

Abstract
Tandem affinity purification (TAP) is a generic approach for the purification of protein complexes. The key advantage of TAP is the engineering of dual affinity tags that, when attached to the protein of interest, allow purification of the target protein along with its binding partners through two consecutive purification steps. The tandem tag used in the original method consists of two IgG-binding units of protein A from Staphylococcus aureus (ProtA) and the calmodulin-binding peptide (CBP), and it allows for recovery of 20–30% of the bait protein in yeast. When applied to higher eukaryotes, however, this classical TAP tag suffers from low yields. To improve protein recovery in systems other than yeast, we describe herein the development of a three-tag system comprised of CBP, streptavidin-binding peptide (SBP) and hexa-histidine. We illustrate the application of this approach for the purification of human Bruton's tyrosine kinase (Btk), which results in highly efficient binding and elution of bait protein in both purification steps (>50% recovery). Combined with mass spectrometry for protein identification, this TAP strategy facilitated the first nonbiased analysis of Btk interacting proteins. The high efficiency of the SBP-His6 purification allows for efficient recovery of protein complexes formed with a target protein of interest from a small amount of starting material, enhancing the ability to detect low abundance and transient interactions in eukaryotic cell systems.
Keywords: tandem affinity purification, protein complex, calmodulin-binding peptide, streptavidin-binding peptide, His-tag, Btk
Introduction
Tandem affinity purification (TAP) is a methodology for the isolation of protein complexes from endogenous sources.1–3 The key advantage of this method involves tagging the protein of interest with a dual affinity handle that allows the protein-of-interest, along with its interacting partners, to be purified in two consecutive steps. The major benefit in comparison with single-step purification is the greatly improved specificity of the pull-down, leading to greater sample purity, greater reproducibility, and in turn simplifying the subsequent identification (and validation) of proteins interacting with the target. Whereas the method was originally developed in yeast, it has been successfully adapted to other cells and organisms.4–7 In combination with mass spectrometry for protein identification, TAP has become an indispensable tool for systematic identification of target-associated protein complexes,8 which have been exploited on a large scale to map the yeast interactome.9
The original TAP tag consists of two IgG-binding units of protein A of Staphylococcus aureus (ProtA) and the calmodulin-binding peptide (CBP), separated by a tobacco etch virus (TEV) protease cleavage site.1,2 Sequential purification of the tagged protein is carried out using IgG matrix and calmodulin resin, respectively. A TEV cleavage site is required because ProtA can only be released from IgG under denaturing conditions and the engineering of this enzyme cleavage site overcomes this limitation, allowing proteolytic release under native conditions. CBP binds to calmodulin in a calcium-dependent manner and can be released by adding chelating agents such as EGTA. In yeast, the ProA-CBP tandem tag allows roughly 20–30% of the target protein to be recovered,10 and in most cases, sufficient amount of complexes for protein identification by mass spectrometry can be obtained from 2 L of culture.11 When the standard TAP tag is applied to higher eukaryotes, however, the recovery of the tagged protein is often much lower (i.e., ∼1%12–15), necessitating a much larger amount of starting material to provide sufficient protein for subsequent analyses. As cultivation and handling of mammalian and insect cells is very costly and labor intensive, the low recovery of the ProA-CBP tag in higher eukaryotes has limited the use of the original TAP method in these systems.
To make the method more practical in non-yeast systems, a number of alternative TAP tags using different affinity handles have been developed,12–26 and some of them have shown improved protein recovery (Table I). In addition to the tags developed by individual laboratories to meet their own needs, Stratagene developed a tandem tag consisting of CBP and streptavidin-binding peptide (SBP) and created the commercial InterPlay mammalian TAP system based on it. The major advantage of this system is that it supports a purification protocol without the need of proteolytic enzyme, as both tags can be eluted from their respective resins under mild conditions. However, protein purification is very case-dependent: each protein behaves differently when expressed in a heterologous system based on its individual structural/functional properties, endogenous interactors and the effects its overexpression has on the cell phenotype. Furthermore, simple considerations of protein folding mean that no affinity purification system will work for all targets.
Table I.
Reported Recovery Rates for Several TAP Tag Combinations
To overcome such limitations with the analysis of our target protein of interest, Btk tyrosine kinase, we added a His-tag to the C-terminus of the target protein and used it in place of the CBP for the second round of purification. Btk is a non-receptor tyrosine kinase, mutations in the gene for which cause x-linked immunodeficiency agammaglobulinemia (XLA) in humans and x-linked immunodeficiency (XID) in mice.27,28 Since its original discovery in the setting of XLA/XID, significant progress has been made to understand the mechanisms of Btk activation in the immune system and its role in lymphocyte development and regulation.29 Nevertheless, a unbiased analysis of Btk interactors by TAP-mass spectrometry has never been reported. To discover novel regulatory mechanisms for this protein, we utilized a newly engineered TAP construct to map Btk interactors from mammalian cells. This approach resulted in a much greater yield of the target protein and its interacting partners and could serve as a useful approach to complement existing tagging strategies for purification of other protein targets.
Results
Expression and extraction of human Btk
The human Btk is 659 amino acids in length, and the fusion (with N-terminal or flanking affinity tags, Figure 1) has an anticipated molecular weight of 85 kDa. As confirmed by Western blot, Btk was expressed in transfected HEK-293 cells with the majority extracted to the soluble portion (Fig. 2).
Figure 1.
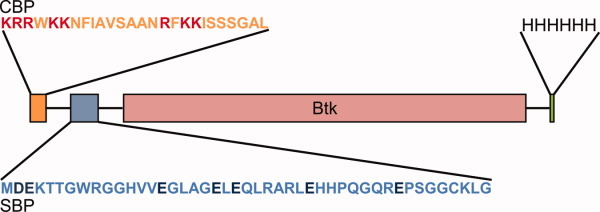
Schematic representation of the affinity-tagged human Btk. Btk was N-terminally fused to the CBP-SBP tandem tag system and a C-terminal His-tag engineered to expand protein purification options. The positively and negatively charged residues in CBP and SBP, respectively, are highlighted. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.
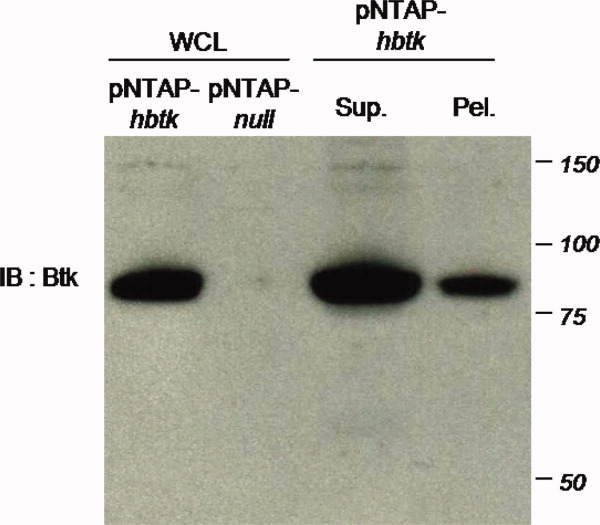
Western blot analysis of human Btk expression and extraction. Whole cell lysate (WCL) of pNTAP-hBtk- or empty vector- (pNTAP-null) transfected HEK-293 cells were separated along with supernatant (sup) or pellet (pel) extraction from pNTAP-hBtk transfected cells, followed by immunoblotting for Btk. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Streptavidin-based first round of purification with calmodulin-based second round
The Btk fusion protein bound to the streptavidin resin efficiently as indicated by its absence from cell lysate supernatant after overnight incubation [Fig. 3(A), SBP-Unb.]. To estimate protein recovery, biotin eluate was subjected to a serial dilution (twofold, fourfold, sixfold, and eightfold) and the diluted samples were electrophoresed with the cleared cell lysate (note: the volume of biotin eluate was 12.5% that of the cell lysate supernatant). Western blotting demonstrates that the band intensity of the cell lysate supernatant is comparable with that of the sixfold diluted biotin eluate [Fig. 3(B), compare lanes “6” and “WCL”]. This result suggests that about 75% of the target protein present in the cleared cell lysate was recovered after this initial purification.
Figure 3.
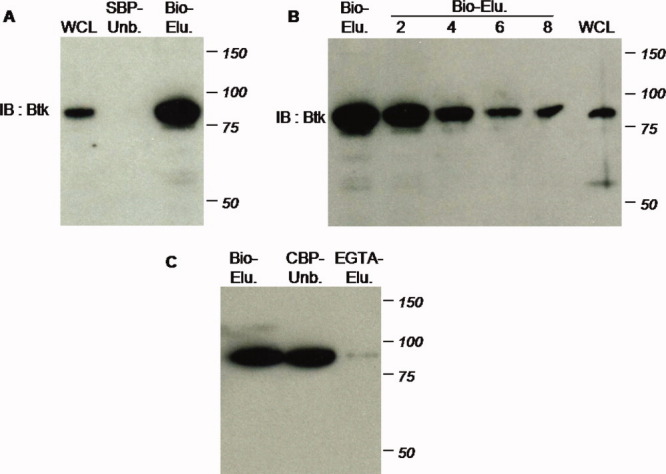
Dual streptavidin- and calmodulin-based purification. A: First step streptavidin-based purification recovers substantial Btk protein: supernatant of whole cell lysate (WCL); unbound portion (SBP-Unb.); and product of 2 mM biotin eluate (Bio-Elu.). B: Protein recovery as illustrated by serial dilution of biotin eluate: undiluted (Bio-Elu.); twofold, fourfold, sixfold, and eightfold dilution; and supernatant of whole cell lysate (WCL). C: Second phase of TAP purification via calmodulin binding is ineffective to recover Btk: biotin eluate resulted from the first round of purification (Bio-Elu.); unbound portion following incubation of streptavidin eluate with calmodulin resin (CBP-Unb.); product of 5 mM EGTA elution (EGTA-Elu.). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
We next attempted to perform the second step of TAP using the CBP tag. However, the majority of Btk remained in the unbound portion after overnight incubation [Fig. 3(C)], indicating that the efficiency of binding to the calmodulin resin was very poor for this fusion protein. Accordingly, almost no Btk was detected in the EGTA eluate.
Nickel-based second round of purification
To overcome this limitation of poor recovery, we designed the construct described above and in Figure 1. The C-terminally His-tagged Btk was first purified with streptavidin resin following the same procedure as for the non-His-tagged version. The newly added His-tag allows the target protein to be subsequently purified by immobilized metal ion affinity chromatography (IMAC). Most of the Btk present in the biotin eluate binds to the Ni-NTA resin after 2 h incubation (as indicated by the miniscule amount of Btk left in the unbound portion) and is subsequently released with 300 mM imidazole [Fig. 4(A)]. The volume of the final eluate was 25% of that of the streptavidin eluate applied to the nickel resin. To assess target recovery of this purification step, a small aliquot of imidazole eluate was diluted fourfold and electrophoresed in parallel with the biotin eluate. Western blotting for Btk found that the intensities of the two bands are indistinguishable [Fig. 4(B)], suggesting that most of the target protein present in the biotin eluate was recovered after IMAC. To evaluate the recovery of Btk-associated proteins, the eluates from single and double purification were also analyzed by silver staining. As shown in Figure 4(C), the tandem purification greatly enriches for the bait protein (asterisk, as determined by mass spectrometry) and reduces background nonspecific binding.
Figure 4.
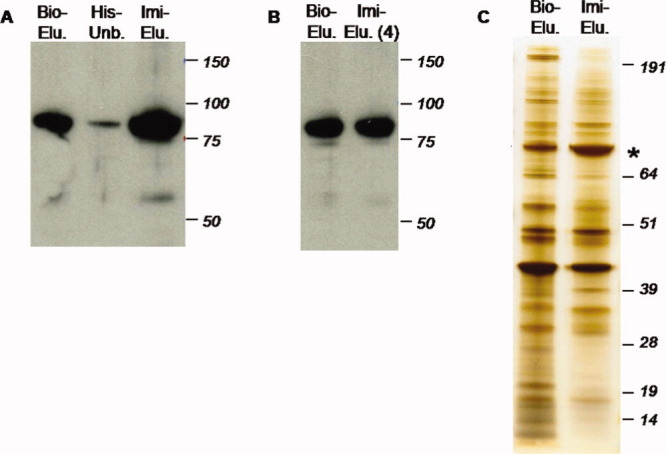
Dual streptavidin- and nickel-based purification. A: Eluate from first step streptavidin-based purification (Bio-Elu.); unbound portion (His-Unb.); and product of 300 mM imidazole eluate (Imi-Elu.). B: Protein recovery, including Biotin eluate (Bio-Elu.) and four-fold diluted imidazole eluate (Imi-Elu. 4). C: Silver staining of partially and fully purified Btk complex: biotin eluate from the first round of purification (Bio-Elu.) and imidazole eluate from the second round of purification (Imi-Elu.). Asterisk indicates Btk as determined by mass spectrometry. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Protein identification by tandem mass spectrometry
Tandem affinity purified protein samples (from Btk-transfected cells and empty vector-transfected negative control) were separated by SDS-PAGE and visualized by Oriole staining [Fig. 5(A)]. Immediately apparent from the gel image is the efficiency of Btk (indicated by asterisk) enrichment and the specificity of the optimized TAP procedure to recover Btk-associated proteins [Fig. 5(B)]. Both gel lanes were cut into ∼16 slices, digested with trypsin and analyzed by high mass accuracy LC/MS/MS on an Orbitrap. As described in Methods, rigorous criteria were used for protein identification and the reported list of Btk interacting proteins excluded any identifications made from the negative control lane (TAP-null). The TAP purification and mass spectrometry analyses were performed three independent times, and proteins included as Btk interactors were observed in two or more biological replicates (Table II).
Figure 5.
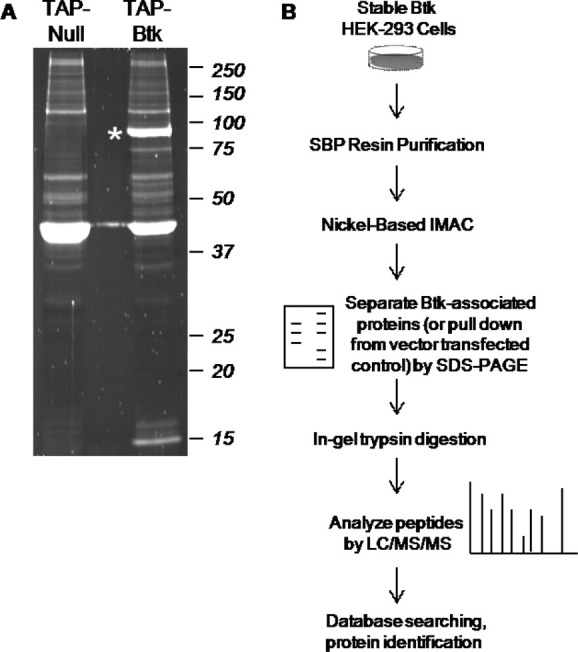
Methodology for analysis of Btk-associated proteins using new TAP vector combined with mass spectrometry. A: Oriole staining of tandem affinity purified Btk complex: negative control of tandem affinity purification (TAP-Null) and tandem affinity purified Btk complex (TAP-Btk). Asterisk indicates Btk. B: Experimental workflow for TAP purification and protein identification.
Table II.
Btk Interacting Proteins Identified by TAP and Mass Spectrometry
| UniProt/IPI | Protein Name | MWa | Xcorrb | Known Functionc |
|---|---|---|---|---|
| P23396/IPI00011253 | 40S ribosomal protein S3 | 26,671 | 90.3 | Protein translation machinery |
| Q9Y4W6/IPI00001091 | AFG-like protein | 88,528 | 40.2 | ATP-dependent protease |
| P68133/IPI00021428 | Alpha skeletal actin | 42,023 | 216.4 | Contraction |
| P12814/IPI00013508 | Alpha-actinin | 102,993 | 60.2 | Myofilament (actin) anchoring protein |
| P25705/IPI00440493 | ATP synthase alpha subunit | 59,713 | 210.3 | ATP production in mitochondria |
| P06576/IPI00303476 | ATP synthase beta subunit | 56,524 | 140.3 | ATP production in mitochondria |
| Q9BYX7/IPI00888712 | Beta-actin | 41,988 | 178.3 | Cytoskeleton |
| B4DMH3/IPI00798401 | Coronin-1C | 49,347 | 30.3 | Cytokinesis, cell motility |
| O00571/IPI00215637 | DDX3X, ATP-dependent RNA helicase | 73,198 | 30.2 | Nucleotide-binding, RNA processing |
| Q16643/IPI00003406 | Drebrin | 71,381 | 30.3 | Cell migration |
| Q05639/IPI00014424 | Elongation factor 1 alpha | 50,438 | 30.2 | Promotes t-RNA binding to ribosome |
| P21333/IPI00302592 | Filamin-A | 279,841 | 30.2 | Anchors cytoskeleton to membrane |
| P04406/IPI00219018 | Glyceraldehyde-3-phosphate dehydrogenase | 36,030 | 40.2 | Glycolytic enzyme |
| Q86YZ3/IPI00398625 | Hornerin | 282,226 | 40.3 | Epidermis formation |
| P13647/IPI00009867 | Keratin, type II | 62,340 | 250.3 | Cytoskeletal structure |
| Q9H1R3/IPI00221127 | Myosin light chain kinase 2 | 64,644 | 40.2 | Regulates contraction of striated muscle |
| P28331/IPI00604664 | NADH-ubiquinone oxidoreductase | 80,945 | 50.3 | Mitochondrial respiratory chain |
| P23246/IPI00010740 | SFPQ, splicing factor | 76,101 | 110.2 | Nucleotide-binding, spliceosome formation |
| P02549/IPI00220741 | Spectrin, alpha chain | 279,998 | 40.2 | Cytoskeletal structure |
| P40939/IPI00031522 | Trifunctional enzyme, alpha subunit | 82,947 | 70.3 | Fatty acid metabolism |
| P68366/IPI00007750 | Tubulin, alpha 4A chain | 49,892 | 30.2 | Major constituent of microtubules |
| P68371/IPI00007752 | Tubulin, beta 2C chain | 49,799 | 38.3 | Major constituent of microtubules |
| Q12792/IPI00183508 | Twinfilin | 43,890 | 40.3 | Actin binding, polymerization |
Molecular weight, as calculated from amino acid sequence.
As determined by SEQUEST from highest single run in which protein was identified.
Determined from UniProt annotation.
Discussion
Multistep purification has various applications for the preparation of recombinantly expressed proteins. The major advantage of this approach when compared with single step purification is improved specificity of recovery: hence, the major application for TAP has been to examine protein interactions, where confounding effects of nonspecificity are a major concern for discovering new functions of a protein. TAP systems based on the CBP-SBP combination are an essential tool used by many laboratories for the successful isolation of various protein complexes.30–32 However, the percentage of overall recovery of the target protein has been unclear from many of these studies. In our hands, the N-terminal CBP-SBP tagged Btk fusion bound to the streptavidin resin efficiently, allowing 70–80% of the target protein to be recovered. This is in agreement with previous reports, which stated that SBP-mediated purification recovered 75–90% of the bait proteins.15,26 In contrast, the CBP-SBP-Btk fusion bound poorly to the second (calmodulin) resin, resulting in a paltry protein yield and effectively defeating the purpose of dual tagging.
Inefficiency of the calmodulin affinity chromatography step has been observed by others and was postulated to be responsible for the low recovery of the original ProA-CBP tag in higher eukaryotes.13,15 The poor binding could be due to several factors. First, as with all recombinant protein experiments, we cannot rule out the possibility that the CBP is inaccessible due to Btk-specific protein folding; related to this possibility, however, we observe that the SBP module, which is sandwiched between the CBP module and the target protein, works well for isolation. Second, endogenous calmodulin could bind to the CBP moiety and prevent its binding to the resin.1 Third, mammalian cells express a greater number of endogenous calmodulin-binding proteins that will compete with the fusion during purification.13,16 A final possibility specific to this tag combination, 16% of residues on adjacent SBP are negatively charged (Fig. 1), which may interfere with CBP binding to calmodulin resin due to electrostatic interactions between the tags.
To circumvent the ineffective purification of the Btk fusion via calmodulin, we added a His-tag to the C-terminus of Btk to be used with IMAC in lieu of CBP-calmodulin for the second round of purification. This approach has the advantage of retaining the SBP and CBP modules in the vector for use in future applications (with Btk or other targets of interest), effectively creating a purification system with three independent options for two-step enrichment of a target protein. This modified approach was very successful for the purification of human Btk, allowing for subsequent mass spectrometry-based analysis of its interacting partners. It is noteworthy that the amount of nickel resin is critical for the His-tag-mediated purification. We initially used 50 μL of resin under which conditions we had difficulty eluting the bound protein even with high concentration of imidazole. For the test case of Btk, protein production from eight 150-mm dishes of cells plus 10 μL of Ni-NTA resin was sufficient for recovery of recombinant protein while also allowing efficient elution. Our western blotting results indicate >80% of the fusion present in the biotin eluate from the first round of purification was recovered in the second step with the improved SBP-His combination (Fig. 4), putting the overall yield at >50%, which is among the highest reported for any TAP tag system. The high recovery makes further concentration of the purified sample unnecessary, which avoids potential loss during such a step. In fact, only 10% of the final purified protein complex was sufficient for protein identification by mass spectrometry.
Although both SBP and His-tag are used as components in various tandem tags, their use together is rare. Our investigations show that they are quite compatible with each other having the advantage that the entire purification can be performed in one buffer system. Furthermore, the Ni-NTA resin is relatively less expensive and has a higher capacity compared with the antibodies required for epitope tag-based purification, and binding can be carried out under denaturing conditions if desired. Finally, this approach minimally modifies the fusion protein construct and effectively adds a third option for purification (as both the CBP and SBP modules are unmodified) of target molecules. The His tag was placed on the C-terminus, in this study, to minimize possible steric interactions with the other tag(s) during purification, although a separate construct with the His on the N-terminus would very likely identify new interacting proteins missed in the current strategy.
Having optimized this purification system for Btk, we sought to apply it to map the proteins interacting with this kinase in human cells. Btk is known to play a fundamental role in lymphocyte development and B cell maturation. B cell receptor stimulation leads to Btk activation after which the cytoplasmic kinase normally translocates to the cytoskeleton, calcium storage, and nuclear domains within the cell. Although many protein regulators of Btk have been described in specific contexts, an unbiased analysis of its interactors was lacking. Our investigations found specific interaction of Btk with numerous filament and cytoskeletal-associated proteins, including myosin light chain, filament actin, twinfilin, keratin, and tubulin, several of which (including tubulin and keratin) are also implicated in nuclear structure (Table II). Interaction of Btk with multiple components of the cytoskeleton is quite intriguing and provides a mechanism for how Btk participates in signaling processes at various subcellular locations. Another Btk interactor identified in this study, spectrin, which also has a PH domain required for membrane anchoring, has been shown to play a key role in cytoskeletal signaling during B cell diseases.33 Previous studies had demonstrated global changes in glycolytic enzyme (including GAPDH) levels by microarray during the process of T helper cell development,34 although the mechanism was unclear. It is interesting to note that Btk interacts with a number of metabolic proteins including ATP synthase subunits α and β and GADPH. It will be intriguing for future studies to explore how Btk plays a role in this aspect of T helper cell development, and whether direct interactions with these proteins (as uncovered in our study) is involved. Figure 5(A) shows clearly that all protein bands examined in this study are at or below a 1:1 ratio with Btk, ruling out a concern of identification of proteins solely on their abundance in the cell. Nevertheless, caveats to our investigation include the uncertain role these interactors have in Btk signaling in the setting of the B cell; however, we reason these proteins are more likely to be reasonable targets when compared with those isolated from non-mammalian cells. Furthermore, Btk is activated following B cell receptor stimulation, a situation not replicated in our analyses. Future studies will be required to understand how this physiological stimulus affects the subproteome of Btk interacting molecules.
Materials and Methods
Materials
HEK-293 cells were obtained from American Type Culture Collection (Manassas, VA). The eukaryotic TAP expression vector pNTAP was purchased from Stratagene (La Jolla, CA). Oligonucleotides were ordered from Eurofins MWG Operon (Huntsville, AL). All reagents were obtained from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA) unless otherwise stated. PCR reagents, DNA polymerase, restriction enzymes, T4 DNA ligase, competent E. coli, Geneticin, NuPAGE 4–12% Bis-Tris gel, Seeblue prestained standard, and SilverXpress silver staining kit were purchased from Invitrogen (Carlsbad, CA). QIAquick PCR purification kit, QIAprep spin miniprep kit, PolyFect transfection reagent, and Ni-NTA agarose were purchased from Qiagen (Valencia, CA). Complete Mini EDTA-free protease inhibitor cocktail tablets was purchased from Roche (Indianapolis, IN). Streptavidin Sepharose and calmodulin Sepharose were purchased from GE Healthcare (Pittsburgh, PA). Biotin was purchased from USB (Cleveland, OH). Nitrocellulose membrane, Oriole fluorescent gel stain, and Precision Plus Protein standards were purchased from Bio-Rad Laboratories (Hercules, CA). Btk (C-20), a goat polyclonal antibody raised against the C-terminal region of human Btk, and donkey anti-goat IgG-HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). SuperSignal West Pico chemiluminescent substrate was purchased from Thermo Fisher Scientific (Rockford, IL). Sequencing grade modified trypsin was purchased from Promega (Madison, WI). Microcon centrifugal filter devices were purchased from Millipore (Billerica, MA).
Methods
Construction of expression vectors
The cDNA for the full-length human btk was the kind gift of Dr. Owen Witte (UCLA). The coding fragment was PCR-amplified using the forward primer 5′-CGCGGATCCCATGGCCGCAGTGATTCTGGAGA GCATC-3′ and the reverse primer 5′-ACGCGTC GACCTAGGATTCTTCATCCATGACATCTAGAAT-3′. The forward and reverse primers contain recognition sites for BamHI and SalI (underlined), respectively. After purification and digestion, the PCR product was cloned into similarly digested pNTAP expression vector. The resultant recombinant plasmid was named pNTAP-hBtk. Cloning into the pNTAP vector allows the target protein to be N-terminally fused to the CBP and SBP tandem affinity tag (Fig. 1). Using the same forward primer and a newly designed reverse primer 5′-ACGCGTCGACT CAATGATGATGATGATGATGGGATTCTTCATCCA TGACATCTAG-3′ (SalI site was underlined) and following the same procedures, the cDNA encoding a C-terminally hexahistidine-tagged human Btk was amplified and inserted into the same pNTAP vector. The new construct was named pNTAP-hBtkHis. In this case, the fusion has a C-terminal His-tag in addition to the N-terminal tag (Fig. 1). The presence and identity of the insert in either construct was verified by diagnostic restriction digestion and DNA sequencing.
Cell culture and stable transfection
HEK-293 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin, at 37°C, 5% CO2. Cells in 100-mm dishes were transfected with 8 μg of recombinant plasmid (pNTAP-hBtk or pNTAP-hBtkHis) or pNTAP (empty vector, served as a negative control) using PolyFect transfection reagent according to the manufacturer's instructions. Stable cell lines were selected with Geneticin (500 μg/mL), which was added 48 h after transfection, for 2 weeks.
Test of protein expression and extraction
Transiently transfected HEK-293 cells (harvested from a 100-mm culture dish) were resuspended in 0.2 mL of lysis buffer (150 mM NaCl, 25 mM Tris, and 0.1% Nonidet P-40, pH 8.0) and disrupted by three rounds of freeze-thawing. Soluble and insoluble portions were separated by centrifugation. Small aliquots of whole cell lysate, supernatant, and pellet (resuspended in 8M urea) were analyzed by SDS-PAGE and immunoblotting.
Streptavidin-based purification
For protein complex isolation, eight 150-mm dishes of HEK-293 cells stably expressing human Btk (with N-terminal or flanking affinity tags) were pelleted by centrifugation at 3000g (∼2.0 g cells can be obtained) and resuspended in 8 mL of lysis buffer containing protease inhibitor cocktail. Cells were disrupted via six cycles of sonication (15 s each with cooling between cycles), and insoluble cell debris was removed by centrifugation at 14,000g for 20 min. For every 1 mL of cleared lysate, 4 μL 0.5M EDTA, and 0.7 μL 14.4M β-mercaptoethanol were added. The lysate was subsequently aliquoted into 1 mL portions and each aliquot was incubated with ∼30 μL of streptavidin resin (110 μL resin suspension) at 4°C overnight on a rotation shaker. The resin was collected by centrifugation (2,000g for 5 min) and washed twice with 1 mL of streptavidin binding buffer (lysis buffer with 2 mM EDTA and 10 mM β-mercaptoethanol). The bound protein complex was eluted by incubation with 125 μL of streptavidin binding buffer containing 2 mM biotin for 4 h, and fractions from each aliquot were pooled.
Calmodulin-based purification
For Btk fusion with N-terminal tag alone, biotin eluate (∼1.1 mL), supplemented with 1 mM magnesium acetate, 1 mM imidazole, and 2 mM CaCl2, was incubated with 100 μL calmodulin Sepharose (200 μL resin slurry) at 4°C overnight. The resin was collected by centrifugation and washed twice with calmodulin binding buffer (lysis buffer supplemented with 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, and 10 mM β-mercaptoethanol). Protein complex was liberated with 275 μL of calmodulin elution buffer (lysis buffer supplemented with 1 mM magnesium acetate, 1 mM imidazole, 5 mM EGTA, and 10 mM β-mercaptoethanol).
Nickel-based purification
For Btk fusion with the C-terminal His-tag, biotin eluate (∼1.1 mL) was incubated with 10-μL Ni-NTA agarose (20-μL resin slurry). Binding was allowed to proceed for 2 h at 4°C with rotation. The resin was collected and washed twice with lysis buffer containing 20 mM imidazole. Bound proteins were eluted with 275 μL of lysis buffer containing 300 mM imidazole.
SDS-PAGE and Western blot analysis
SDS-PAGE separated proteins were visualized by silver or Oriole staining using SilverXpress silver staining kit or Oriole fluorescent gel stain according to the instructions from the respective manufacturers. For Westerns, protein samples were resolved on 4–12% Bis-Tris SDS-PAGE gels and transferred to a nitrocellulose membrane. Protein transfer was confirmed by Ponceau staining. After blocking, the membrane was immunoblotted with goat polyclonal antibody against human Btk (diluted 1:500), followed by donkey anti-goat IgG conjugated to horseradish peroxidase (diluted 1:5000). Blots were developed with SuperSignal West Pico chemiluminescent substrate and detected by autoradiography.
In-gel tryptic digest and mass spectrometric analysis
The volume of the final imidazole eluate was reduced to half using Microcon centrifugal filter devices to achieve a two-fold concentration (subsequent experiments simply used 120 μL instead of 275 μL elution buffer to eliminate this step). The overall scheme for analysis of Btk interactors is shown in Figure 5(B). Approximately 1/10 (i.e., 12 μL) of the concentrated sample was loaded onto a SDS-gel for protein separation and Oriole staining. The gel lanes for isolated Btk complex and negative control (cells transfected with empty vector and subjected to the same purification procedure) were each sectioned into 16 slices in such a way that, when visible, protein bands constituted a single slice. Proteins in the gel slices were reduced, alkylated, and digested with 1 μg of modified trypsin (in 50 μL of 50 mM ammonium bicarbonate) at 37°C overnight. The reaction was terminated by adding 5 μL of 5% trifluoroacetic acid. The peptide mixture was recovered from the gel slices with 0.1% trifluoroacetic acid in 50% acetonitrile solution, separated by reverse phase LC (Eksigent) and analyzed by MS/MS on a Thermo Orbi-trap.35 Spectra were acquired in data-dependent mode (top six ions selected) with dynamic exclusion. Data were searched against the human IPI database (version 3.51; 74,024 entries) using the SEQUEST algorithm in the Bioworks software (Thermo). All proteins were identified on the basis of ≥3 peptides, and each spectra used for identification had deltaCN > 0.1 and met the following Xcorr criteria: >2 (+1), >3 (+2), >4 (+3), and >5 (+4). Searches required full tryptic cleavage, no missed cleavages, and were performed with the differential modifications of carbamidomethylation on cysteine and methionine oxidation. Mass tolerance was 1 Da for precursor and 0.5 Da for product ions. Rate of false positive identification, as estimated by reversed database searching, was <1%.
Acknowledgments
The authors thank Dr. Owen Witte (UCLA) for the cDNA of human Btk.
Glossary
Abbreviations:
- Btk
Bruton's tyrosine kinase
- CBP
calmodulin-binding peptide
- IMAC
immobilized metal ion affinity chromatography
- ProtA
protein A of Staphylococcus aureus
- TAP
tandem affinity purification
- TEV
tobacco etch virus.
References
- 1.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 2.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 3.Honey S, Schneider BL, Schieltz DM, Yates JR, Futcher B. A novel multiple affinity purification tag and its use in identification of proteins associated with a cyclin-CDK complex. Nucleic Acids Res. 2001;29:1–9. doi: 10.1093/nar/29.4.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox DM, Du M, Guo X, Siu KW, McDermott JC. Tandem affinity purification of protein complexes from mammalian cells. Biotechniques. 2002;33:267–268. 270. doi: 10.2144/02332bm02. [DOI] [PubMed] [Google Scholar]
- 5.Forler D, Kocher T, Rode M, Gentzel M, Izaurralde E, Wilm M. An efficient protein complex purification method for functional proteomics in higher eukaryotes. Nat Biotechnol. 2003;21:89–92. doi: 10.1038/nbt773. [DOI] [PubMed] [Google Scholar]
- 6.Gully D, Moinier D, Loiseau L, Bouveret E. New partners of acyl carrier protein detected in Escherichia coli by tandem affinity purification. FEBS Lett. 2003;548:90–96. doi: 10.1016/s0014-5793(03)00746-4. [DOI] [PubMed] [Google Scholar]
- 7.Rivas S, Romeis T, Jones JD. The Cf-9 disease resistance protein is present in an approximately 420-kilodalton heteromultimeric membrane-associated complex at one molecule per complex. Plant Cell. 2002;14:689–702. doi: 10.1105/tpc.010357. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Gingras AC, Aebersold R, Raught B. Advances in protein complex analysis using mass spectrometry. J Physiol. 2005;563:11–21. doi: 10.1113/jphysiol.2004.080440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 10.Seraphin B, Puig O, Bouveret E, Rutz B, Caspary F. Tandem affinity purification to enhance interacting protein identification. In: Golemis EA, Adams PD, editors. Protein-protein interactions: a molecular cloning manual. New York: Cold Spring Harbor Laboratory Press; 2002. pp. 313–328. [Google Scholar]
- 11.Dziembowski A, Seraphin B. Recent developments in the analysis of protein complexes. FEBS Lett. 2004;556:1–6. doi: 10.1016/s0014-5793(03)01357-7. [DOI] [PubMed] [Google Scholar]
- 12.Drakas R, Prisco M, Baserga R. A modified tandem affinity purification tag technique for the purification of protein complexes in mammalian cells. Proteomics. 2005;5:132–137. doi: 10.1002/pmic.200400919. [DOI] [PubMed] [Google Scholar]
- 13.Schimanski B, Nguyen TN, Gunzl A. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell. 2005;4:1942–1950. doi: 10.1128/EC.4.11.1942-1950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang P, Sampson HM, Krause HM. A modified tandem affinity purification strategy identifies cofactors of the Drosophila nuclear receptor dHNF4. Proteomics. 2006;6:927–935. doi: 10.1002/pmic.200500230. [DOI] [PubMed] [Google Scholar]
- 15.Burckstummer T, Bennett KL, Preradovic A, Schutze G, Hantschel O, Superti-Furga G, Bauch A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat Methods. 2006;3:1013–1019. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- 16.Knuesel M, Wan Y, Xiao Z, Holinger E, Lowe N, Wang W, Liu X. Identification of novel protein-protein interactions using a versatile mammalian tandem affinity purification expression system. Mol Cell Proteomics. 2003;2:1225–1233. doi: 10.1074/mcp.T300007-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Zeghouf M, Li J, Butland G, Borkowska A, Canadien V, Richards D, Beattie B, Emili A, Greenblatt JF. Sequential peptide affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J Proteome Res. 2004;3:463–468. doi: 10.1021/pr034084x. [DOI] [PubMed] [Google Scholar]
- 18.Rubio V, Shen Y, Saijo Y, Liu Y, Gusmaroli G, Dinesh-Kumar SP, Deng XW. An alternative tandem affinity purification strategy applied to Arabidopsis protein complex isolation. Plant J. 2005;41:767–778. doi: 10.1111/j.1365-313X.2004.02328.x. [DOI] [PubMed] [Google Scholar]
- 19.Tsai A, Carstens RP. An optimized protocol for protein purification in cultured mammalian cells using a tandem affinity purification approach. Nat Protoc. 2006;1:2820–2827. doi: 10.1038/nprot.2006.371. [DOI] [PubMed] [Google Scholar]
- 20.Tagwerker C, Flick K, Cui M, Guerrero C, Dou Y, Auer B, Baldi P, Huang L, Kaiser P. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Mol Cell Proteomics. 2006;5:737–748. doi: 10.1074/mcp.M500368-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Giannone RJ, McDonald WH, Hurst GB, Huang Y, Wu J, Liu Y, Wang Y. Dual-tagging system for the affinity purification of mammalian protein complexes. Biotechniques. 2007;43:296–298. doi: 10.2144/000112550. 300 passim. [DOI] [PubMed] [Google Scholar]
- 22.Gloeckner CJ, Boldt K, Schumacher A, Roepman R, Ueffing M. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics. 2007;7:4228–4234. doi: 10.1002/pmic.200700038. [DOI] [PubMed] [Google Scholar]
- 23.Kyriakakis P, Tipping M, Abed L, Veraksa A. Tandem affinity purification in Drosophila: the advantages of the GS-TAP system. Fly (Austin) 2008;2:229–235. doi: 10.4161/fly.6669. [DOI] [PubMed] [Google Scholar]
- 24.Gloeckner CJ, Boldt K, Schumacher A, Ueffing M. Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP tag. Methods Mol Biol. 2009;564:359–372. doi: 10.1007/978-1-60761-157-8_21. [DOI] [PubMed] [Google Scholar]
- 25.Lehmann R, Meyer J, Schuemann M, Krause E, Freund C. A novel S3S-TAP-tag for the isolation of T-cell interaction partners of adhesion and degranulation promoting adaptor protein. Proteomics. 2009;9:5288–5295. doi: 10.1002/pmic.200900294. [DOI] [PubMed] [Google Scholar]
- 26.Glatter T, Wepf A, Aebersold R, Gstaiger M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol Syst Biol. 2009;5:1–14. doi: 10.1038/msb.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 28.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, Jenkins NA, Witte ON. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 29.Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, Christensson B, Berglof A, Vihinen M, Nore BF, Smith CI. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228:58–73. doi: 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 30.Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, Heidmann T, Greene WC. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci USA. 2006;103:15588–15593. doi: 10.1073/pnas.0604524103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei X, Shimizu T, Lai ZC. Mob as tumor suppressor is activated by Hippo kinase for growth inhibition in Drosophila. EMBO J. 2007;26:1772–1781. doi: 10.1038/sj.emboj.7601630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradley CM, Jones S, Huang Y, Suzuki Y, Kvaratskhelia M, Hickman AB, Craigie R, Dyda F. Structural basis for dimerization of LAP2alpha, a component of the nuclear lamina. Structure. 2007;15:643–653. doi: 10.1016/j.str.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Gorman EB, Chen L, Albanese J, Ratech H. Patterns of spectrin expression in B-cell lymphomas: loss of spectrin isoforms is associated with nodule-forming and germinal center-related lymphomas. Mod Pathol. 2007;20:1245–1252. doi: 10.1038/modpathol.3800962. [DOI] [PubMed] [Google Scholar]
- 34.Hamalainen HK, Tubman JC, Vikman S, Kyrola T, Ylikoski E, Warrington JA, Lahesmaa R. Identification and validation of endogenous reference genes for expression profiling of T helper cell differentiation by quantitative real-time RT-PCR. Anal Biochem. 2001;299:63–70. doi: 10.1006/abio.2001.5369. [DOI] [PubMed] [Google Scholar]
- 35.Paulsson AK, Franklin S, Mitchell-Jordan SA, Ren S, Wang Y, Vondriska TM. Post-translational regulation of calsarcin-1 during pressure overload-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;48:1206–1214. doi: 10.1016/j.yjmcc.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]