

Abstract
OBJECTIVE
β-Cell turnover and its potential to permit β-cell regeneration in adult primates are unknown. Our aims were 1) to measure β-cell turnover in adult nonhuman primates; 2) to establish the relative contribution of β-cell replication and formation of new β-cells from other precursors (defined thus as β-cell neogenesis); and 3) to establish whether there is an adaptive increase in β-cell formation (attempted regeneration) in streptozotocin (STZ)-induced diabetes in adult nonhuman primates.
RESEARCH DESIGN AND METHODS
Adult (aged 7 years) vervet monkeys were administered STZ (45–55 mg/kg, n = 7) or saline (n = 9). Pancreas was obtained from each animal twice, first by open surgical biopsy and then by euthanasia. β-Cell turnover was evaluated by applying a mathematic model to measured replication and apoptosis rates.
RESULTS
β-Cell turnover is present in adult nonhuman primates (3.3 ± 0.9 mg/month), mostly (∼80%) derived from β-cell neogenesis. β-Cell formation was minimal in STZ-induced diabetes. Despite marked hyperglycemia, β-cell apoptosis was not increased in monkeys administered STZ.
CONCLUSIONS
There is ongoing β-cell turnover in adult nonhuman primates that cannot be accounted for by β-cell replication. There is no evidence of β-cell regeneration in monkeys administered STZ. Hyperglycemia does not induce β-cell apoptosis in nonhuman primates in vivo.
In health, the number of β-cells (collectively referred to as the “β-cell mass”) is sufficient to permit the required rate of basal and meal-stimulated insulin secretion to maintain glycemic control. Both type 1 and 2 diabetes are characterized by deficit of β-cell mass and insufficient insulin secretion (1–6). Type 1 and 2 diabetes can be effectively treated by restoring β-cell mass through pancreas transplantation (7,8). However, because of the limited availability of donor organs and the adverse effects of long-term immunosuppression, alternative strategies to reverse the deficit in β-cell mass in diabetes are required. One such strategy is to foster restoration of β-cells from putative endogenous sources.
Complete or partial reversal of diabetes by β-cell regeneration has been reported in rodents under several conditions. An acute deficit in β-cell mass induced by the β-cell toxin streptozotocin (STZ) in newborn rats is followed by recovery of β-cell mass (9). However, neither glucose intolerance nor diabetes induced by a partial pancreatectomy in adult humans or dogs resolves spontaneously (10–14). Likewise, diabetes due to a β-cell deficit induced by a β-cell toxin in nonhuman primates or pigs does not resolve spontaneously (15,16). The differences in the observed potential for β-cell regeneration in juvenile rodents and adult humans are likely due to the age of study subjects because adult rodents have also been shown to have a much reduced capacity for β-cell regeneration (17).
The current study sought to establish 1) whether β-cell turnover is present in adult nonhuman primates, and if so to quantify its rate; 2) whether the primary source of new β-cell formation in adult nonhuman primates is replication of existing β-cells or formation of new β-cells independently of β-cell replication (defined thus as β-cell neogenesis); and 3) whether there is any evidence of an adaptive increase in β-cell formation (regeneration) in an STZ model of type 1 diabetes in nonhuman primates.
RESEARCH DESIGN AND METHODS
Study design.
β-Cell turnover was quantified by a recently developed model (18). If β-cell mass and β-cell turnover are at nonsteady state, this model requires measurement of β-cell mass, β-cell replication, and β-cell apoptosis at multiple different time points. Alternatively if β-cell mass and β-cell turnover are at steady state, then a single time point is sufficient.
To ensure steady state, we obtained pancreas first by an open surgical biopsy and then subsequently at euthanasia from each study animal. The first surgical sample was obtained 1 month after administration of STZ (n = 7) or saline (n = 9), and the second euthanasia sample was obtained between 2 and 6 months later as shown in Fig. 1.
FIG. 1.

Study protocol. Male vervet monkeys were administered STZ (STZ group, n = 7) or saline (control group, n = 9). Pancreas open biopsy was conducted in all monkeys 1 month after STZ was given. The same monkeys were then euthanized 2 to 6 months after the treatment, and the whole pancreas was collected.
Study animals.
All procedures involving animals were conducted in compliance with state and federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by the Institutional Animal Care and Use Committee at Wake Forest University. Sixteen male vervet monkeys (Chlorocebus aethiops, 7.1 ± 0.4 years) were obtained from a multigenerational pedigreed colony at University of California Los Angeles Vervet Research Colony and transported to the animal facility of Wake Forest University. All monkeys were fed a standard chow diet (LabDiet 5037, Purina LabDiet, Richmond, IN) and acclimatized to place and staff before study. The animals were randomly divided into a control group (n = 9) and a group administered STZ (n = 7) (Table 1).
TABLE 1.
Baseline characteristics of the monkeys
| Control group | STZ group | P value | |
|---|---|---|---|
| n | 9 | 7 | |
| Age (years) | 7.0 ± 0.4 | 7.3 ± 0.9 | 0.7 |
| Body weight (kg) | 7.4 ± 0.3 | 7.0 ± 0.3 | 0.3 |
| Glucose (mg/dL)* | 65 ± 3 | 68 ± 5 | 0.7 |
| HbA1c (%) | 5.0 ± 0.1 | 5.1 ± 0.1 | 0.5 |
| Insulin (pmol/L)* | 229 ± 30 | 226 ± 66 | 0.9 |
| C-peptide (pmol/L)* | 346 ± 42 | 434 ± 152 | 0.5 |
| Total pancreas volume (cm3) | 6.9 ± 0.7 | 6.5 ± 0.6 | 0.6 |
| Pancreas parenchymal volume (cm3) | 5.4 ± 0.5 | 5.2 ± 0.5 | 0.8 |
*Overnight fasting.
Throughout the study period, body weight and an overnight-fasted blood sample were procured monthly after sedation with ketamine (10–15 mg/kg i.m., Ketaset, Fort Dodge Animal Health, Fort Dodge, IA). The blood sample was processed for measurement of plasma glucose, insulin and C-peptide concentrations, and HbA1c. Plasma glucose (Sigma-Aldrich, St. Louis, MO) was assessed by enzymatic colorimetric methods. The inter- and intra-assay coefficients of variation % are <5% for glucose. Insulin and C-peptide were determined using ELISA (Mercodia, Uppsala, Sweden), with inter- and intra-assay coefficients of variation <10%. HbA1c was measured by high-performance liquid chromatography (Primus PDQ, Primus Diagnostic, Kansas City, MO).
Induction of diabetes.
To induce diabetes, monkeys were given a single dose of STZ (45–55 mg/kg i.v., Zanosar, SICOR Pharmaceutics, Irvine, CA) under sedation with ketamine. Control animals were manipulated similarly and given similar injections (~0.3 mL) of saline rather than STZ. Plasma glucose was measured 24 and 48 h after the treatment. All monkeys given STZ developed fasting hyperglycemia (>144 mg/dL) within 48 h.
After induction of diabetes, postprandial glucose concentrations were maintained in the range of 300–400 mg/dL by twice daily insulin injections (70% intermediate-acting [e.g., NPH], 30% short-acting [e.g., regular] insulin, Novolin 70/30, Novo Nordisk USA, Princeton, NJ). The dose of insulin was adjusted according to biweekly postprandial whole blood glucose measurements obtained by a tail stick of nonsedated animals and measured by a glucometer. Daily exogenous insulin requirements in the STZ group were 15.6 ± 2.4 units (range 7–23 units). Control monkeys were treated with 0.2 mL of saline injected subcutaneously twice daily at the same time as diabetic monkeys received insulin.
Pancreas computed tomography scans.
To determine pancreas parenchymal volume, abdominal computed tomography (CT) scans were performed at baseline, 1 month after treatment (before pancreas biopsy), and before euthanasia. After induction of anesthesia (as above), an orogastric tube was placed and 20–50 mL of water was infused into the stomach to aid in outlining the pyloric region. CT scans were taken by a Toshiba 32 Scanner with a helical pitch of 27 (Toshiba, Tokyo, Japan). The parameters included 0.5 mm collimation, ∼120 kVp, and 300 mA based on a 7–8 kg monkey with an abdominal depth of 12 cm. A 180-degree scan field of view using small focus was set, and a whole monkey scout was taken in the supine position to ensure correct positioning. Scans were taken pre- and postcontrast (iopromide at 2 mL/kg injected at 2 mL/s into the saphenous vein via a temporary indwelling catheter). Scout scans were taken before contrast, with a 10–15 s delay from injection initiation before starting the postcontrast scan for capture of the later arterial phase at the level of the abdominal slices. A second scan was initiated ∼8 s from the completion of the first scan to repeat the images of the abdomen at 1 min, which represent delayed-enhancement postcontrast. The contiguous 0.5-mm axial images of delayed-enhancement postcontrast CT scans were transferred to a PC workstation and analyzed using Vitrea 2 software (version 3.5, Vital Images, Minnetonka, MN). All images were analyzed at a window level of 40 Hounsfield units (HU) and window width of 300 HU. Pancreas was identified and hand-outlined on the basis of the typical landmarks (splenic vein, superior mesenteric artery). Then the total and parenchymal pancreas area (cm2) on each image was computed as previously described (19). Pancreas volume per section was calculated as the product of each pancreas area and the CT section thickness. Total and parenchymal volumes of pancreas were computed by summing the volume from each section that included a piece of pancreas tissue.
Both the total and parenchymal pancreas volumes were comparable in controls and monkeys given STZ (total pancreas 6.9 ± 0.7 vs. 6.5 ± 0.6 cm3 controls vs. STZ; parenchymal pancreas 5.4 ± 0.5 vs. 5.2 ± 0.5 cm3 controls vs. STZ) (Table 1). The mean pancreas parenchymal volume of the three measurements in each monkey was used to calculate β-cell mass. The total and parenchymal pancreas volumes were ∼30 and ∼45% lower than the pancreas weight measured at euthanasia (6.7 ± 0.4, 5.3 ± 0.3 cm3, and 9.4 ± 0.5 g, respectively, assuming 1 cm3 = 1 g), consistent with findings in humans (19), most likely because of the impossibility of removing all fat from the pancreas by dissection (fat extends into interlobular areas). Nonetheless, the mean pancreas parenchymal volume in each monkey correlated with total pancreas weight (r2 = 0.82, P < 0.0001, Fig. 2).
FIG. 2.

A: There was no difference in pancreas parenchymal volume measured by CT scan between control (n = 9) and STZ (n = 7) groups at baseline, and no change in pancreas parenchymal volume was seen during the study. All 16 animals had CT scans to quantify pancreas parenchymal volume on all three occasions (for details, see research design and methods) (all P > 0.4). B: There was a good correlation between mean pancreas parenchymal volume and pancreas weight (r2 = 0.82, P < 0.0001), and the final pancreas parenchymal volume and pancreas weight (r2 = 0.5, P < 0.01).
Intravenous glucose tolerance test.
To assess β-cell function, an intravenous glucose tolerance test (IVGTT) was performed at baseline, 1 month after treatment (before pancreas biopsy), and immediately before euthanasia. Animals were sedated with ketamine (10–15 mg/kg i.m.) after an overnight fast. After baseline blood sampling, 1.5 mL/kg of 50% dextrose was administered via a butterfly catheter in a peripheral vein (saphenous vein commonly chosen). Blood was sampled at −5, 0, 5, 10, 20, 30, and 60 min for analysis of plasma glucose, insulin, and C-peptide concentrations in the baseline study, whereas the IVGTTs before biopsy and euthanasia were followed by 67 mg/kg of arginine infused over 30 s after the 30-min sample, and the sampling grid was −5, 0, 5, 10, 20, 25, 30, 31, 33, 35, 37, and 40 min.
β-Cell function and insulin sensitivity.
To assess β-cell function, the minimal model (20) was applied to IVGTT samples collected 0–30 min after glucose bolus injection, i.e., before the arginine stimulus. In detail, β-cell function was evaluated by applying the minimal model of C-peptide secretion and kinetics (21). Because no information is available on C-peptide kinetics in vervet monkeys, the parameters of C-peptide kinetics were fixed to values provided by Van Cauter et al. (22). Indexes of responsivity to glucose of first phase, Φ1 (10−9), second phase, Φ2 (min−1 10−9), and basal secretion, Φb (min−1 10−9) were quantified. To evaluate insulin sensitivity, SI (min−1 per pmol/L), and glucose effectiveness, SG (min−1), the glucose minimal model (23) was applied. Because glucose concentration exceeded the renal excretion threshold in monkeys given STZ, a glucose minimal model was integrated with the glucose kidney excretion model (24), with glomerular filtration rate, ke1 (min−1), and renal threshold of glucose, ke2 (mg/dL), fixed to human population values (i.e., ke1 = 0.0005 min−1 in controls and ke1 = 0.000676 min−1 in monkeys given STZ; ke2 = 180 mg/dL both in controls and in monkeys given STZ) (25).
Pancreas biopsy.
After an overnight fast, monkeys were sedated with ketamine (10–15 mg/kg i.m.) and atropine (0.04 mg/kg, AtroJect SA, IVX Animal Health Inc., St. Joseph, MO) and then intubated and anesthetized with isoflurane gas (IsoFlo, Abbott Laboratories, North Chicago, IL). The monkeys that received STZ were given regular insulin (Novolin R, Novo Nordisk USA) at anesthesia induction. A midline surgical approach was used starting at the umbilicus and extending cranially once visualization of the abdominal cavity was achieved. The pancreas was isolated, and a small section of the pancreas (0.44 ± 0.02 g) was removed from the caudal edge (distal from the vasculature). Omentum was advanced and sutured over the area to aid in rapid healing. The incision was closed in three layers (linea, subcutaneous, and skin), and monkeys were monitored postoperatively. Postoperative analgesia was provided. The biopsy sample was immediately fixed in neutral-buffered 10% formalin for 24 h at 4°C and then placed into 70% alcohol for another 24 h at 4°C before processing and embedding in paraffin.
Euthanasia and pancreas collection.
On the day of euthanasia and pancreas collection, insulin (or saline) treatment was administered as throughout the study. Blood samples were collected under ketamine sedation, and then animals were euthanized via overdose with intravenous pentobarbital. The pancreas was immediately harvested intact, fat was trimmed, and pancreas was weighed; then, 3- to 4-mm slices of the pancreas from tail to head (total 10 slices) were immediately fixed in neutral-buffered 10% formalin for 24 h at 4°C and placed into 70% alcohol for another 24 h at 4°C before processing and embedding in paraffin.
Immunohistochemistry, immunofluorescence, and morphometric analysis.
Sections of pancreas (4 μm) were stained as follows: for insulin (alkaline phosphatase staining) and hematoxylin for β-cell mass measurements (light microscopy); terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) and insulin for apoptosis assessment (immunofluorescence); Ki67 and insulin for assessment of proliferation (immunofluorescence); with the cocktail of antibodies for glucagon, somatostatin, and pancreatic polypeptide and insulin (immunofluorescence), and for GLUT-2 and insulin (immunofluorescence).
The following primary antibodies were used: guinea pig anti-insulin, 1:100 (Zymed Laboratories, San Francisco, CA); mouse antiglucagon, 1:500 (Dako, Carpenteria, CA); mouse anti-Ki67, 1:50 (Dako); mouse antiglucose transporter 2 (GLUT2), 1:50 (R&D Systems, Minneapolis, MN); rat antisomatostatin, 1:100 (Millipore, Temecula, CA); and goat antipancreatic polypeptide, 1:100 (Novus Biologicals, Littleton, CO). Secondary antibodies labeled with Cy3 and FITC were obtained from Jackson Laboratories (Jackson ImmunoResearch Laboratories, West Grove, PA) and used at dilutions at 1:200. For insulin alkaline phosphatase staining, Vecstatin ABC kit from Vector Laboratories (Burlingame, CA) was used. For the TUNEL staining, an in situ cell death detection kit (KIT AP; Roche Diagnostics, Indianapolis, IN) was used. Immunofluorescence-stained sections were coverslipped with Vectashield with DAPI (nuclear stain).
For determination of the fractional β-cell area, the entire pancreatic section stained with insulin and hematoxylin was imaged at 40× magnification (4× objective). For the ratio of the β-cell area, the pancreas parenchymal area was digitally quantified using Image Pro Plus software (Media Cybernetics, Silver Spring, MD) as previously described (2). β-Cell mass was determined as a product of the fractional pancreas parenchymal area (%) and pancreas parenchymal mass (g), assuming 1 cm3 of parenchymal volume is 1 g.
To measure individual β-cell size, 10 islets per case, stained for insulin and hematoxylin, were selected at random and imaged at 400× (40× objective). β-Cell size was determined as mean individual β-cell cross-sectional area. First, the diameter of each measured β-cell was determined. The distance from one edge of the β-cell nucleus, across that nucleus to the nearest edge of the adjacent β-cell nucleus, was determined using Image Pro Plus software. Next, a similar measurement was taken, but at 90 degrees to the first, and the mean of the two diameters was taken as the β-cell diameter for that individual β-cell. From this, the β-cell area was calculated using the formula Area = πr2, assuming a circular cross-sectional area. Twenty-five individual β-cell areas per monkey were calculated.
To determine β-cell replication, sections of pancreas were double-stained for insulin and Ki67 by immunofluorescence. At least 100 islets per section were examined, and the number of total β-cells and the number of β-cells labeled with Ki67 were counted. The number of replicating β-cells was expressed as a percentage of the total number of β-cells per case. In control monkeys, we analyzed one section per monkey, but because of the smaller number of β-cells in the STZ group, we analyzed five sections (∼500 islets) per case for euthanasia pancreas of the STZ group. A total of 28,025 β-cells in the control group and 8,488 β-cells in the STZ group were counted.
To determine the frequency of β-cell apoptosis, we double-stained sections for insulin and TUNEL by immunofluorescence and analyzed a minimum of 100 islets per section. The number of total β-cells and the number of TUNEL-labeled β-cells were quantified. Because of the low frequency of apoptosis, we quantified TUNEL in five sections per control case and 5–10 sections per STZ case. The number of apoptotic β-cells was expressed as a percentage of the total number of β-cells per case. A total of 127,570 β-cells in the control group and 19,634 β-cells in the STZ group were examined. The number of β-cells evaluated in each individual monkey is reported in Supplementary Table 1.
Calculations of β-cell turnover.
β-Cell turnover was established applying the model proposed recently (18). The model described β-cell mass as the balance between β-cell formation (either by β-cell replication or by other sources of β-cells) and loss (through β-cell apoptosis or necrosis):
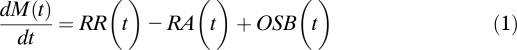 |
where dM(t)/dt [mg/month] is the mass derivative, i.e., rate of change in β-cell mass, M(t) [mg]; RR(t) [mg/month] and RA(t) [mg/month] are, respectively, rates of β-cell replication and apoptosis derived from frequencies of β-cell replication and apoptosis (fraction of replicated [apoptotic] β-cells) each multiplied by the respective conversion factors (26) and by β-cell mass; OSB(t) [mg/month] indicates the contribution of other sources of β-cells, i.e., from sources different from β-cell replication, to β-cell mass. Conversion factors for β-cell replication and apoptosis were assumed to be the same as in rats (26).
At steady state, i.e., dM(t)/dt = 0 and the rates are time-independent, that is, RR(t) = RR, RA(t) = RA, and OSB(t) = OSB, Eq. (1) becomes:
Statistical analysis.
Data are presented as mean ± SEM. Statistical comparisons were carried out using the Student t test with a P value < 0.05 taken as significant. A simple regression was carried out for the correlation analysis.
RESULTS
Induction of diabetes.
Before STZ treatment, fasting plasma glucose levels were comparable in control and STZ groups (65 ± 3 vs. 68 ± 5 mg/dL, control vs. STZ groups, P = 0.7, Table 1). Two days after STZ injection, all monkeys in the STZ group were hyperglycemic (257 ± 55 mg/dL), and they remained diabetic throughout the study (Fig. 3A). As anticipated, HbA1c was increased in the STZ versus control group (8.2 ± 0.3 vs. 4.9 ± 0.1%, 1 month after STZ or saline, P < 0.0001, Fig. 3B). Body weight remained comparable during the study in control and STZ groups (Fig. 3C).
FIG. 3.

Fasting plasma glucose (A), HbA1c levels (B), and body weight (C) during the study. One month after the treatment, all monkeys in the STZ group developed severe diabetes, and this marked hyperglycemia remained during the study (A and B). There was no significant change in body weight during the study in both control and STZ groups (C). *P < 0.01 vs. control.
IVGTT data.
Glucose, insulin, and C-peptide concentrations during baseline IVGTT and IVGTT plus arginine before biopsy and euthanasia are shown in Fig. 4 and Table 2. At baseline IVGTT, glucose levels were similar in the two groups, but, as expected, they were significantly higher (P < 0.01) in monkeys given STZ during the two following IVGTTs (Fig. 4A). Insulin and C-peptide concentrations were comparable in controls and monkeys given STZ at baseline, whereas they were almost zero after STZ treatment even after the arginine infusion (Fig. 4B and C), thus preventing the use of minimal models to quantify β-cell function and insulin sensitivity in monkeys given STZ.
FIG. 4.

Plasma glucose (A), insulin (B), and C-peptide (C) concentrations during IVGTT followed by arginine (t = 30 min) 1 month after STZ or saline treatment. The marked deficit in insulin secretion in response to glucose and arginine after STZ is consistent with the ∼90% deficit in β-cell mass induced by STZ (Fig. 5).
TABLE 2.
Responsivity to glucose of first phase (Φ1), second phase (Φ2), basal insulin secretion (Φb), insulin sensitivity (SI), and glucose effectiveness (SG) in controls and monkeys administered STZ at baseline, biopsy, and euthanasia studies
| Control group |
STZ group |
|||||
|---|---|---|---|---|---|---|
| Baseline | Biopsy | Euthanasia | Baseline | Biopsy | Euthanasia | |
| Φ1 [109] | 125 ± 29 | 289 ± 66+ | 145 ± 43 | 107 ± 30 | 0 ± 0*+ | 0 ± 0*† |
| Φ2 [109 min−1] | 8 ± 3 | 9 ± 4 | 4 ± 3 | 7 ± 6 | 0 ± 0 | 0 ± 0 |
| Φb [109 min−1] | 6.4 ± 0.9 | 9.3 ± 1.9 | 5.2 ± 1.1¶ | 8.4 ± 2.3 | 0.8 ± 0.2*+ | 0.8 ± 0.3*† |
| SI [10−4 L pmol−1 min−1] | 1.4 ± 0.2 | 1.6 ± 0.4 | 1.3 ± 0.5 | 1.9 ± 0.6 | 0 ± 0*+ | 0 ± 0*† |
| SG [min−1] | 2.5 ± 0.2 | 2.5 ± 0.3 | 2.2 ± 0.2 | 2.7 ± 0.4 | 2.7 ± 0.2 | 2.7 ± 0.3 |
*Control vs. STZ, P < 0.05.
+Baseline vs. biopsy, P < 0.05.
†Baseline vs. euthanasia, P < 0.05.
¶Biopsy vs. euthanasia, P < 0.05.
β-Cell function and insulin sensitivity.
Indices of β-cell function and insulin sensitivity are shown in Table 2. During baseline IVGTT, parameters were comparable in controls and monkeys given STZ. After STZ treatment, the fasting insulin and C-peptide concentrations were at or near the limit of detection so that Φ1, Φ2, and SI were not distinguishable from zero.
β-Cell mass.
β-Cell mass remained comparable in the control group during the 5-month period of observation (93 ± 17 vs. 89 ± 9 mg, biopsy vs. euthanasia, Fig. 5). As expected, the STZ group had a marked (∼90%) deficit in β-cell mass 1 month after STZ (10 ± 5 vs. 93 ± 17 mg, STZ vs. control, P < 0.001), the deficit being stable during the subsequent 5 months (Fig. 5). No difference was found between the mean individual β-cell size in controls versus monkeys given STZ (108 ± 2 vs. 101 ± 5 μm2, P = ns). There was also no apparent difference in the distribution of small islets around ducts or scattered β-cells in the monkeys given STZ compared with controls. The residual β-cells in the islets were found mainly in relatively β-cell–depleted islets rather than as increased numbers of small aggregates.
FIG. 5.

A: Islet morphology of control (a–d) and STZ (e–h) groups. As expected, there was no change in islet morphology between biopsy and euthanasia pancreas in control group. In contrast, there was a marked reduction in the number of β-cells per islet in STZ group at biopsy and euthanasia. B: β-Cell mass during the study. The β-cell mass was ∼90% decreased at biopsy (1 month after the treatment) in the STZ group compared with that of the control group. This decreased β-cell mass did not recover during the study. Glu, glucagon; Ins, insulin. (A high-quality digital representation of this figure is available in the online issue.)
β-Cell replication and apoptosis.
Consistent with adult humans, the frequency of detected β-cell replication was very low in control monkeys (0.050 ± 0.023%) (Fig. 6A). There was no detected increase in β-cell replication in response to the β-cell deficit induced by STZ during the sampling interval (0.050 ± 0.023 and 0.013 ± 0.008%, P = 0.2 control vs. STZ at euthanasia) (Fig. 6A). Also consistent with adult humans, the frequency of β-cell apoptosis was very low in the control monkeys (0.013 ± 0.003%). The frequency of β-cell apoptosis did not appear to be influenced by the glucose concentrations of ∼300–400 mg/dL in the monkeys given STZ because we did not find any TUNEL-positive β-cells in the pancreas of monkeys given STZ.
FIG. 6.

Frequency of β-cell replication (A) and apoptosis (B) in control and STZ groups. Despite the marked hyperglycemia, no increase in frequency of β-cell apoptosis was observed in the STZ group compared with the control group (B). GLUT2 staining did not reveal degranulated β-cells (i.e., insulin-negative GLUT2-positive islet cells); however, in the STZ group, GLUT2 immunoreactivity was decreased in cell membranes and increased in cytoplasm of β-cells (C). When examining islets for insulin immunoreactivity (green) and glucagon, somatostatin and pancreatic polypeptide combined (red), all endocrine cells in islets in both controls and monkeys given STZ were accounted for (D). (A high-quality digital representation of this figure is available in the online issue.)
β-Cell turnover.
Having established that β-cell mass, replication, and apoptosis were at steady state over the period of observation, we applied the model to compute β-cell turnover to the euthanasia samples where the sample size was much larger than from the biopsy sample. This analysis revealed ongoing β-cell turnover in adult nonhuman primates (3.3 ± 0.9 mg/month, Fig. 7).
FIG. 7.

β-Cell turnover in control group, showing the relative contributions of β-cell replication, formation of β-cells from other sources than replication (neogenesis), and β-cell apoptosis. In controls there was a minimal change in β-cell mass during the study, but the mathematic model revealed active β-cell turnover with the major contribution being from neogenesis rather than β-cell replication. In the absence of measurable apoptosis in monkeys given STZ, β-cell turnover could not be reliably computed, but given the absence of any change in β-cell mass, β-cell turnover can reasonably be assumed to not be adaptively increased to a clinically meaningful extent.
In control monkeys, the measured rate of β-cell apoptosis (3.3 ± 0.9 mg/month) exceeded the measured rate of β-cell replication (0.7 ± 0.3 mg/month), implying that for steady-state β-cell mass to prevail, the majority of newly forming β-cells are derived independently of β-cell replication, i.e., from β-cell neogenesis (2.6 ± 0.8 mg/month) (Fig. 7A). In a similar manner, we also assessed β-cell turnover by adapting the model to compute the turnover of β-cell number rather than mass. The rates of β-cell apoptosis and replication were (3.9 ± 1.0) · 106 and (0.9 ± 0.4) · 106 β-cells per month, with a computed rate of neogenesis of (3.0 ± 1.0) · 106 β-cells per month. By use of this approach, the computed fractional rate of β-cell turnover in control monkeys was ∼4% β-cells per month.
Given the low frequency of β-cell apoptosis events in monkeys administered STZ, we were unable to securely use the model in this group, but simply note the lack of an adaptive increase in β-cell mass or β-cell replication (vide supra) implying lack of a meaningful adaptive increase in β-cell regeneration.
GLUT2 in β-cells and other endocrine cell types.
Because we found no evidence of hyperglycemia-induced β-cell apoptosis in monkeys given STZ, the extent and distribution of GLUT2 protein in pancreatic sections of STZ and control pancreas were evaluated. As expected, in control samples GLUT2 immunoreactivity was abundant in the membrane of β-cells (Fig. 6C). In contrast, GLUT2 immunoreactivity was decreased in membranes of β-cells in monkeys given STZ (Fig. 6C). We did not identify islet cells expressing GLUT2 that did not stain for insulin in either control or STZ pancreas, suggesting that degranulated β-cells were not present in detectable numbers in either controls or monkeys given STZ. This conclusion was supported by immunostaining for insulin and a mixture of antibodies against glucagon, somatostatin, and pancreatic polypeptide that collectively accounted for the remaining endocrine cells in the islets of monkeys given STZ (Fig. 6D).
DISCUSSION
We report that β-cell turnover continues in adult nonhuman primates, with newly formed β-cells derived primarily from sources of β-cells independent of duplication of existing β-cells, defined as such here as β-cell neogenesis. However, we found no evidence of increased β-cell turnover in an STZ model of type 1 diabetes.
Measurement of β-cell turnover by a model-based approach was used in this study in nonhuman primates because lineage tracing is not readily accomplished in primates. The attributes and limitations of the model have been discussed in detail (18). Conversion of the frequency of β-cell replication and apoptosis to the respective rates of replication and apoptosis was accomplished by conversion factors developed by a combination of in vivo time lapse imaging and subsequent immunohistochemistry (26). These conversion factors were developed in rat islets and to the extent that they differ in nonhuman primates, the current study may be in error. Given the low frequency of both β-cell apoptosis and replication, relatively large errors in quantification of β-cell turnover may arise unless a substantial number of cells are evaluated. To this end, we quantified β-cell replication and apoptosis in ∼36,500 and ∼147,000 β-cells, respectively.
We define β-cell neogenesis in this study as β-cells arising from sources other than replication detected by Ki67 in insulin-positive β-cells. One possible source of β-cell neogenesis using this definition would be duplication of degranulated β-cells that were not detected by insulin immunostaining. Also, if β-cells dedifferentiated and lost insulin immunoreactivity before duplication, by this definition these cells would be considered as arising from neogenesis. In the absence of lineage tracing in nonhuman primates, we cannot fully exclude either of these possible sources of β-cells. However, we were able to account for the endocrine cells in monkeys given STZ by immunostaining for a combination of insulin, glucagon, somatostatin, and pancreatic polypeptide. Although some β-cells did have less intense immunofluorescence for insulin in monkeys given STZ versus controls consistent with some degree of insulin degranulation, they were still detectable. A similar pattern is present in patients with type 1 diabetes who have β-cells still present, whereas complete loss of insulin immunoreactivity has been reported in apparent β-cells in some rodent models of diabetes (27).
Ideally to model changes in β-cell turnover, multiple measurements of β-cell mass and turnover would be acquired in the same individuals at different time points. Because it is currently not possible to quantify β-cell mass or β-cell turnover noninvasively, we elected to obtain two samples of pancreas from the same individual on two occasions, first by surgical open biopsy and the second at euthanasia. The time between the first and second samples was varied between individuals as illustrated in Fig. 1 to permit a range of measures of β-cell mass and turnover after acute induction of a deficit in β-cell mass by STZ.
As expected, in the control group, there was no change in β-cell mass during the study period. Also the frequency of β-cell replication (by Ki67) and apoptosis (by TUNEL) were comparable in control pancreas secured by the samples at various time points during the study, implying steady-state β-cell turnover. Given the greater sample size available when the pancreas was resected at euthanasia, we focused on this sample to evaluate β-cell turnover. The results of that analysis imply that β-cell mass is renewed and sustained by ongoing β-cell turnover in nonhuman primates, with a fractional β-cell turnover of ∼4% per month, implying a β-cell average life span of ∼3 years. Consistent with recent data in the adult rat using the same model (18), the majority of new β-cell formation in adult nonhuman primates is derived independently of replication of existing β-cells, defined as such in this article as β-cell neogenesis. However, the extent of ongoing β-cell turnover in nonhuman primates quantified here is much less than found in the rat. Recent lineage tracing data implying β-cell neogenesis from ductal precursors in mice are supported by present findings (28). After documenting and quantifying β-cell turnover in adult nonhuman primates, our secondary goal was to establish whether β-cell turnover is sustained in an STZ model of type 1 diabetes. Moreover, we sought to establish whether there was any evidence of an adaptive increase in β-cell formation in response to β-cell deficiency and diabetes in this model.
The deficit in β-cell mass in the monkeys given STZ was comparable to that in established type 1 diabetes in humans (1,3). To accomplish effective β-cell regeneration in the STZ nonhuman primate model, β-cell formation would need to be adaptively increased to a rate that exceeds loss of β-cells through normal aging. However, there was no increase in β-cell mass or amelioration of diabetes in the adult nonhuman primates during the 6 months after induction of diabetes. There was no increase in β-cell replication in monkeys given STZ compared with the control monkeys. Given the absence of a recordable frequency of β-cell apoptosis in monkeys given STZ, we were not able to reliably compute β-cell turnover, and therefore neogenesis, under these conditions. However, given the lack of an increment in β-cell mass in monkeys studied twice over time after STZ, and the rarity of β-cell apoptosis, it can be reasonably surmised that any adaptive increase in β-cell neogenesis in response to diabetes in this model was trivial.
These data are consistent with available data in humans with established type 1 diabetes in whom a low number of β-cells are often still detected, with minimal β-cell replication and a detectable but low frequency of β-cell apoptosis (3,29). We previously proposed that the latter implies ongoing β-cell formation, but presumably at a rate that is insufficient to accomplish β-cell regeneration (a net increase in β-cell mass), a conclusion that has recently gained independent support (29). In either type 1 diabetes or the nonhuman primate administered STZ, effective β-cell regeneration would require formation of new β-cells at a rate that exceeds natural attrition. It is unclear to what extent β-cell neogenesis can adaptively increase, and if so how it is regulated. In a rat model of type 2 diabetes, we observed a marked increase in β-cell neogenesis that delayed loss of β-cell mass in response to a shortened β-cell life span through increased apoptosis (18). The lack of an adaptive increased rate of β-cell neogenesis in nonhuman primates compared with rats might be a species difference. Another possibility is that STZ induces toxicity in the precursor cells that give rise to β-cell neogenesis. Alternatively, the high glucose values present in this study may have constrained effective new β-cell formation. In a prior study of individuals with long-standing type 1 diabetes, we did observe more residual β-cells in those individuals with better glycemic control before death, although it is plausible that the better control was due to residual insulin secretion (3). Notably, in the current study we deliberately included adult monkeys, whereas most studies of β-cell regeneration have been undertaken in juvenile rodents with a much higher capacity for new β-cell formation (19).
The absence of increased β-cell apoptosis in the nonhuman primate administered STZ exposed to glucose values of ∼300–400 mg/dL for up to 6 months is in contrast with the reported actions of high glucose concentrations to induce increased β-cell apoptosis (30). However, most studies that report actions of high glucose concentrations to induce β-cell apoptosis have been undertaken in animal models with other mechanisms of β-cell toxicity present or in isolated islets that have a high rate of β-cell apoptosis consequent upon anoxia. In the current study, we noted that plasma membrane localization of GLUT2 transporter protein in β-cells of STZ nonhuman primates was decreased, consistent with prior reports in rodents with diabetes (31–33). Thus, it is likely that β-cells adaptively protect themselves from long-term glucose toxicity by decreasing access of glucose to the β-cell. Of course, this in turn would likely compound impaired β-cell function (34,35). In a recent study that examined the actions of comparable glucose concentrations on the islet in vivo in rats, there was no increase in β-cell apoptosis, but rather an adaptive increase in β-cell replication and mass in response to hyperglycemia (36). Collectively these studies imply that in vivo glucotoxicity may act more through its actions to impair β-cell function than induce β-cell apoptosis.
In conclusion, we report that β-cell turnover is present in adult nonhuman primates with the predominant source of new β-cells being derived from a source independent of duplication of existing differentiated β-cells. The rate of β-cell turnover in adult nonhuman primates is much slower than in rodents, consistent with the reported long life span of β-cells in humans (37,38). There was no measurable adaptive increase in β-cell mass or formation in STZ-induced diabetes in nonhuman primates, again in contrast with the regeneration reported in young rodents. The failure of β-cell regeneration after a single dose of STZ in nonhuman primates mirrors that in humans with established type 1 diabetes. The STZ monkey model may represent a more clinically relevant model to test strategies to foster β-cell regeneration in type 1 diabetes than rodents administered STZ. Given the possible importance of ongoing local cytokines on fostering stem cell recruitment, ideally a model with spontaneous autoimmune-mediated β-cell destruction in a primate would be available for such investigations.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by funding from the National Institutes of Health (DK-059579, DK-077967), the Juvenile Diabetes Research Foundation (7-2005-1152), the Larry L. Hillblom Foundation, and the Manpei Suzuki Diabetes Foundation.
No potential conflicts of interest relevant to this article were reported.
Y.S., E.M., and A.E.B. researched data, contributed to discussion, and reviewed and edited the article. R.G. researched data. K.K. and M.F. researched data, contributed to discussion, and reviewed and edited the article. L.Z. and P.C. researched data. T.G. and G.M.T. researched data, contributed to discussion, and reviewed and edited the article. C.C. contributed to discussion and reviewed and edited the article. J.D.W. researched data, contributed to discussion, and reviewed and edited the article. P.C.B. contributed to discussion, wrote the article, and reviewed and edited the article.
The authors thank their colleagues in the Larry Hillblom Islet Research Center for excellent suggestions. The authors acknowledge Inderroop Singh and David Kirakossian, Larry Hillblom Islet Research Center at UCLA, for assistance, and Bonnie Lui, Larry Hillblom Islet Research Center at UCLA, for administrative assistance.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db09-1368/-/DC1.
REFERENCES
- 1.Butler AE, Galasso R, Meier JJ, Basu R, Rizza RA, Butler PC. Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia 2007;50:2323–2331 [DOI] [PubMed] [Google Scholar]
- 2.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102–110 [DOI] [PubMed] [Google Scholar]
- 3.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 2005;48:2221–2228 [DOI] [PubMed] [Google Scholar]
- 4.Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care 2006;29:717–718 [DOI] [PubMed] [Google Scholar]
- 5.Meier JJ, Butler PC. Insulin Secretion. Philadelphia, Elsevier Saunders, 2005 [Google Scholar]
- 6.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009;58:773–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dean PG, Kudva YC, Stegall MD. Long-term benefits of pancreas transplantation. Curr Opin Organ Transplant 2008;13:85–90 [DOI] [PubMed] [Google Scholar]
- 8.Nath DS, Gruessner AC, Kandaswamy R, Gruessner RW, Sutherland DE, Humar A. Outcomes of pancreas transplants for patients with type 2 diabetes mellitus. Clin Transplant 2005;19:792–797 [DOI] [PubMed] [Google Scholar]
- 9.Wang RN, Bouwens L, Klöppel G. Beta-cell proliferation in normal and streptozotocin-treated newborn rats: site, dynamics and capacity. Diabetologia 1994;37:1088–1096 [DOI] [PubMed] [Google Scholar]
- 10.Matveyenko AV, Veldhuis JD, Butler PC. Mechanisms of impaired fasting glucose and glucose intolerance induced by an approximate 50% pancreatectomy. Diabetes 2006;55:2347–2356 [DOI] [PubMed] [Google Scholar]
- 11.Kendall DM, Sutherland DE, Najarian JS, Goetz FC, Robertson RP. Effects of hemipancreatectomy on insulin secretion and glucose tolerance in healthy humans. N Engl J Med 1990;322:898–903 [DOI] [PubMed] [Google Scholar]
- 12.Kumar AF, Gruessner RW, Seaquist ER. Risk of glucose intolerance and diabetes in hemipancreatectomized donors selected for normal preoperative glucose metabolism. Diabetes Care 2008;31:1639–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menge BA, Tannapfel A, Belyaev O, et al. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes 2008;57:142–149 [DOI] [PubMed] [Google Scholar]
- 14.Menge BA, Schrader H, Breuer TG, et al. Metabolic consequences of a 50% partial pancreatectomy in humans. Diabetologia 2009;52:306–317 [DOI] [PubMed] [Google Scholar]
- 15.Kjems LL, Kirby BM, Welsh EM, et al. Decrease in beta-cell mass leads to impaired pulsatile insulin secretion, reduced postprandial hepatic insulin clearance, and relative hyperglucagonemia in the minipig. Diabetes 2001;50:2001–2012 [DOI] [PubMed] [Google Scholar]
- 16.Litwak KN, Cefalu WT, Wagner JD. Streptozotocin-induced diabetes mellitus in cynomolgus monkeys: changes in carbohydrate metabolism, skin glycation, and pancreatic islets. Lab Anim Sci 1998;48:172–178 [PubMed] [Google Scholar]
- 17.Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009;58:1312–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manesso E, Toffolo GM, Saisho Y, et al. Dynamics of beta-cell turnover: evidence for beta-cell turnover and regeneration from sources of beta-cells other than beta-cell replication in the HIP rat. Am J Physiol Endocrinol Metab 2009;297:E323–E330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saisho Y, Butler AE, Meier JJ, et al. Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin Anat 2007;20:933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cobelli C, Toffolo GM, Dalla Man C, et al. Assessment of beta-cell function in humans, simultaneously with insulin sensitivity and hepatic extraction, from intravenous and oral glucose tests. Am J Physiol Endocrinol Metab 2007;293:E1–E15 [DOI] [PubMed] [Google Scholar]
- 21.Toffolo G, De Grandi F, Cobelli C. Estimation of beta-cell sensitivity from intravenous glucose tolerance test C-peptide data. Knowledge of the kinetics avoids errors in modeling the secretion. Diabetes 1995;44:845–854 [DOI] [PubMed] [Google Scholar]
- 22.Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992;41:368–377 [DOI] [PubMed] [Google Scholar]
- 23.Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol 1979;236:E667–E677 [DOI] [PubMed] [Google Scholar]
- 24.Dalla Man C, Rizza RA, Cobelli C. Meal simulation model of the glucose-insulin system. IEEE Trans Biomed Eng 2007;54:1740–1749 [DOI] [PubMed] [Google Scholar]
- 25.Johansen K, Svendsen PA, Lørup B. Variations in renal threshold for glucose in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1984;26:180–182 [DOI] [PubMed] [Google Scholar]
- 26.Saisho Y, Manesso E, Gurlo T, et al. Development of factors to convert frequency to rate for beta-cell replication and apoptosis quantified by time-lapse video microscopy and immunohistochemistry. Am J Physiol Endocrinol Metab 2009;296:E89–E96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes 2006;55:3238–3245 [DOI] [PubMed] [Google Scholar]
- 28.Inada A, Nienaber C, Katsuta H, et al. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A 2008;105:19915–19919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keenan HA, Sun JK, Levine J, et al. Residual insulin production and pancreatic ß-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010;59:2846–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 2008;29:351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jörns A, Tiedge M, Ziv E, Shafrir E, Lenzen S. Gradual loss of pancreatic beta-cell insulin, glucokinase and GLUT2 glucose transporter immunoreactivities during the time course of nutritionally induced type-2 diabetes in Psammomys obesus (sand rat). Virchows Arch 2002;440:63–69 [DOI] [PubMed] [Google Scholar]
- 32.Jörns A, Klempnauer J, Tiedge M, Lenzen S. Recovery of pancreatic beta cells in response to long-term normoglycemia after pancreas or islet transplantation in severely streptozotocin diabetic adult rats. Pancreas 2001;23:186–196 [DOI] [PubMed] [Google Scholar]
- 33.Frese T, Bazwinsky I, Mühlbauer E, Peschke E. Circadian and age-dependent expression patterns of GLUT2 and glucokinase in the pancreatic beta-cell of diabetic and nondiabetic rats. Horm Metab Res 2007;39:567–574 [DOI] [PubMed] [Google Scholar]
- 34.Thorens B, Guillam MT, Beermann F, Burcelin R, Jaquet M. Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J Biol Chem 2000;275:23751–23758 [DOI] [PubMed] [Google Scholar]
- 35.McCulloch DK, Koerker DJ, Kahn SE, Bonner-Weir S, Palmer JP. Correlations of in vivo beta-cell function tests with beta-cell mass and pancreatic insulin content in streptozocin-administered baboons. Diabetes 1991;40:673–679 [DOI] [PubMed] [Google Scholar]
- 36.Fontés G, Zarrouki B, Hagman DK, et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia 2010;53:2369–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cnop M, Hughes SJ, Igoillo-Esteve M, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2010;53:321–330 [DOI] [PubMed] [Google Scholar]
- 38.Perl S, Kushner JA, Buchholz BA, et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 2010;95:E234–E239 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.