

Abstract
Cardiac trabeculation is a crucial morphogenetic process by which clusters of ventricular cardiomyocytes extrude and expand into the cardiac jelly to form sheet-like projections. Although it has been suggested that cardiac trabeculae enhance cardiac contractility and intra-ventricular conduction, their exact function in heart development has not been directly addressed. We found that in zebrafish erbb2 mutants, which we show completely lack cardiac trabeculae, cardiac function is significantly compromised, with mutant hearts exhibiting decreased fractional shortening and an immature conduction pattern. To begin to elucidate the cellular mechanisms of ErbB2 function in cardiac trabeculation, we analyzed erbb2 mutant hearts more closely and found that loss of ErbB2 activity resulted in a complete absence of cardiomyocyte proliferation during trabeculation stages. In addition, based on data obtained from proliferation, lineage tracing and transplantation studies, we propose that cardiac trabeculation is initiated by directional cardiomyocyte migration rather than oriented cell division, and that ErbB2 cell-autonomously regulates this process.
Keywords: Cardiac trabeculation, Endocardium, SPIM, ErbB2, Zebrafish
INTRODUCTION
Cardiac trabeculae are highly organized sheet-like muscular structures that form as a result of the extrusion and expansion of differentiated cardiomyocytes into the lumen of the ventricular chambers (Ben-Shachar et al., 1985; Sedmera et al., 1997; Sedmera et al., 2000; Moorman and Christoffels, 2003; Stankunas et al., 2008). The lack of cardiac trabecular formation or a failure of the trabeculae to undergo compaction causes embryonic lethality or cardiomyopathy in adult humans, respectively (Jenni et al., 1999). These data imply that cardiac trabeculation is a highly regulated morphogenetic process and suggest that subtle perturbations could lead to severe physiological defects.
One central question in organ development is how the morphogenetic movements of the different cells are regulated to form a complex functional structure. Cell labeling experiments have revealed some interesting aspects of the cell lineage and growth pattern of the vertebrate heart. For example, lineage analyses in avian and mouse embryos have shown that the compact and trabecular cardiomyocytes are clonally related (Mikawa et al., 1992; Meilhac et al., 2003; Meilhac et al., 2004a; Meilhac et al., 2004b). These studies also suggest a gradient of cardiomyocyte proliferation across the ventricular wall, one that is greater in the outer than inner layers. It is also evident from these studies that the daughter cells of two independent cardiomyocyte progenitors do intermingle to some extent. However, this intermixing was less frequently observed when retroviral labeling was performed at later stages. Although these studies provide an overall picture of the cardiomyocyte lineage and its growth pattern, little is known about the cellular mechanisms that initiate cardiac trabeculation. For instance, whether and how directional migration and/or oriented cell division of the cardiomyocytes are involved in the initiation of cardiac trabeculation has not been established.
Recently, several proteins have been shown to regulate cardiac trabeculation. Angiopoietin 1, which is produced by cardiomyocytes, appears to signal through its endocardially localized receptor Tie2 to regulate endocardial angiogenesis. Mouse embryos that lack the activity of angiopoietin 1 or Tie2 exhibit a much less complex ventricular endocardium. This defective endocardium in turn affects ventricular cardiomyocyte development as the mutant hearts are clearly devoid of trabeculae (Suri et al., 1996). Interestingly, endocardial-derived neuregulin 1 (Nrg1) appears to be crucial for cardiac trabeculation by signaling to cardiomyocytes through its receptor complex ErbB4 and ErbB2 to drive trabecular initiation. Targeted deletion of Nrg1, Erbb4 or Erbb2 results in the absence of trabecular formation in mouse (Gassmann et al., 1995; Lee et al., 1995; Meyer and Birchmeier, 1995; Kramer et al., 1996; Jones et al., 2003). Nrg-ErbB signaling plays an important role for multiple cellular functions, including cell proliferation, differentiation, migration and survival. However, its exact function in cardiac trabeculation still remains to be elucidated. In this study, we reveal a dual requirement for ErbB2 signaling in cardiac trabeculation. We present evidence that, in addition to its role in cardiomyocyte proliferation, ErbB2 is also required for regulating cardiomyocyte migration (delamination) to initiate cardiac trabeculation. As a probable consequence of the absence of cardiac trabeculation, erbb2 mutants develop progressive cardiac dysfunction. Their fractional shortening is reduced over time and cardiac conduction maturation is suppressed, suggesting an important physiological function of cardiac trabeculae.
MATERIALS AND METHODS
Zebrafish strains
Embryos and adult fish were raised and maintained under standard laboratory conditions (Westerfield, 2000). The mutant and transgenic lines used in this study are as follows: erbb2st61, erbb2st50 (Lyons et al., 2005) (erbb2st61 mutants were used throughout this study and erbb2st50 mutants appear to show the same cardiac phenotypes as erbb2st61 mutants), Tg(cmlc2:GFP)twu26 (Huang et al., 2003), Tg(cmlc2:dsRed)s879, Tg(cmlc2:ras-GFP)s883 (D'Amico et al., 2007), Tg(cmlc2:gCaMP)s878 (Arnaout et al., 2007), Tg(flk1:GFP)s843 (Jin et al., 2005), Tg(gata1:dsRed)sd2 (Traver et al., 2003), Tg(cmlc2:CreER)pd12, Tg(bactin2:loxP-DsRed-STOP-loxP-GFP)s928 (Kikuchi et al., 2010).
Cell transplantation
Cells were removed from donor embryos at mid-blastula stages and transplanted along the blastoderm margin of age-matched host embryos. The host embryos were allowed to develop until 5 days post-fertilization (dpf) before monitoring the compact versus trabecular distribution of the donor-derived cells. Donor embryos obtained from in-crossing erbb2st61 heterozygotes were genotyped by as described previously (Lyons et al., 2005).
BrdU labeling and histochemistry
Wild-type and erbb2 mutant animals were incubated in embryo medium containing 10 mg/ml BrdU (Sigma) for ∼24 hours. The animals were then fixed and stained with anti-BrdU (Roche) and anti-dsRed (Clontech) antibodies on 10 μm frozen sections. The proliferation `index' was determined by the number of BrdU-positive cells divided by the surface area of the ventricle.
AG4178 and 4-hydroxytamoxifen treatment and morpholino and DNA injections
Larvae were treated with 5 μM AG4178 (Calbiochem) in 1% DMSO containing embryo medium from 60 until 120 hours post-fertilization (hpf). Control embryos were incubated in 1% DMSO in embryo medium.
To induce Cre-mediated recombination, embryos obtained from crossing Tg(cmlc2:CreER)pd12 fish with Tg(bactin2:loxP-DsRed-STOP-loxP-GFP)s928 reporter animals were treated with 0.0625 μM 4-hydroxytamoxifen at 48 hpf for 4 hours.
To knockdown ErbB2 levels, approximately 570 pg of a splice morpholino (5′-GTCCGCCTCCATCGATTATTCCTCC-3′) against erbb2 was injected into one-cell-stage embryos as previously published (Lyons et al., 2005). The mismatched oligo (5′-GTCCGCCACCATCTATTATGCCACC-3′) (Lyons et al., 2005) was used as a control.
An uncut DNA construct harboring a cmlc2:H2B-mCherry reporter cassette was co-injected at a concentration of 20 ng/μl with the I-SceI enzyme (1 U/μl) into one-cell-stage Tg(cmlc2:GFP) or Tg(cmlc2:ras-GFP) embryos.
Measurement of fractional shortening
Assessment of fractional shortening was performed as previously described (Shu et al., 2003; Hassel et al., 2009). Statistical significance was evaluated with a one-sided unpaired Student's t-test. Differences were considered significant if the probability value was P<0.05 and highly significant if the probability value was P<0.01.
Optical mapping
Optical mapping of zebrafish hearts was performed as previously described (Chi et al., 2008). In brief, individual zebrafish were immobilized with 1% low-melting agarose. Cardiac contraction was suppressed with 10 mM 2,3-butanedione monoxime (Sigma) before imaging. The images were acquired with a Nikon Te-2000u microscope (Nikon) at a rate of 250 frames per second using a high-speed CMOS camera (MiCam Ultima, SciMedia). Data were analyzed and isochronal maps of the activation sequences were generated using the Brain Vision Analyzer software V.1001 (SciMedia).
RESULTS
Cardiac trabeculae start to form in the outer curvature of the ventricle at 3 dpf
To investigate the timing and morphogenetic events associated with cardiac trabeculation, we utilized two cardiomyocyte specific lines Tg(cmlc2:GFP) and Tg(cmlc2:ras-GFP), which express GFP or membrane-bound GFP in all differentiated cardiomyocytes. Around 72 hours post-fertilization (72 hpf, 3 dpf), differentiated cardiomyocytes started to protrude out of the otherwise mono-layered ventricular myocardium, generating small clusters of cells (Fig. 1A, arrow). This process appeared to mark the initiation of cardiac trabeculation in the developing zebrafish heart. The prospective trabecular cardiomyocytes were no longer epithelial-like in shape, suggesting an epithelial-to-mesenchymal transformation (EMT) during cardiac trabeculation.
Fig. 1.
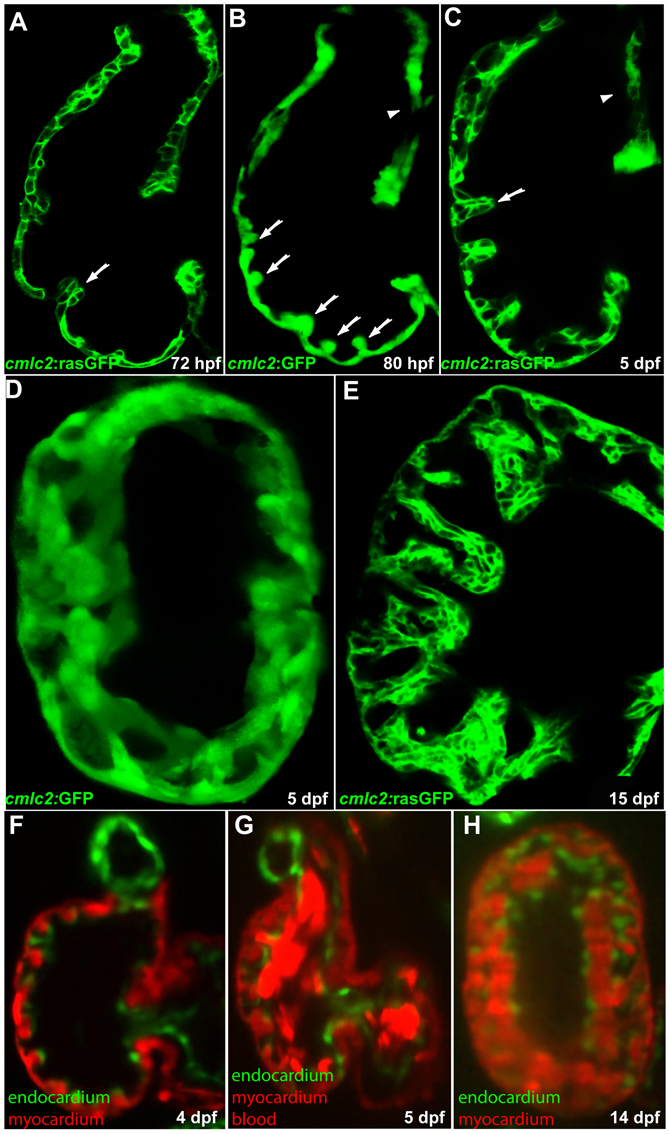
Cardiac trabeculation in zebrafish. (A) Cardiac trabeculae start to form in the ventricle around 72 hpf (arrow). (B) By 80 hpf, cardiac trabeculation has become more pronounced (arrows), with trabeculae distributed exclusively along the luminal side of the outer curvature of the ventricle. (C) The ventricle becomes extensively trabeculated by 5 dpf, but the inner curvature (arrowhead) remains smooth. (D) Cardiac trabeculae form ridges into the lumen as shown by 3D reconstruction. (E) The trabeculae continue to expand as shown at 15 dpf. (F-H) SPIM imaging shows that cardiac trabeculation increases in complexity (see corresponding movies in the supplementary material).
As the heart further developed, the extent and complexity of cardiac trabeculation significantly increased. By 80 hpf, cardiac trabeculation became more pronounced, with radially arranged projections distributed along the luminal side of the ventricle. These newly emerged trabeculae were well separated by compact myocardium and could only be identified in some areas of the outer curvature (Fig. 1B, arrows). By 5 dpf, the entire length of the outer curvature had developed extensive trabeculation. Each finger-like projection at this stage comprised about 7-9 cardiomyocytes of different shapes (Fig. 1C, arrow). Cardiac trabeculae are not simply finger-like projections as seen from a single confocal optical section, but rather resemble ridge or sheet-like structures that span the ventricular lumen. This structure can best be appreciated with a 3D projection of confocal images of a 5 dpf ventricle (Fig. 1D). Cardiac trabeculations continued to expand and, by 15 dpf, the trabecular projections were significantly larger in size. The compact layer, however, still remained one cell thick and no sign of compaction was observed up to this stage (Fig. 1E). As the trabeculae grow in size, the complexity of cardiac trabeculation also increases significantly, as shown by SPIM imaging (Huisken and Stainier, 2009) (Fig. 1F-H; see Movies 1 and 2 in the supplementary material).
erbb2 mutants lack cardiac trabeculation but develop normal cardiac valves
Prior studies have shown that mouse embryos deficient in Neuregulin 1 (Nrg1), ErbB2 or ErbB4 do not develop cardiac trabeculation and consequently die at mid-gestation (Gassmann et al., 1995; Lee et al., 1995; Meyer and Birchmeier, 1995; Kramer et al., 1996; Jones et al., 2003). Based on this phenotype, as well as the expression patterns of these factors, it has been proposed that endocardium-derived Nrg1, acting through the myocardially expressed ErbB4-ErbB2 heterodimer, is essential for cardiac trabeculation (Gassmann et al., 1995; Lee et al., 1995; Meyer and Birchmeier, 1995). However, the precise mechanisms by which Nrg1-ErbB signaling regulates cardiac trabeculation remain an open question. As a first step, we wanted to test whether the role of Nrg1-ErbB signaling in cardiac trabeculation was conserved. To this end, we examined the progression of cardiac trabeculation in zebrafish erbb2 mutants. At 3 dpf, although a radially arranged pattern of cardiac trabeculation became apparent in wild-type larvae, no sign of trabeculation was evident in erbb2 mutants (Fig. 2A,B). Likewise, 7 dpf erbb2 mutant larvae did not exhibit any trabeculae. By contrast, wild-type larvae at this stage exhibited extensive trabeculation. These data suggest that the lack of trabecular formation in erbb2 mutants is unlikely to be due to a developmental delay, but rather indicate a positive requirement for ErbB2 in trabecular initiation. To further test that the failure of cardiac trabeculation is due to a loss of ErbB2 activity, we knocked down erbb2 levels by injecting erbb2 morpholino (MO) into one-cell-stage embryos; all individuals (n=24) injected with the erbb2 MO had significantly reduced trabeculation at 5 dpf (Fig. 2F), whereas individuals injected with a mismatch MO had normal trabeculation (Fig. 2E; n=23). Although Nrg1 and its receptor genes Erbb2 and Erbb4 appear to be expressed uniformly in mouse endocardium and myocardium, respectively (Lee et al., 1995), the absence of cardiac trabeculation in the atrium might be owing to the fact that the endocardium in the atrium is not closely apposed to the myocardium as it is in the ventricle (see Movie 3 in the supplementary material).
Fig. 2.

ErbB2 signaling is essential for cardiac trabeculation in zebrafish. (A,B) Trabeculae start to form in wild-type larvae (A) at 3 dpf but are absent in erbb2 mutants (B). (C,D) The wild-type ventricle (C) is extensively trabeculated at 7 dpf, whereas trabeculae remain absent in erbb2 mutants (D) at the same stage. (E,F) Injection of an antisense morpholino oligonucleotide (MO) designed against erbb2 caused a significant reduction in cardiac trabecular formation (F), whereas a mismatch control MO had no effect (E). (G,H) Cardiac valve formation (arrows) appears to be unaffected in erbb2 mutants (H) compared with wild-type siblings (G).
ErbB2 has been previously implicated in endocardial EMT during cardiac valve development in mouse (Camenisch et al., 2002), thus we wanted to address whether the absence of cardiac trabeculation in zebrafish erbb2 mutants could be secondary to cardiac valve malformations. To test this idea, we crossed the erbb2 mutation into the flk1:GFP transgenic line (Jin et al., 2005), which expresses GFP in all endothelial cells including the endocardial cells. At 5 dpf, both the superior and inferior valve leaflets were formed in erbb2 mutants as in wild type (Fig. 2G,H), suggesting that ErbB2 is dispensable for cardiac valve morphogenesis in zebrafish. Taken together, our data indicate an essential and direct role for ErbB2 in cardiac trabeculation but not in cardiac valve formation.
To delineate the temporal requirement for ErbB2 in cardiac trabeculation, we treated Tg(cmlc2:GFP) larvae with the pharmacological inhibitor AG1478 to temporally block ErbB2 activity (Levitzki and Gazit, 1995; Busse et al., 2000). AG1478 treatment was shown to fully recapitulate the myelination defects observed in zebrafish erbb2 mutants (Lyons et al., 2005). Cardiac trabecular formation was completely abolished in 5 dpf larvae treated with 5 μM AG1478 from 60, 64 and 68 hpf through to 120 hpf (5 dpf); however, embryos treated with AG1478 from 72-120 hpf exhibited trabeculae, although reduced compared with controls (Fig. 3A,B,F; data not shown). In a separate set of experiments, we treated larvae with AG1478 starting at 60 hpf and the drug was then extensively washed out at 120 hpf. These larvae were then allowed to develop until 7 dpf. As shown in Fig. 3D,E, the trabeculae that formed in these larvae were significantly smaller than control or even 5 dpf larvae (compare Fig. 3D,E with 3A; see also 3F). Altogether, these data indicate that ErbB2 signaling occurring from 68-72 hpf is essential for cardiac trabeculation; this requirement for ErbB2 function occurs only a few hours prior to the appearance of trabeculae, indicating an immediate and direct effect of this signaling pathway in the formation of trabeculae.
Fig. 3.

ErbB2 signaling from 68-72 hpf is required for the initiation of cardiac trabeculation. (A,B) Extensive cardiac trabeculation in wild-type control (A) at 5 dpf and absence of cardiac trabeculation in wild-type larvae (B; data not shown) treated with 5 μM AG1478 from 60, 63 or 68 to 120 hpf. (C-E) Some cardiac trabeculae formed in wild-type larvae that were allowed to develop until 7 dpf after treatment with 5 μM AG1478 from 60-120 hpf. (F) Quantification of cardiac trabecular formation in control and AG1478-treated larvae, as determined by measuring the surface area of the trabecular myocardium and the compact myocardium (larvae treated as in A-E).
Cardiac trabeculation probably involves directional cardiomyocyte delamination
Given the diverse functions of ErbB2 in regulating multiple cellular processes, including cell migration and proliferation (Mahanthappa et al., 1996; Meintanis et al., 2001), we wanted to determine how ErbB2 signaling regulates cardiac trabeculation. It is conceivable that the nascent trabeculae arise from directional cardiomyocyte migration (delamination) and/or oriented cell division, by which one daughter cell remains in the compact layer while the other forms the trabecular layer. To distinguish between these possibilities, we examined cardiomyocyte proliferation in wild-type and erbb2 mutant larvae during the initial stages of cardiac trabeculation. To label all the cardiomyocytes that proliferated during these stages, larvae were soaked in BrdU for approximately 24 hours starting at 60, 84 and 108 hpf (n=10 for each stage). In wild-type ventricles, cardiomyocyte proliferation increased significantly over time and the number of BrdU-positive cardiomyocytes increased from 0.6 cell/μm2 (see Materials and methods) in 84 hpf larvae to 2.3 cells/μm2 in 132 hpf larvae (n=10; Fig. 4A-C). By contrast, the erbb2-deficient cardiomyocytes remained quiescent during these stages (n=10; Fig. 4D-F). Therefore, ErbB2-mediated cardiomyocyte proliferation was probably required to generate a sufficient number of cells to populate the trabeculae. This observation raised the question of whether cardiomyocyte proliferation was the major driving force for trabecular initiation. If so, one might expect that the nascent trabecular cardiomyocytes would all be BrdU-positive if wild-type larvae were exposed to BrdU from 60-84 hpf and subsequently fixed for immunohistochemistry. Interestingly, in such experiments, not all nascent trabecular cardiomyocytes were positive for BrdU incorporation (n=12; Fig. 5A,B).
Fig. 4.

ErbB2 is required for cardiomyocyte proliferation. (A-C) Cardiomyocyte proliferation, as assessed by BrdU incorporation in cardiomyocytes (yellow), increases over time in wild-type animals from 3-5 dpf. (D-F) Cardiomyocyte proliferation appears to be absent in erbb2 mutants during these stages.
Fig. 5.
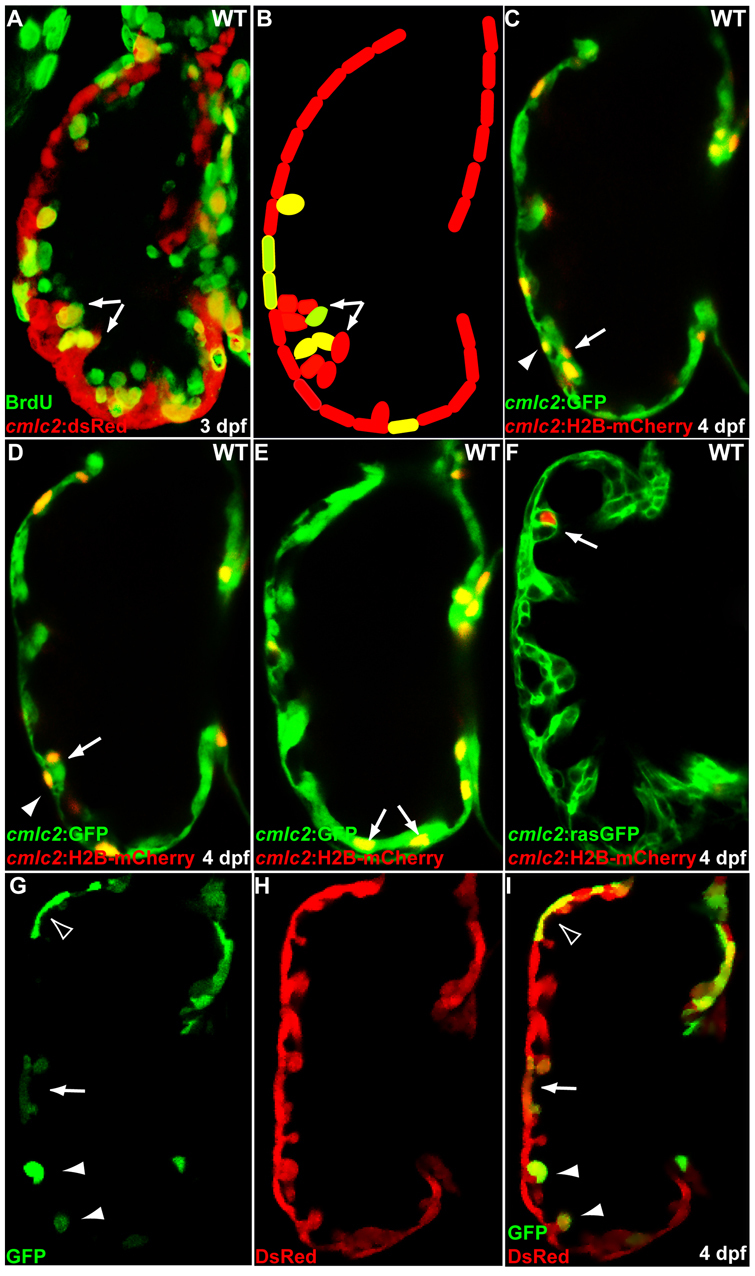
Initiation of cardiac trabeculation appears to involve directional cardiomyocyte migration (delamination) rather than oriented cell division. (A,B) Tg(cmlc2:dsRed) embryos at 60 hpf were exposed to BrdU for 24 hours. BrdU labeling shows that not all trabecular cells (arrows) are BrdU-positive. (C-F) cmlc2:H2B-mCherry reporter construct was injected into one-cell-stage Tg(cmlc2:GFP) (C-E) or Tg(cmlc2:rasGFP) (F) embryos and the distribution of mCherry-positive cardiomyocytes was monitored at 4 dpf. (C,D) Two optical sections of the same heart show that the lineage marker mCherry labels compact layer cardiomyocytes (arrowheads) as well as some of their immediate trabecular neighbors (arrows). (E,F) Trabecular cardiomyocytes (arrows), but not their compact layer neighbors, are labeled in another heart. (G-I) Genetic clones generated by crossing Tg(cmlc2:CreER) fish with Tg(bactin2:loxP-DsRed-STOP-loxP-GFP) reporter animals were found to contain both compact and trabecular cardiomyocytes (arrow), only compact cardiomyocytes (empty arrowhead) or only trabecular cardiomyocytes (arrowheads).
As a parallel approach to investigate mechanisms responsible for trabecular initiation, a cmlc2:H2B-mCherry reporter construct was injected into one-cell-stage Tg(cmlc2:GFP) or Tg(cmlc2:ras-GFP) embryos and the distribution of H2B-mCherry-positive cardiomyocytes was monitored in 4 dpf larvae. As shown in Fig. 5C,D, H2B-mCherry-positive cells were found in both compact cardiomyocytes and their immediate trabecular neighbors (n=4 embryos). There were, however, several cases (n=3 embryos) in which H2B-mCherry-positive cardiomyocytes were exclusively detected in the trabecular layer, but not in the adjacent area of the compact layer (Fig. 5E,F; see Movie 4 in the supplementary material). Thus, it is probable that cardiomyocytes emerge in the trabecular layer by migration rather than oriented cell division.
To further test this hypothesis, we crossed Tg(cmlc2:CreER)pd12 zebrafish with the Tg(bactin2:loxP-DsRed-STOP-loxP-GFP) reporter line and treated 48 hpf embryos with 0.0625 μM 4-hydroxytamoxifen for 4 hours. Upon 4-hydroxytamoxifen treatment, Cre-mediated recombination excised the floxed stop cassette, permanently marking recombinant cells and their descendants with GFP. Because the Tg(cmlc2:CreER)pd12 line facilitates low frequency recombination when used with this reporter line (data not shown), only small clusters of randomly distributed GFP-positive cardiomyocytes were observed (Fig. 5G,H). Of the 30 imaged clones, 12 contained cells in both compact and trabecular layers (arrow), whereas the others contained cells exclusively in the compact layer (empty arrowheads, n=10) or the trabecular layer (arrowheads, n=8) (Fig. 5G-I). Taken together, these data support the hypothesis that cardiac trabeculation is initiated by directional cardiomyocyte migration rather than oriented cell division.
A cell-autonomous role for ErbB2 in cardiomyocytes to regulate migration into the trabecular layer
Next, we wanted to determine whether ErbB2 was also required for cardiomyocyte migration in addition to its role in regulating cardiomyocyte proliferation. Thus, we examined the behavior of erbb2 mutant cardiomyocytes in wild-type ventricles by performing cell transplantation to generate chimeric hearts. In a first set of experiments, we tested this approach by transplanting blastomeres from Tg(cmlc2:dsRed) wild-type donors into Tg(cmlc2:GFP) wild-type hosts (Fig. 6A). The host embryos were allowed to develop until 5 dpf, at which stage we determined the distribution (compact versus trabecular layer) of the transplanted cells. In this experimental set up, the transplanted dsRed-positive wild-type cells did not show any bias in their distribution in the compact versus trabecular layer; approximately 45% of the donor-derived cells were found in the trabeculae and the rest in the compact layer (Fig. 6A,D; n=5 samples, 90 cells analyzed). We then transplanted homozygous erbb2 mutant cells into Tg(cmlc2:GFP) embryos (Fig. 6B; n=4 samples, 70 cells analyzed). As shown in Fig. 6B,D, the chimeric ventricles were extensively trabeculated with the trabeculae derived exclusively from wild-type cells. The dsRed-positive erbb2 mutant cells failed to contribute to the trabeculae and were found exclusively in the compact layer. A similar failure of erbb2 mutant cells to contribute to the trabeculae was also observed in the chimeric ventricles, in which wild-type cells were transplanted into Tg(cmlc2:dsRed); erbb2 mutant hosts. The cells that populated the trabeculae in such chimeric ventricles were all GFP-positive and thus wild type for erbb2 (Fig. 6C,D; n=2 samples, 46 cells analyzed). Taken together, these data indicate that ErbB2 is required in the cardiomyocytes to regulate cardiac trabeculation.
Fig. 6.

ErbB2 is required cell-autonomously in cardiomyocytes for the initiation of cardiac trabeculation. (A,B) Chimeric ventricles resulting from transplanting wild-type (A) or erbb2 mutant (B) blastomeres from Tg(cmlc2:dsRed) donors into Tg(cmlc2:GFP) wild-type hosts. Wild-type donor cells were detected in both the compact and trabecular layers, whereas erbb2 mutant donor cells failed to incorporate into the trabeculae (arrows). (C) Chimeric ventricles resulting from transplanting wild-type blastomeres from Tg(cmlc2:GFP) donors into Tg(cmlc2:dsRED); erbb2 mutant hosts. Note that the trabeculae only contain wild-type cells. (D) Quantification of the distribution of the wild-type and mutant donor-derived cells in the compact versus trabecular layers in the chimeric ventricles, assessed by measuring the surface area of the donor-derived cells.
Defective ErbB2 signaling and lack of trabeculation leads to progressive cardiac dysfunction
Because targeted deletion of Erbb2 leads to cardiac trabeculation defects and early embryonic lethality in mouse (Lee et al., 1995), we speculated that ErbB2 signaling and trabeculation of the ventricular myocardium were essential to maintain cardiac performance during later stages of development. In order to test this hypothesis, we assessed cardiac contractility, as determined by atrial as well as ventricular fractional shortening (FS), and heart rate of wild-type and erbb2 mutant larvae at 3, 5 and 10 dpf. At 3 dpf, right when trabeculation is initiated, atrial and ventricular contractility was indistinguishable between wild-type and erbb2 mutant hearts (atrium: wild type, 31.5±5.8%; erbb2–/–, 25.4±5.5%; ventricle: wild type, 24.8±4.9%; erbb2–/–, 21.7±5.4%; heart rate: wild type, 144±8.2 beats/minute; erbb2–/–, 135.4±10.8 beats/minute; n=8 of each) (Fig. 7A). However, by 5 dpf, erbb2 mutant ventricles showed a significantly reduced FS compared with their wild-type siblings (ventricle: wild type, 28.7±4%; erbb2–/–, 9.3±3.6%; P<0.005, n=8 of each), which declined even further at 10 dpf (ventricle: wild type, 20.5±2%; erbb2–/–, 8.8±5%; P<0.00005, n=11 of each) (Fig. 7A). This decrease in FS in erbb2 mutants over time strongly indicates a progressive ventricular systolic dysfunction. Noteworthy, ∼20% of the erbb2 mutants showed an FS that was lower than 3%. Additionally, although systolic function of the atrium was not affected at 5 dpf (atrium: wild type, 18.8±1.7%; erbb2–/–, 22.6±6.9%; n=6 of each), atrial contractility was found to be severely reduced in erbb2 mutant larvae compared with wild type at 10 dpf (atrium: wild type, 27.9±3.0%; erbb2–/–, 13.1±5.6%; P<0.004, n=6 of each) (Fig. 7B). Interestingly, at the time when erbb2 mutant hearts started showing reduced ventricular contractility (5 dpf), they also developed bradycardia (wild type, 178.5±1.7 beats/minute; erbb2–/–, 162.8±10.5 beats/minute; P<0.004, n=8 of each) (Fig. 7C). Heart beat further declined by 10 dpf (wild type, 156±5.6 beats/minute; erbb2–/–, 127.2±21.5 beats/minute; P<0.003, n=10 of each) (Fig. 7C), with ∼10% of the erbb2 mutants showing a heart rate even lower than 80 beats per minute.
Fig. 7.

erbb2 mutants display progressive cardiac dysfunction. (A,B) The ventricular fractional shortening of 3 dpf erbb2 mutants is not significantly different from that of wild type (A). A ventricular fractional shortening defect manifests itself at ∼5 dpf (A) and an atrial fractional shortening defect appears at ∼10 dpf (B). Data are presented as mean ± s.d. *P<0.05. (C) erbb2 mutant hearts begin to develop bradycardia at ∼5 dpf. (D-G) Optical mapping of wild-type (D,F) and erbb2 mutant (E,G) hearts at 3 (D,E) and 10 (F,G) dpf. erbb2 mutants at 10 dpf (G) fail to develop the mature conduction system observed in their wild-type siblings (F).
To start addressing whether an absence of cardiac trabeculae and ErbB2 signaling in zebrafish could also result in a cardiac conduction defect, we performed optical mapping on 3 and 10 dpf wild-type and erbb2 mutant hearts. To do so, we crossed the fish into the Tg(cmlc2:gCaMP) line (Arnaout et al., 2007). At 3 dpf, when trabeculae start to form in wild type, optical mapping revealed that the activation patterns in wild-type and erbb2 mutant ventricles were indistinguishable (n=5). In these larvae, the calcium activation wave broke through the AV (atrioventricular) junction into the ventricle at one single point (Fig. 7D,E, arrows) and subsequently spread laterally across the ventricular myocardium to the outflow tract (Fig. 7D,E). By contrast, following the formation of trabeculae, the calcium wave in 10 dpf wild-type hearts appeared to break into the ventricle at two distinct sites at the AV junction (Fig. 7F, arrows). By focusing at the endocardial surface, we observed that the calcium wave spread radially through the trabeculae into the outer curvature of the heart (n=4; Fig. 7F). This activation pattern is very similar to what has been observed in adult zebrafish hearts (Sedmera et al., 2003). erbb2 mutant hearts at 10 dpf (n=5), however, displayed a ventricular calcium activation pattern that spread laterally across the myocardium from the AV junction to the outflow tract, reminiscent of the activation pattern observed in 3 dpf wild-type hearts (Fig. 7G). In contrast to the nrg1 MO-injected animals, which were reported to show a loss of AV delay (Milan et al., 2006), erbb2 mutant hearts displayed an AV delay similar to their wild-type siblings (data not shown). Altogether, these data suggest an important role for cardiac trabeculae and ErbB2 signaling in the maturation of the ventricular conduction system. Similar findings have recently been reported from a detailed analysis of null and hypomorphic Nrg1 mutant mice (Lai et al., 2010).
Taken together, these findings strongly indicate that ErbB2 signaling and cardiac trabeculation are essential to adapt cardiac excitation and function to the changing needs during development. Thus, animals lacking ErB2 function are compromised in their ability to form trabeculae and go on to develop fatal heart failure.
DISCUSSION
In this study, we revealed an evolutionarily conserved role for ErbB2 signaling in cardiac trabeculation. Our transplantation data indicate that ErbB2 is required cell-autonomously in cardiomyocytes in order for cardiac trabeculation to occur. During this process, ErbB2 is required for cardiomyocyte proliferation and it also plays a role in regulating myocardial migration (delamination) to initiate cardiac trabeculation. In addition, we presented the first in vivo evidence that erbb2 mutants exhibit progressive cardiac dysfunction with reduced fractional shortening and conduction disturbances, suggesting an important physiological function for the cardiac trabeculae.
Role of ErbB2 in cardiomyocyte proliferation and migration during cardiac trabeculation
Previous studies in mouse showed that ErbB2 (as well as Nrg1 and ErbB4) is required for cardiac trabeculation (Gassmann et al., 1995; Lee et al., 1995; Kramer et al., 1996). Erbb2-deficient mice develop severe cardiac defects with a complete absence of trabeculae in the developing ventricles (Lee et al., 1995). This cardiac trabecular defect could be rescued by introducing rat Erbb2 expression in the cardiomyocytes (Morris et al., 1999; Woldeyesus et al., 1999). Similarly, loss of ErbB2 function results in a complete absence of cardiac trabeculae in zebrafish. However, we did not observe any cardiac valve defects in the zebrafish mutants, as reported with the mouse Erbb2 mutants (Camenisch et al., 2002). Because ErbB2 does not appear to be functionally compensated by other ErbB2-related factors in zebrafish (Reischauer et al., 2009), this difference in the involvement of ErbB2 in endocardial cushion morphogenesis could suggest that it gained a new function during cardiac valve development in amniotes and/or that the streamlining of cardiac valve development in zebrafish (Scherz et al., 2008) led to the loss of an essential function for ErbB2 in this process.
During cardiac trabeculation, cardiomyocytes are highly proliferative. BrdU labeling experiments indicate that cardiomyocyte proliferation in wild-type animals increases substantially from 3 to 5 dpf. Loss of ErbB2 activity, however, resulted in a complete failure of cardiomyocyte proliferation during cardiac trabeculation. This loss of cardiomyocyte proliferation in erbb2 mutants is somewhat reminiscent of the reduction of postnatal cardiomyocyte proliferation observed in the Erbb4 mutant mice (Bersell et al., 2009), suggesting a general requirement for ErbB signaling in cardiomyocyte proliferation. Our observations might appear contradictory to a recent finding that ventricular cardiomyocyte proliferation in mouse Nrg1 mutants is comparable with that of wild type (Grego-Bessa et al., 2007). As the compact layer of the developing mouse heart comprises several layers of cardiomyocytes as opposed to a single layer in zebrafish, other factors like Bmp and Fgf proteins are also probably involved in cardiomyocyte proliferation in mouse (Chen et al., 2004; Lavine et al., 2005). Thus, the difference in proliferation between wild-type and Nrg1 mutant mice might not be as pronounced as in zebrafish.
Our transplantation studies showed that the erbb2 mutant cells that were transplanted into wild-type hearts were exclusively detected within the compact layer. This failure of erbb2 mutant cells to incorporate into the trabeculae cannot be simply explained by the fact that ErbB2 controls cardiomyocyte proliferation. If ErbB2 was required only for cardiomyocyte proliferation, one would expect that a subset of the erbb2 mutant cells would contribute to the trabeculae as the wild-type cells in the chimeric ventricles generated enough cells to allow cardiac trabeculation to occur. The failure of erbb2 mutant cells to populate the trabeculae suggests that (1) cardiomyocyte proliferation and migration are coupled processes so that the failure of erbb2 mutant cells to proliferate could prevent them from delaminating from the compact layer to form the trabeculae, or (2) ErbB2 regulates cardiomyocyte proliferation and migration separately. If cardiomyocyte proliferation and migration were coupled processes, one would expect that all nascent trabecular cells would be BrdU-positive if the larvae were exposed to BrdU during the initiation stages of cardiac trabeculation. However, BrdU did not label all trabecular cells when 60 hpf wild-type embryos were treated with BrdU for 24 hours. In addition, our lineage tracing experiments showed that, in several cases, cardiac trabecular cells, but not the neighboring compact layer cells, were labeled by the lineage marker, suggesting that cardiac trabeculation is primarily driven by directional cardiomyocyte migration (delamination).
Overall, our data from the proliferation, lineage tracing and transplantation assays strongly support the idea that ErbB2 has a dual requirement for cardiomyocyte proliferation and migration during cardiac trabeculation.
The role for ErbB2 and trabeculation in cardiac function
Mice that lack Nrg1-ErbB2 signaling die at approximately E10.5 (Lee et al., 1995; Meyer and Birchmeier, 1995; Kramer et al., 1996), thus preventing detailed functional analysis of ErbB2-deficient hearts and thereby hindering insights into the role of ErbB2 during early cardiac physiology. Zebrafish larvae, however, do not rely on a functional cardiovascular system during the first 7-10 days of development and therefore constitute a good model to study the consequences of otherwise embryonic lethal mutations on early cardiac function (Sehnert et al., 2002). In this study, we utilized zebrafish erbb2 mutants to start to examine the functional consequences of the lack of cardiac trabeculation. Our work provides the first in vivo evidence that ErbB2 signaling is required for the establishment of a post-embryonic cardiac conduction system, as well as the adaptation of cardiac contractility to increasing functional demands during development.
erbb2 mutant zebrafish die at approximately 12 dpf. Interestingly, starting from the stage when the ventricle is extensively trabeculated in wild-type larvae (5 dpf), erbb2 mutants display a significantly decreased ventricular contractility, as well as bradycardia. These phenotypes become more pronounced as the hearts develop further, indicative of progressive heart failure. Earlier studies in mouse had suggested that trabeculae mechanically guarantee the maintenance of blood flow throughout early cardiogenesis and before the expansion of the compact zone. It was also suggested that the lack of trabeculation as a morphological feature is probably the primary cause for the embryonic lethality of the Erbb2-deficient mice (Lee et al., 1995; Bartman and Hove, 2005). However, the combination of the decreased ventricular contractility and bradycardia observed in zebrafish erbb2 mutants suggests that these hearts suffer from more than mechanical defects arising from disrupted blood flow. Indeed, our optical mapping studies show that erbb2 mutants fail to develop the mature conduction system observed in their wild-type siblings.
Deletion of Erbb2 in the adult mouse heart leads to ventricular systolic dysfunction, whereas trabeculation is indistinguishable from that of wild type, suggesting that ErbB2 signaling might also be involved in the regulation of cardiomyocyte contractility (Crone et al., 2002). In addition, treatment of mammary carcinoma patients with the ERBB2 inhibitory antibody trastuzumab can lead to cardiomyopathy with decreased cardiac contractility (Dillman, 1999; Ewer et al., 1999), whereas short-term activation of ERBB2 signaling by administration of recombinant NRG1 could improve cardiac performance in cardiomyopathy patients (Jabbour et al., 2010). Together, these data suggest that ErbB2 signaling, besides its role in cardiomyocyte behavior and proliferation, might also modulate cardiac contractility (Liu et al., 2006). In addition, the failure of the zebrafish erbb2 mutant hearts to increase cell mass could also contribute to the observed decrease in cardiac contractility.
During cardiogenesis, the early conduction system undergoes a refinement, resulting in ventricular action potential propagation that proceeds from apex-to-base rather than the embryonic base-to-apex propagation. Our optical mapping studies indicate that the erbb2 mutant hearts remain in an embryonic state of cardiac excitation. The persistence of the base-to-apex activation pattern observed in the erbb2 mutant hearts suggests that the ventricular conduction system does not form in these animals, an interpretation consistent with the findings that Nrg1 induces the expression of fast conduction markers during cardiac development (Rentschler et al., 2002). Although our data cannot discriminate between ErbB2 being directly involved in the initiation of conduction system development and the observed defects being secondary to ErbB2 involvement in cardiac trabeculation, ErbB2 is clearly required for the functional maturation of the cardiac ventricle.
Supplementary Material
Acknowledgments
We thank Dr William Talbot for fish stocks and reagents. We also thank Ana Ayala and Milly Alva for excellent fish care. J.L. was supported by a postdoctoral fellowship from the American Heart Association and is currently supported by an NIH (National Institutes of Health) training grant (Principle investigator: Shaun R. Coughlin, T32 HL007731). M.B. is supported by an NIH training grant (Principle investigator: Harold Bernstein, T32 HL007544). D.H. is supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (HA 5819/1-1). This work was funded by grants from the NIH (R01081674 to K.D.P.; R01 HL093566 to T.M.; R01 HL54737 to D.Y.R.S.) as well as the Packard foundation (D.Y.R.S.). Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.053736/-/DC1
References
- Arnaout R., Ferrer T., Huisken J., Spitzer K., Stainier D. Y., Tristani-Firouzi M., Chi N. C. (2007). Zebrafish model for human long QT syndrome. Proc. Natl. Acad. Sci. USA 104, 11316-11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman T., Hove J. (2005). Mechanics and function in heart morphogenesis. Dev. Dyn. 233, 373-381 [DOI] [PubMed] [Google Scholar]
- Ben-Shachar G., Arcilla R. A., Lucas R. V., Manasek F. J. (1985). Ventricular trabeculations in the chick embryo heart and their contribution to ventricular and muscular septal development. Circ. Res. 57, 759-766 [DOI] [PubMed] [Google Scholar]
- Bersell K., Arab S., Haring B., Kuhn B. (2009). Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257-270 [DOI] [PubMed] [Google Scholar]
- Busse D., Doughty R. S., Ramsey T. T., Russell W. E., Price J. O., Flanagan W. M., Shawver L. K., Arteaga C. L. (2000). Reversible G(1) arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J. Biol. Chem. 275, 6987-6995 [DOI] [PubMed] [Google Scholar]
- Camenisch T. D., Schroeder J. A., Bradley J., Klewer S. E., McDonald J. A. (2002). Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat. Med. 8, 850-855 [DOI] [PubMed] [Google Scholar]
- Chen H., Shi S., Acosta L., Li W., Lu J., Bao S., Chen Z., Yang Z., Schneider M. D., Chien K. R., et al. (2004). BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 131, 2219-2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi N. C., Shaw R. M., Jungblut B., Huisken J., Ferrer T., Arnaout R., Scott I., Beis D., Xiao T., Baier H., et al. (2008). Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLoS Biol. 6, e109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone S. A., Zhao Y. Y., Fan L., Gu Y., Minamisawa S., Liu Y., Peterson K. L., Chen J., Kahn R., Condorelli G., et al. (2002). ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 8, 459-465 [DOI] [PubMed] [Google Scholar]
- D'Amico L., Scott I. C., Jungblut B., Stainier D. Y. (2007). A mutation in zebrafish hmgcr1b reveals a role for isoprenoids in vertebrate heart-tube formation. Curr. Biol. 17, 252-259 [DOI] [PubMed] [Google Scholar]
- Dillman R. O. (1999). Perceptions of Herceptin: a monoclonal antibody for the treatment of breast cancer. Cancer Biother. Radiopharm. 14, 5-10 [DOI] [PubMed] [Google Scholar]
- Ewer M. S., Gibbs H. R., Swafford J., Benjamin R. S. (1999). Cardiotoxicity in patients receiving transtuzumab (Herceptin): primary toxicity, synergistic or sequential stress, or surveillance artifact? Semin. Oncol. 26, 96-101 [PubMed] [Google Scholar]
- Gassmann M., Casagranda F., Orioli D., Simon H., Lai C., Klein R., Lemke G. (1995). Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378, 390-394 [DOI] [PubMed] [Google Scholar]
- Grego-Bessa J., Luna-Zurita L., del Monte G., Bolos V., Melgar P., Arandilla A., Garratt A. N., Zang H., Mukouyama Y. S., Chen H., et al. (2007). Notch signaling is essential for ventricular chamber development. Dev. Cell 12, 415-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel D., Dahme T., Erdmann J., Meder B., Huge A., Stoll M., Just S., Hess A., Ehlermann P., Weichenhan D., et al. (2009). Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat. Med. 15, 1281-1288 [DOI] [PubMed] [Google Scholar]
- Huang C. J., Tu C. T., Hsiao C. D., Hsieh F. J., Tsai H. J. (2003). Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev. Dyn. 228, 30-40 [DOI] [PubMed] [Google Scholar]
- Huisken J., Stainier D. Y. (2009). Selective plane illumination microscopy techniques in developmental biology. Development 136, 1963-1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbour A., Hayward C. S., Keogh A. M., Kotlyar E., McCrohon J. A., England J. F., Amor R., Liu X., Li X. Y., Zhou M. D., et al. (2010). Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur. J. Heart Fail. (In press). [DOI] [PubMed] [Google Scholar]
- Jenni R., Rojas J., Oechslin E. (1999). Isolated noncompaction of the myocardium. N. Engl. J. Med. 340, 966-967 [DOI] [PubMed] [Google Scholar]
- Jin S. W., Beis D., Mitchell T., Chen J. N., Stainier D. Y. (2005). Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 132, 5199-5209 [DOI] [PubMed] [Google Scholar]
- Jones F. E., Golding J. P., Gassmann M. (2003). ErbB4 signaling during breast and neural development: novel genetic models reveal unique ErbB4 activities. Cell Cycle 2, 555-559 [PubMed] [Google Scholar]
- Kikuchi K., Holdway J. E., Werdich A. A., Anderson R. M., Fang Y., Egnaczyk G. F., Evans T., Macrae C. A., Stainier D. Y., Poss K. D. (2010). Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464, 601-605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R., Bucay N., Kane D. J., Martin L. E., Tarpley J. E., Theill L. E. (1996). Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc. Natl. Acad. Sci. USA 93, 4833-4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D., Forrai A., Liu X., Wolstein O., Michalicek J., Ahmed I., Garratt A. N., Birchmeier C., Zhou M., Hartley L., et al. (2010). Neuregulin 1 sustains the gene regulatory network in both trabecular and nontrabecular myocardium. Circ. Res. 107, 715-727 [DOI] [PubMed] [Google Scholar]
- Lavine K. J., Yu K., White A. C., Zhang X., Smith C., Partanen J., Ornitz D. M. (2005). Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell 8, 85-95 [DOI] [PubMed] [Google Scholar]
- Lee K. F., Simon H., Chen H., Bates B., Hung M. C., Hauser C. (1995). Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378, 394-398 [DOI] [PubMed] [Google Scholar]
- Levitzki A., Gazit A. (1995). Tyrosine kinase inhibition: an approach to drug development. Science 267, 1782-1788 [DOI] [PubMed] [Google Scholar]
- Liu X., Gu X., Li Z., Li X., Li H., Chang J., Chen P., Jin J., Xi B., Chen D., et al. (2006). Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. J. Am. Coll. Cardiol. 48, 1438-1447 [DOI] [PubMed] [Google Scholar]
- Lyons D. A., Pogoda H. M., Voas M. G., Woods I. G., Diamond B., Nix R., Arana N., Jacobs J., Talbot W. S. (2005). erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr. Biol. 15, 513-524 [DOI] [PubMed] [Google Scholar]
- Mahanthappa N. K., Anton E. S., Matthew W. D. (1996). Glial growth factor 2, a soluble neuregulin, directly increases Schwann cell motility and indirectly promotes neurite outgrowth. J. Neurosci. 16, 4673-4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilhac S. M., Kelly R. G., Rocancourt D., Eloy-Trinquet S., Nicolas J. F., Buckingham M. E. (2003). A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development 130, 3877-3889 [DOI] [PubMed] [Google Scholar]
- Meilhac S. M., Esner M., Kelly R. G., Nicolas J. F., Buckingham M. E. (2004a). The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev. Cell 6, 685-698 [DOI] [PubMed] [Google Scholar]
- Meilhac S. M., Esner M., Kerszberg M., Moss J. E., Buckingham M. E. (2004b). Oriented clonal cell growth in the developing mouse myocardium underlies cardiac morphogenesis. J. Cell Biol. 164, 97-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meintanis S., Thomaidou D., Jessen K. R., Mirsky R., Matsas R. (2001). The neuron-glia signal beta-neuregulin promotes Schwann cell motility via the MAPK pathway. Glia 34, 39-51 [PubMed] [Google Scholar]
- Meyer D., Birchmeier C. (1995). Multiple essential functions of neuregulin in development. Nature 378, 386-390 [DOI] [PubMed] [Google Scholar]
- Mikawa T., Borisov A., Brown A. M., Fischman D. A. (1992). Clonal analysis of cardiac morphogenesis in the chicken embryo using a replication-defective retrovirus: I. Formation of the ventricular myocardium. Dev. Dyn. 193, 11-23 [DOI] [PubMed] [Google Scholar]
- Milan D. J., Giokas A. C., Serluca F. C., Peterson R. T., MacRae C. A. (2006). Notch1b and neuregulin are required for specification of central cardiac conduction tissue. Development 133, 1125-1132 [DOI] [PubMed] [Google Scholar]
- Moorman A. F., Christoffels V. M. (2003). Cardiac chamber formation: development, genes, and evolution. Physiol. Rev. 83, 1223-1267 [DOI] [PubMed] [Google Scholar]
- Morris J. K., Lin W., Hauser C., Marchuk Y., Getman D., Lee K. F. (1999). Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 23, 273-283 [DOI] [PubMed] [Google Scholar]
- Reischauer S., Levesque M. P., Nusslein-Volhard C., Sonawane M. (2009). Lgl2 executes its function as a tumor suppressor by regulating ErbB signaling in the zebrafish epidermis. PLoS Genet. 5, e1000720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentschler S., Zander J., Meyers K., France D., Levine R., Porter G., Rivkees S. A., Morley G. E., Fishman G. I. (2002). Neuregulin-1 promotes formation of the murine cardiac conduction system. Proc. Natl. Acad. Sci. USA 99, 10464-10469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz P. J., Huisken J., Sahai-Hernandez P., Stainier D. Y. (2008). High-speed imaging of developing heart valves reveals interplay of morphogenesis and function. Development 135, 1179-1187 [DOI] [PubMed] [Google Scholar]
- Sedmera D., Pexieder T., Hu N., Clark E. B. (1997). Developmental changes in the myocardial architecture of the chick. Anat. Rec. 248, 421-432 [DOI] [PubMed] [Google Scholar]
- Sedmera D., Pexieder T., Vuillemin M., Thompson R. P., Anderson R. H. (2000). Developmental patterning of the myocardium. Anat. Rec. 258, 319-337 [DOI] [PubMed] [Google Scholar]
- Sedmera D., Reckova M., deAlmeida A., Sedmerova M., Biermann M., Volejnik J., Sarre A., Raddatz E., McCarthy R. A., Gourdie R. G., et al. (2003). Functional and morphological evidence for a ventricular conduction system in zebrafish and Xenopus hearts. Am. J. Physiol. Heart Circ. Physiol. 284, H1152-H1160 [DOI] [PubMed] [Google Scholar]
- Sehnert A. J., Huq A., Weinstein B. M., Walker C., Fishman M., Stainier D. Y. (2002). Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 31, 106-110 [DOI] [PubMed] [Google Scholar]
- Shu X., Cheng K., Patel N., Chen F., Joseph E., Tsai H. J., Chen J. N. (2003). Na,K-ATPase is essential for embryonic heart development in the zebrafish. Development 130, 6165-6173 [DOI] [PubMed] [Google Scholar]
- Stankunas K., Hang C. T., Tsun Z. Y., Chen H., Lee N. V., Wu J. I., Shang C., Bayle J. H., Shou W., Iruela-Arispe M. L., et al. (2008). Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev. Cell 14, 298-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri C., Jones P. F., Patan S., Bartunkova S., Maisonpierre P. C., Davis S., Sato T. N., Yancopoulos G. D. (1996). Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87, 1171-1180 [DOI] [PubMed] [Google Scholar]
- Traver D., Paw B. H., Poss K. D., Penberthy W. T., Lin S., Zon L. I. (2003). Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 4, 1238-1246 [DOI] [PubMed] [Google Scholar]
- Westerfield M. (2000). The Zebrafish Book: a Guide for the Laboratory Use of Zebrafish (Danio rerio). Eugene: University of Oregon Press; [Google Scholar]
- Woldeyesus M. T., Britsch S., Riethmacher D., Xu L., Sonnenberg-Riethmacher E., Abou-Rebyeh F., Harvey R., Caroni P., Birchmeier C. (1999). Peripheral nervous system defects in erbB2 mutants following genetic rescue of heart development. Genes Dev. 13, 2538-2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.