 5’-TCATCATCGAATTGGCACTC-3’.
Proliferation assay
5×104 Th1 cells were stimulated with plate bound anti-CD3 and anti-CD28 in 96-well plates. 60 hr later, BrdU was added for 12 hr. Incorporation of BrdU was measured by ELISA according to manufacturer’s instructions (Roche).
Apoptosis assay
Apoptosis was determined with an Annexin V-PE apoptosis detection kit (BD) or by TUNEL assay with the In situ Cell Death Detection kit (Roche). Stained cells were analyzed by FACS.
Immunoblotting
Total cellular lysates were prepared using RIPA buffer (1% Triton-X 100, 1% sodium deoxycholate, 0.1%SDS, 0.15 M NaCl, 0.01 M sodium phosphate, pH 7.2). Membranes were incubated with the following primary antibodies: anti-Atg7, AMPKα, AKT, phospho-AKT, S6K, phospho-S6K (Cell Signaling) and LC3 (MBL). Immunoblotting of actin (AbCam) was used to control for loading. To measure autophagic flow, immunoblots for LC3 were performed in untreated cells and cells treated with the lysosomal inhibitors NH4Cl and leupeptin. Autophagic flow was determined by the ratio of the densitometric value for LC3-II in the presence of inhibitors to that in the absence of inhibitors, as described previously (8, 19).
Fluorescence microscopy
T cells were fixed with 4% paraformaldehyde, blocked and incubated with antibodies against the microtubule-associated protein 1 light chain 3 LC3 (MBL) to detect autophagosomes, and the lysosome-associated membrane protein type 1 LAMP-1 (The Developmental Studies Hybridoma Bank) to identify the lysosomal compartment. Where indicated, cells were incubated with 100 nM Mitotracker-Green and 100 nM Lysotracker-Red (Molecular Probes) for 30 minutes prior to fixation in the presence of 100 μM leupeptin to detect mitochondria and lysosomes, respectively. Images were taken in a Zeiss Axiovert inverted microscope with deconvolution software.
Electron microscopy
Cells were fixed in 2.5% gluteraldehyde in 100 mM sodium cacodylate, pH 7.43 and post-fixed in 1% osmium tetroxide in 100 mM sodium cacodylate, pH 7.43, followed by 1% uranyl acetate (20). After ethanol dehydration and embedding in LX112 resin (LADD Research Industries), ultrathin sections were cut on a Reichert Ultracut E and stained with uranyl acetate followed by lead citrate. All grids were viewed on a JEOL 100CX II transmission electron microscope at 80 kV. Morphometric analysis was performed using ImageJ software (National Institutes of Health) in 15-20 different micrographs for each condition after thresholding. Autophagic vacuoles were identified by visual inspection of the micrographs using previously established criteria (8, 21, 22). Briefly, autophagic vacuoles (vesicles <0.5 μm) were classified as autophagosomes when they met two or more of the following criteria: double membranes (complete or at least partially visible), absence of ribosomes attached to the cytosolic side of the membrane, luminal density similar to cytosol, and identifiable organelles or regions of organelles in their lumen. Vesicles of similar size but with a single membrane (or less than 40% of the membrane visible as double), luminal density lower than the surrounding cytosol, multiple single membrane-limited vesicles containing light or dense amorphous material were classified as autophagolysosomes. For morphometric purposes, both types of autophagic vesicles were pooled together in this study as we did not find differences in the maturation of these compartments between resting and stimulate cells.
Statistical analysis
Differences between multiple groups were analyzed by ANOVA with a Tukey post-test. Comparisons between specific pairs of groups were analyzed with a t-test.
Results
Macroautophagy is induced during activation in effector T helper cells
Macroautophagy has been shown to be active in naïve T cells and regulate T cell homeostasis and survival (12, 23). In many cell types macroautophagy is activated in response to different cellular stresses. In order to determine if macroautophagy could also be regulated in a similar manner in T cells, we measured macroautophagy activity following T cell activation in effector T helper cells. In vitro differentiated primary mouse Th1 cells were activated with plate bound anti-CD3 and anti-CD28 for 6 h and autophagosome formation was assessed by immunofluorescence using antibodies against LC3, a commonly used marker for autophagic vacuoles (8). Analysis of the stimulated effector T helper cells revealed that, although there was a basal level of macroautophagic activity in T cells, macroautophagy was activated in response to TCR and costimulation engagement. The number of vesicles that stained for LC3 was significantly higher in activated T cells (Fig. 1A and B). These results were corroborated by measuring the conversion of LC3-I into its PE-conjugated form, LC3-II, which also showed a significant increase in activated T cells (Fig. 1C and D). To determine if active degradation of autophagosomes occurred in these conditions, formation of LC3+ vacuoles and LC3 conjugation were also monitored in the presence of inhibitors of lysosomal proteolysis (NH4Cl and leupeptin), as comparison to untreated cells allows measuring autophagic flow (19). Cells that were stimulated in the presence of the inhibitors of lysosomal degradation showed significantly higher numbers of LC3+ vacuoles (Fig. 1A-B) and LC3-II (Fig. 1C-D) when compared to cells that were activated in the absence of inhibitors, supporting enhanced active degradation of autophagosome cargo in activated T cells. Furthermore, detection of LAMP1 in many LC3+ vacuoles indicated formation of autophagolysosomes (autophagosome/lysosome fusion) in activated T cells.
Figure 1. Macroautophagy is induced during activation in effector T helper cells.

A. Murine CD4+ were polarized to Th1 cells for 6 days and then stimulated with plate bound anti-CD3 and anti-CD28 in the presence or absence of 20 mM NH4Cl and 100 μM leupeptin (Leup). Immunofluorescence analysis was performed using antibodies against LC3 and LAMP-1. Arrows indicate the presence of LC3+ autophagic vacuoles. B. Quantification of the number of LC3 puncta per cell in resting (Rest) and stimulated (Stim) cells from 3 independent experiments (mean+SEM). C and D. Lysates from resting or plate bound anti-CD3 and anti-CD28 stimulated T cells in the presence or absence of NH4Cl and leupeptin were immunoblotted with antibodies against LC3. The blot is representative of 3 experiments that were quantified using Image J software. Results of the quantification of the levels of LC3-II relative to actin are shown in D as mean+SEM. All experiments shown in A-D were performed 4-6 h following stimulation. E. Th1 cells were labeled with [3H] Leucine for 48 h. Cells were then left resting or stimulated with either plate bound anti-CD3, anti-CD3+anti-CD28, or plate bound anti-CD3+anti-CD28+IL-2 (10 U/mL), and chased in medium containing an excess of unlabeled Leucine for 24hrs. Analysis of proteolysis rates (left) and levels of proteolysis after 24 h of stimulation (right) with or without costimulation are shown. Results are mean+SEM of calculated total levels of proteolysis from five to seven different experiments. *p<0.05; **p<0.01.
Activation of CD4+ T cells has been shown to require the engagement of two different signals: one provided by recognition of MHC-II/peptide complexes (signal 1) by the TCR; and a second signal that results from costimulatory receptors, such as CD28. In the absence of signal 2, T cells do not activate properly and enter into an anergic state, which renders them unresponsive to subsequent antigen encounters (24, 25). Signals through the IL-2 receptor can also lead to full activation and prevent the establishment of clonal anergy in cells activated in the absence of costimulation (26). Active degradation of autophagosome cargo should result in increased proteolysis in activated T cells. Therefore, to determine whether costimulation would enhance autophagic activity, we performed pulse and chase experiments to measure proteolysis of long half-life proteins. Levels of total protein degradation were compared in cells activated with anti-CD3, anti-CD3 and anti-CD28 or anti-CD3 and IL-2. Most of the proteolysis detected in these assays was mediated by lysosomal activity, as close to 80% inhibition was achieved using NH4Cl (Suppl. Fig. 1). These experiments showed that signals provided by CD28 or IL-2 receptor engagement induced a significant increase of the proteolytic activity in effector T cells (Fig. 1E). These results confirmed that the enhanced autophagosome formation and macroautophagy activation observed in stimulated T helper cells resulted in increased degradation of cargo through the autophagic pathway.
Blockade of macroautophagy inhibits T cell activation
Our previous results showed that stimulated T cells activated macroautophagy. In order to further clarify the role of macroautophagy in the regulation of T cell activity, we determined the effect of blocking macroautophagy in activation-induced cytokine production and proliferation. Leupeptin and NH4Cl were used to block lysosomal proteolysis and 3-MA, to inhibit autophagosome formation. Inhibition of autophagy resulted in a dramatic decrease in IL-2 and IFNγ production and profoundly impaired T cell proliferation (Fig. 2) in the absence of any increase in cell death (Suppl. Fig. 2).
Figure 2. Inhibition of macroautophagy inhibits T cell activation.

Mouse Th1 cells were stimulated with plate bound anti-CD3 and anti-CD28 in the presence or absence of 10 mM 3-MA or 20 mM NH4Cl and 100 μM leupeptin, and (A) IL-2 production, (B) IFNγ production and (C) cell proliferation (BrdU incorporation) measured after 24 h. Results are mean+SEM from three to four different experiments. * p<0.05; **p<0.01
In order to avoid possible off-target effects of using these inhibitors and to determine whether decreased cytokine production and proliferation were in fact attributable to macroautophagy blockade, we generated mice genetically deficient in macroautophagy in T cells Atg7F/F-Lck-Cre). As previously reported, total number of thymocytes and peripheral T cells were reduced in these mice. However, differences between control and Atg7-deficient mice were reduced as animals age, suggesting the possibility of compensatory mechanism being activated. For this reason, we only used 3-6 week old mice for our analysis (Suppl. Fig. 3 and data not shown). Naïve CD4+ T cells defective in the macroautophagy machinery also showed decreased IL-2 and IFNγ production and defective activation-induced cell proliferation in response stimulation (Fig. 3A-B). Annexin-V staining confirmed that this effect was not due to higher levels of activation-induced cell death, as we could not detect any significant increase in cell death in activated macroautophagy-deficient T cells (Suppl. Fig. 2). Low levels of activation-induced proliferation prevented us from differentiating polarized populations of Th1 cells in vitro. For that purpose we isolated CD4+ T cells from Atg7F/F-Cre-ER mice and differentiated them in vitro into Th1 cells. Deletion of the floxed Atg7 allele was achieved using 4-OH–tamoxifen (control T cells were also treated with this drug). As previously seen in our experiments using autophagy inhibitors, blocking autophagy through deletion of Atg7 resulted in decreased activation-induced cytokine production (Fig. 3C). This effect was not due to defective secretion, as real time PCR analysis revealed markedly decreased Il2 mRNA production in macroautophagy-deficient Th1 cells (Suppl. Fig. 4). Furthermore, the inhibition of T cell responses when autophagy was blocked did not appear to be caused by increased downregulation of the TCR, as levels of TCR surface expression were not reduced in Atg7-deficient T cells (Suppl. Fig. 5A). Similarly, CD28 signaling did not seem to be affected and T cell activation-induced AKT phosphorylation was maintained in cells where lysosomal proteolysis had been inhibited (Suppl. Fig. 5B). Since macroautophagy has been implicated in the control of levels of oxidized proteins in cells, and ROS are readily produced following T cell activation, we also determined if macroautophagy-control of ROS could be playing a role during T cell activation. We performed experiments in autophagy competent or inhibited Th1 cells that were stimulated in the presence or absence of the antioxidant N-acetylcysteine. We found that addition of antioxidants did not rescue the defect seen in IL-2 production in autophagy-compromised cells (Suppl. Fig. 5C). In fact, N-acetylcysteine appeared to decrease IL-2 production in our control cells, indicating that ROS production may be needed for T cell activation (27).
Figure 3. Genetic blockade of macroautophagy inhibits T cell activation.

A. Naïve CD4+ T cells from Atg7F/F-Lck-Cre mice or control mice were stimulated with plate bound anti-CD3 and anti-CD28 antibodies. IL-2 production was determined by ELISA (A), and proliferation was measured by BrdU incorporation (B). Results show the mean +SEM of three independent experiments. C. CD4+ T cells from Atg7F/F Cre-ER mice or control litter mates were polarized to Th1 cells and then treated with 2 μM 4-OH-tamoxifen for 48 h prior to stimulation with plate bound anti-CD3 and anti-CD28 antibodies. IL-2 production was determined by ELISA. Data is shown as mean+SEM of three independent experiments. *p<0.05; **p<0.01.
Macroautophagy activation is required to ensure sufficient energy production during T cell activation
T cell activation is a highly metabolic and dynamic process that requires a large amount of energy (16). The fact that blockade of macroautophagy was enough to inhibit T cell proliferation in activated cells, supports that activation of macroautophagy under these conditions is not merely required to isolate particular cytosolic components through autophagosome sequestration, but that breakdown of cargo –and likely reutilization of their constituents– is necessary. In fact, macroautophagy is not only involved in the renovation of the cell proteome and organelle homeostasis but it can also regulate the generation of energy through the utilization of the amino acids produced from the degradation of proteins in the lysosomes (3, 17). In order to test this hypothesis, we measured how ATP production might be affected by the inhibition of autophagy. T cells obtain ATP as a result of breakdown of glucose, amino acids and lipids during oxidative phosphorylation. During T cell activation there is a metabolic switch to ensure sufficient energy to promote proliferation and effector functions. This energetically demanding process leads to activation of glycolysis as a result of ligation of CD28 and up-regulation of the expression of the glucose transporter Glucose transporter 1 with subsequent increase in glucose uptake. However, T cells can still conserve their function under glucose restrictive conditions indicating that other mechanisms may also cooperate with glycolysis and contribute to support the increase in ATP consumption that occurs during T cell activation (28). Macroautophagy has been shown to play a critical role in energy homeostasis. In order to further clarify the role of autophagy in the regulation of energy homeostasis in T cells we measured ATP production in cells were autophagy had been inhibited. We found that the increase in ATP production induced by TCR+CD28 engagement was inhibited when autophagy was blocked (Fig. 4A and Suppl. Fig 6A). This defective production of ATP correlated with a decrease in the production of lactate and in fatty acid utilization and AMPK phosphorylation in cells treated with lysosomal inhibitors (Suppl. Fig. 6 and 7). Autophagy has been recently shown to contribute to mobilization of intracellular lipid stores (29) and, consequently, the lack of fatty acid utilization could also be in part due to the reduced lipolysis observed in cells with compromised autophagy. It is interesting to note that we also found that phosphorylation of the ribosomal protein S6 kinase S6K1, an mTOR substrate, was downregulated in these cells, suggesting the possibility that a decreased capability to recycle essential intracellular macromolecules through degradation might have induced inhibition of mTOR activity (Suppl. Fig. 7). Supplementation of our cultures with methyl pyruvate, a cell-permeable intermediate of glucose metabolism that has been shown to maintain the viability of nutrient-depleted cells with impaired autophagy (30), partially restored ATP production and IL-2 production in activated T cells with compromised macroautophagy (Fig. 4B-D). Overall these results support that T cells require functional macroautophagy to accommodate to the energetic requirements of activation.
Figure 4. Methyl-pyruvate restores activation-induced IL-2 production in autophagy-deficient T cells.
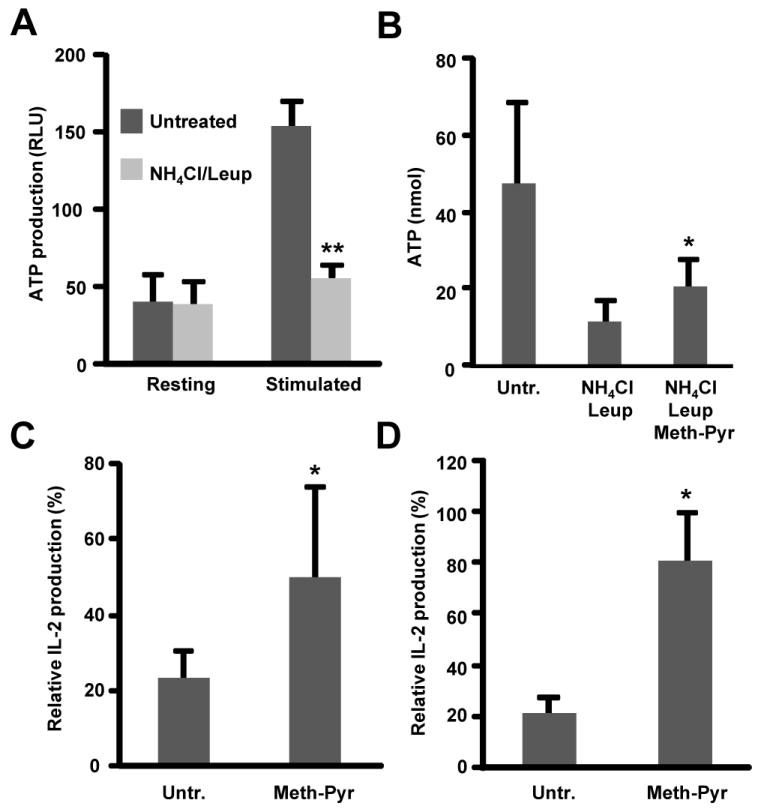
A and B. ATP levels were measured in lysates from resting or plate bound anti-CD3+anti-CD28 stimulated Th1 cells incubated in the presence or absence of NH4Cl and leupeptin (Leup) with or without (Untr.) 5 mM methyl-pyruvate (Meth-Pyr). Results show mean+SEM of three independent experiments. *p<0.05; **p<0.01. C. Th1 cells were stimulated with plate bound anti-CD3 and anti-CD28 in the presence or absence of NH4Cl and leupeptin. 24 h post stimulation, supernatants were collected and IL-2 expression was determined by ELISA. D. Th1 cells from Atg7F/F-Cre-ER mice or control litter mates were treated with 2 μM 4-OH-tamoxifen for 48 h and then stimulated with plate bound anti-CD3 and anti-CD28 antibodies in the presence or absence of 5 mM methyl-pyruvate. IL-2 production was determined by ELISA. In C and D, results are mean+SEM of three independent experiments and show relative IL-2 production in autophagy-deficient cells compared with control cells in untreated or methyl-pyruvate-treated cells. *p<0.05.
Upregulation of macroautophagy during T-cell activation results in differential sequestration of autophagosomal cargo
The presence of autophagosomal structures in T cells was confirmed by performing electron microscopy on CD4+ Th1 cells that were stimulated in the presence or absence of NH4Cl/leupeptin or vinblastine. Inhibiting lysosomal proteolysis prevents degradation of the cytosolic components sequestered inside the autophagic vesicles and allows for visualization of autophagosomal cargo. In addition, vinblastine depolymerizes microtubules and therefore prevents the delivery of autophagosomes to the lysosomal compartment (8). Quantification of the number of autophagic vacuoles in stimulated versus resting conditions revealed a marked increase in the number of autophagic vacuoles per cell and a decrease in their size upon T cell activation (Fig. 5A-D). Despite the smaller size of these autophagic vesicles in activated T cells, their higher abundance led to a net increase in the percentage of cellular area occupied by autophagic vesicles (Fig. 5E). Interestingly, analysis of the nature of the cargo found in the interior of autophagosomes in resting and activated T cells revealed a differential sequestration of cargo in both conditions (Fig. 5F). In basal conditions, numerous mitochondria were frequently enclosed in autophagosomes (examples shown in Fig. 5B, left). However, upon stimulation the cargo consisted almost exclusively of cytosolic material (see Fig. 5B, right). The lack of organelles inside autophagosome vesicles in stimulated T cells was not due to their rapid degradation inside this compartment, as both inhibition of lysosomal proteolysis or autophagosome/lysosomes fusion did not change the characteristics of these compartments. Lower mitochondria sequestration was not due to a decreased number of mitochondria in activated cells, as the relative cell content of these organelles did not decrease, but rather increased (Suppl. Fig. 8). These results were corroborated by analysis of colocalization of the lysosomal and mitochondrial compartments by immunofluorescence. A clear colocalization of specific tracking dyes for mitochondria and lysosomes indicated the association of these two cellular compartments in resting T cells (as evidence of mitochondria degradation in lysosomes), which did not occur in activated cells (Suppl. Fig. 9A). Furthermore, measurement of degradation of proteins in the cytosolic soluble fraction (organelle-free) using metabolic labeling showed increased degradation of the proteins in this fraction in activated T cells compared to resting cells or cells activated in the presence of inhibitors of lysosomal activity (Suppl. Fig. 9B). These results point to a specific regulation of cargo selection by macroautophagy that excludes mitochondria from degradation during T cell activation and favors instead sequestration of soluble cytosolic content.
Fig 5. Ultrastructural analysis of the autophagic compartments in resting and stimulated T cells.

Resting T Cells and cells stimulated for 4 h with plate bound anti-CD3 and anti-CD28 were processed for electron microscopy analysis. A. Lower magnification fields to show representative cells. Black arrows: autophagic vacuoles with distinguishable content; white arrows: autophagic vacuoles with electro lucent content or content of comparable density to the surrounding cytosol. B. Higher magnification fields to show individual autophagic vacuoles and their content. Bar: 1 μm C-F. Morphometric analysis was performed in 15-20 micrographies as the ones shown in panel A, corresponding to cells from two different experiments. The number of autophagic vacuoles (AVs) per field (C), vacuole average size (D), percentage of cellular area occupied by autophagic vacuoles (E) and percentage of vacuoles with soluble or particulate cargo (F) is shown. Values are mean+SEM. Differences between resting and stimulated cells are significant for ** p< 0.01 and *** p<0.001.
Discussion
Constant turnover of proteins is required to maintain cellular homeostasis. However, the proteolytic systems of the cell must also be able to respond to different stimuli and adapt the cell proteome to the requirements of new environmental conditions. Although macroautophagy was initially identified as a lysosomal-mediated degradative mechanism activated mainly in response to starvation, evidence has recently accumulated that has led to the identification of many other processes where macroautophagy plays an essential role (3-5). Here, we show that macroautophagy is activated in peripheral CD4+ T cells in response to the engagement of the TCR and costimulatory receptors. In activated T cells, exclusion of mitochondria and other organelles from autophagic vacuoles leads to the degradation of cargo different to the one present in basal autophagosomes. Furthermore, induction of macroautophagy is essential to support T cell activation. When macroautophagy is blocked, activated T cells show profound defects in their proliferative responses and their ability to secret cytokines. This effect is, at least in part, due to the defects in energy metabolisms caused by macroautophagy blockade.
Recently, macroautophagy has been reported to regulate growth factor withdrawal-induced cell death in a Th-2 cell line, as autophagy-deficient D10 cells became more resistant to apoptosis induced by culture in media without growth factors (12). Autophagy has also been suggested to control cell death induced by IFNγ signaling in T helper cells (31). Our results show that induction of macroautophagy during effector Th1 cell activation does not initiate or control activation-induced cell death, as levels of apoptosis in macroautophagy-deficient T cells following stimulation were similar to those found in control cells. It has been recently shown that in activated CD8+ T cells the extent of macroautophagy activation is controlled by the interaction of a complex formed by the Fas associated death domain protein and caspase 8 with Atg5, Atg12 and Atg16L, which prevents the induction of Receptor (TNFRSF)-interacting serine/threonine kinase 1-dependent necrosis (32). This fine regulation of macroautophagy in activated T cells would allow for a regulatory effect of macroautophagy in T cell activation independent of its possible ability to induce cell death in other conditions (12).
Whereas basal autophagy may likely contribute to T cell homeostasis and protein and organelle renewal (13), our results indicate that macroautophagy is activated in stimulated cells, where this proteolytic process plays a role different from basal cell homeostasis or death regulation. Pharmacological or genetic blockade of macroautophagy have been reported to preferentially affect activation-induced proliferation in CD8+ T cells (23). Our data reveals that macroautophagy also regulates proliferative responses on T helper cells. Furthermore, our results show that T helper cells activated in the presence of the inhibitors of lysosomal proteolysis NH4Cl and leupeptin, as well as Atg7-deficient T cells, also have a profound defect in their ability to produce effector cytokines. This impairment is due to deficient transcription and not to possible interference with cytokine secretion.
Macroautophagy has been shown to be important for the degradation of existing proteins to provide amino acids for synthesis of new proteins to guarantee survival during stress conditions. Activation of T cells is a highly demanding bioenergetic process. T cells require increased production of energy to sustain growth, proliferation and the de novo synthesis of effector molecules (16, 33). Increased glycolysis has been shown to play a central role in supporting this new demand of energy (34). Signals transmitted by engagement of CD28 and transduced through the activation of AKT lead not only to the upregulation of the expression for the glucose transporter Glucose transporter 1 and subsequent increase in glucose uptake, but also to an increase in glucose metabolism (35). However, to some extent, T cells still conserve their function even in environments with very low glucose, suggesting that other mechanisms may cooperate with glycolysis and contribute to support the increase bioenergetic consumption that occurs in T cells following activation (28). For instance, activation of β-oxidation by AMPK1 has been shown to overcome the dependence on glycolysis of some cell types (36). Our results show that at least one of the key functions that macroautophagy fulfils in T cells is to guarantee an adequate energy metabolism in activated effector T helper cells. This role of macroautophagy has been previously characterized in several cell types in response to starvation conditions, in which breakdown of non-essential cellular components provides a source of energy and macromolecules building blocks to ensure cell survival (30). Our results indicate that this function of macroautophagy is not limited to ensure survival during cell starvation but that it is also necessary in other processes that require a high demand of energy, such as T cell activation. Interestingly, the inability of anergic T cells to respond to re-stimulation has been shown to correlate with a failure to induce the mechanisms required to increase their metabolic activity (37). Furthermore, T cell activation in the presence of metabolic inhibition mimics activation in the absence of costimulation and renders the T cell anergic (37). Our results show that costimulation is required to induce autophagy-mediated increase in proteolysis in activated T cells, as anergizing stimulation with anti-CD3 in the absence of CD28 engagement fails to do so. This defect can also be reversed through signaling by the IL-2 receptor, which is also able to reverse the anergic phenotype (24-26, 38). Regulation of macroautophagy may therefore control the metabolic activity of T cells and contribute to determine the fate of T cells to be activated and proliferate or become anergic. The molecular mechanisms that may induce macroautophagy in activated T cells are still not clear. Recent work has implicated Jun kinases, which in T cells are fully activated in the presence of costimulatory signals, in the activation of macroautophagy (39-42). JNK1 can phosphorylate Bcl-2 favoring its dissociation from Beclin1, which is then able to activate autophagy (41). The expression of Beclin1 has also been reported to be upregulated in activated Jurkat cells by direct p65-mediated transcription (43). In any case, it is interesting to note that costimulation in T cells leads to the activation of the AKT/mTOR pathway, which has been extensively characterized as an inhibitor of macroautophagy activation by inhibiting the mammalian Atg13 complex (44, 45). Therefore, it is likely that macroautophagy in T cells may be activated through an mTOR-independent mechanisms. mTOR-independent activation of macroautophagy has recently been characterized in several systems in response to accumulation of cytosolic protein aggregates, and it is regulated by calcium/calpain or cAMP/Inositol trisphosphate signaling (46-48).
Basal macroautophagy in T cells has been recently shown to play an important role in T cell development by regulating the turnover of mitochondria (15, 49). Our results corroborate this observation, as we have seen that the cargo of basal autophagosomes in resting T cells is comprised mostly of cellular organelles with a high representation of mitochondria. However, in activated T cells the cargo that is found inside autophagic vacuoles is qualitatively different. Our data shows that most of the autophagosomes found in stimulated cells had a content of similar density as the cytosol, suggesting a selective degradation of soluble cytosolic components. The presence of mitochondria in those autophagosomes was drastically reduced when compared with resting cells. The need for increased ATP production and the important role of the mitochondria in the regulation of calcium signaling (50, 51) could explain why these organelles are spared from turnover during activation, and macroautophagy turns to the degradation of cytosolic soluble components to ensure a sufficient energetic and metabolic output. Although initially thought to be a regulated but non-discriminative process, evidence has mounted in the last several years that indicate that macroautophagy can also be a selective process (52). Degradation of mitochondria by mitophagy and selective degradation of highly ubiquitinated protein aggregates are two examples of this ability of macroautophagy to select cargo (53-56). In fact, several cargo recognizing molecules able to also interact with components of the autophagic machinery, such as p62 or NBR1, have been recently identified (57, 58). How this selectivity is established in activated T cells remains to be characterized. In this case, “selective exclusion” rather than selective recognition seems to take place. Previous studies have shown that changes in mitochondria fusion-fission properties determine their susceptibility for autophagic degradation (59). Although further studies are required, the fact that the average size of mitochondria in stimulated cells was larger than in resting cells may offer a mechanism to preserve these organelles from degradation during T cell stimulation. However, changes during T cell activation in the properties of the surface markers of highly functional mitochondria, in the intracellular location of these mitochondria, in the site of formation of autophagosomes or in the cytoskeleton network that contributes to bring these two compartments together, could also be behind the avoidance of mitochondria from macroautophagy degradation. In addition to amino acids, it is possible that macroautophagy degradation of cytoplasmic soluble regions may also contribute free fatty acids and glycogen as additional sources of energy, since break down of both lipid stores and glycogen deposits by macroautophagy have been described (29, 60).
We present evidence of a novel role for macroautophagy during T cell activation. Regulation of the energy needs of activated T cells is favored by the ability of autophagosomes to exclude form their cargo mitochondria, which are required for signal transduction and activation in response to TCR engagement. It still remains to be determined if macroautophagy may regulate other T cell function during activation, and if situations that result in vivo in compromised autophagy, such as aging or certain metabolic disorders, may also cause limited ability to recycle intracellular macromolecules that may lead to defective T cell responses.
Supplementary Material
Acknowledgments
We thank members of the Macian and Cuervo labs for helpful discussions and technical assistance.
Footnotes
This work was supported by National Institutes of health Grants P01AG031782 (F.M. and A.M.C.), F31AG035533 and T32CA009173 (V.M.H.) and K01DK087776 (R.S.).
Abbreviations used in this paper. 3-MA: 3-methyl adenine; AMPK: AMP-activated kinase; Atg: autophagy-related gene; LAMP-1: lysosomal associated membrane protein 1; LC3: microtubule-associated protein 1 light chain 3; mTOR: mammalian target of rapamycin; ROS: reactive oxygen species; S6K: ribosomal protein S6 kinase.
References
- 1.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12:1535–1541. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B, Cuervo A, Klionsky D. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuervo AM. Autophagy: many pathways to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 6.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 7.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 9.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231–236. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Kasai M, Tanida I, Ueno T, Kominami E, Seki S, Ikeda T, Mizuochi T. Autophagic compartments gain access to the MHC class II compartments in thymic epithelium. J Immunol. 2009;183:7278–7285. doi: 10.4049/jimmunol.0804087. [DOI] [PubMed] [Google Scholar]
- 11.Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, Mizushima NN, Iwasaki A, He YW, Swat W, Virgin HWt. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 12.Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, Jin S, Lu B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- 13.Pua HH, He YW. Maintaining T lymphocyte homeostasis: another duty of autophagy. Autophagy. 2007;3:266–267. doi: 10.4161/auto.3908. [DOI] [PubMed] [Google Scholar]
- 14.Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 16.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 17.Stipanuk MH. Macroautophagy and its role in nutrient homeostasis. Nutr Rev. 2009;67:677–689. doi: 10.1111/j.1753-4887.2009.00252.x. [DOI] [PubMed] [Google Scholar]
- 18.Auteri J, Okada A, Bochaki V, Dice J. Regulation of intracellular protein degradation in IMR- 90 human diploid fibroblasts. J Cell Physiol. 1983;115:159–166. doi: 10.1002/jcp.1041150210. [DOI] [PubMed] [Google Scholar]
- 19.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal Turnover, but Not a Cellular Level, of Endogenous LC3 is a Marker for Autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 20.Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997;272:5606–5615. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- 21.Dunn WA., Jr Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 23.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A Critical Role for the Autophagy Gene Atg5 in T Cell Survival and Proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macian F, Im SH, Garcia-Cozar FJ, Rao A. T-cell anergy. Curr Opin Immunol. 2004;16:209–216. doi: 10.1016/j.coi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. Epub 2001 Dec 2019. [DOI] [PubMed] [Google Scholar]
- 26.Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM, D’Andrea AD, Ritz J, Nadler LM. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science. 1994;266:1039–1042. doi: 10.1126/science.7973657. [DOI] [PubMed] [Google Scholar]
- 27.Williams MS, Kwon J. T cell receptor stimulation, reactive oxygen species, and cell signaling. Free Radic Biol Med. 2004;37:1144–1151. doi: 10.1016/j.freeradbiomed.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 28.Tripmacher R, Gaber T, Dziurla R, Haupl T, Erekul K, Grutzkau A, Tschirschmann M, Scheffold A, Radbruch A, Burmester GR, Buttgereit F. Human CD4(+) T cells maintain specific functions even under conditions of extremely restricted ATP production. Eur J Immunol. 2008;38:1631–1642. doi: 10.1002/eji.200738047. [DOI] [PubMed] [Google Scholar]
- 29.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 31.Feng CG, Zheng L, Jankovic D, Bafica A, Cannons JL, Watford WT, Chaussabel D, Hieny S, Caspar P, Schwartzberg PL, Lenardo MJ, Sher A. The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-gamma-induced cell death. Nat Immunol. 2008;9:1279–1287. doi: 10.1038/ni.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, Morrissette NS, Walsh CM. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA. 2008;105:16677–16682. doi: 10.1073/pnas.0808597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661–4665. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 34.Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem. 1994;269:31484–31490. [PubMed] [Google Scholar]
- 35.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 36.Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene. 2005;24:4165–4173. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 37.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol. 2009;183:6095–6101. doi: 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dure M, Macian F. IL-2 signaling prevents T cell anergy by inhibiting the expression of anergy-inducing genes. Mol Immunol. 2009;46:999–1006. doi: 10.1016/j.molimm.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy. 2010;6:153–154. doi: 10.4161/auto.6.1.10537. [DOI] [PubMed] [Google Scholar]
- 40.Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S, Auberger P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70:1042–1052. doi: 10.1158/0008-5472.CAN-09-3537. [DOI] [PubMed] [Google Scholar]
- 41.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–951. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29:2594–2608. doi: 10.1128/MCB.01396-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci USA. 2009;106:17049–17054. doi: 10.1073/pnas.0903316106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 47.Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 48.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O’Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, Graham DB, Mizushima NN, Xavier R, Virgin HW, Swat W. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009;5:625–635. doi: 10.4161/auto.5.5.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quintana A, Schwarz EC, Schwindling C, Lipp P, Kaestner L, Hoth M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem. 2006;281:40302–40309. doi: 10.1074/jbc.M607896200. [DOI] [PubMed] [Google Scholar]
- 51.Quintana A, Schwindling C, Wenning AS, Becherer U, Rettig J, Schwarz EC, Hoth M. T cell activation requires mitochondrial translocation to the immunological synapse. Proc Natl Acad Sci USA. 2007;104:14418–14423. doi: 10.1073/pnas.0703126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu L, Strandberg L, Lenardo MJ. The selectivity of autophagy and its role in cell death and survival. Autophagy. 2008;4:567–573. doi: 10.4161/auto.5902. [DOI] [PubMed] [Google Scholar]
- 53.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 54.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 55.Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem. 2007;282:5617–5624. doi: 10.1074/jbc.M605940200. [DOI] [PubMed] [Google Scholar]
- 56.Wong ES, Tan JM, Soong WE, Hussein K, Nukina N, Dawson VL, Dawson TM, Cuervo AM, Lim KL. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet. 2008;17:2570–2582. doi: 10.1093/hmg/ddn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 58.Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010;584:1379–1385. doi: 10.1016/j.febslet.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 59.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy in glucose homeostasis. Pathol Res Pract. 2006;202:631–638. doi: 10.1016/j.prp.2006.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.