

Abstract
Recent studies show that silibinin possesses a strong anti-neoplastic potential against many cancers; however, its efficacy and underlying molecular mechanisms in non-small cell lung cancer (NSCLC) are not well-defined. Herein, we assessed silibinin activity on prime endpoints and key molecular targets such as cell number, cell cycle progression and cell cycle regulatory molecules in three cell lines representing different NSCLC subtypes, namely large cell carcinoma cells (H1299 and H460) and a bronchioalveolar carcinoma cell line (H322). Silibinin treatment (10–75 μM) inhibited cell growth and targeted cell cycle progressing causing a prominent G1 arrest in dose- and time-dependent manner. In mechanistic studies, silibinin (50–75 μM) modulated the protein levels of CDKs (4, 6 and 2), cyclins (D1, D3 and E), CDKIs (p18/INK4C, p21/Cip1 and p27/Kip1) in a differential manner in these three cell lines. Consistent with these observations, silibinin caused a reduction in kinase activity of CDK4 and CDK2 in all cell lines except no effect on CDK4 kinase activity in H460 cells, and concomitantly reduced Rb phosphorylation. Together, for the first time, these results identify potential molecular targets and anticancer effects of silibinin in NSCLC cells representing different NSCLC subtypes.
Keywords: chemoprevention, non-small cell lung cancer, silibinin, cell cycle
INTRODUCTION
The number one cause of cancer-related mortalities is lung cancer, which accounts for 1.18 million deaths, worldwide [1]. According to the recent statistics reported by the American Cancer Society, 159,000 deaths are estimated to occur from lung cancer in 2009 alone, in the United States [2]. Despite the available standard therapies, the clinical prognosis of this disease continues to remain poor with an overall 15% survival rate, 5 years after the diagnosis [3,4]. Non-small cell lung cancer (NSCLC) accounts for approximately 80–85% of all lung cancers, suggesting that additional strategies are needed to control this disease. The use of systemic chemotherapeutic drugs and molecular-targeted therapies in the treatment of patients with locally advanced or metastatic NSCLC is limited due to the associated acute and cumulative dose limiting toxicities and acquisition of drug resistance [4,5]. This fact further stresses on the importance of developing new effective alternative chemopreventive therapies, with minimal adverse effects. Prevention and therapeutic intervention by developing new phytochemicals which are nontoxic, cost-effective, and physiologically bioavailable is an emerging field in cancer management [6,7]. One such widely known phytochemical is Silibinin (Fig. 1A), which is isolated from Silybum marianum (L.) Gaertn and is primarily used for its hepatoprotective and antioxidant activity. Silymarin is the major component of the crude form of milk thistle extract, comprising of several flavanolignans including silibinin and its stereoisomers, namely isosilybin A, isosilybin B, silychristin, isosilychristin and silydianin. Both silibinin and silymarin have human consumption and acceptability, and have been used clinically in Europe, USA and Asia [7–10]. Silibinin has been shown to possess strong chemopreventive and anticancer efficacy against various cancer models of skin [11,12], prostate [13–16], lung [17–20], bladder [9], colon [8] etc. Moreover, it has no LD50 reported in laboratory animals and also has been considered exceptionally safe as it has exhibited extremely low toxicity even at acute or chronic administration in both animals and humans, which emphasizes on the importance of utilizing this effective biological agent in cancer chemoprevention studies [7–9].
Figure 1.

Silibinin inhibits the growth of human NSCLC cells in a dose-dependent manner. (A) Chemical structure of silibinin. (B-D) H1299, H460, and H322 cells were treated with DMSO (control) or various doses of silibinin (10–75 μM) for 24, 48 and 72 h. At the end of each treatment time, adherent and non-adherent cells were collected and processed for determination of total cell number (B), live cell number (C), and dead cells (D) as mentioned in Materials and methods. (E) H1299, H460 and H322 cells were treated with either DMSO control or silibinin (50 or 75 μM) for 48 h. The percentage of apoptotic cell population was determined by annexin V and PI staining followed by flow cytometry as described in the Material and methods. The data shown are mean ± SE of four samples for each treatment. These results were similar in three independent experiments. ‡, p<0.01; *, p<0.05; #, p<0.001versus control group; Sb, silibinin.
Existing literature underscores the critical role of silibinin in regulating the various mitogenic signaling cascades involved in controlling cell cycle progression, cell proliferation, and cell survival [13]. In previously conducted preclinical studies, silibinin has been shown to exert its anti-neoplastic actions through several mechanisms which include (i) G1 arrest and apoptosis, along with an increase of p21 and p27 protein levels [7–9,13,21–23], (ii) reduced phosphorylation of retinoblastoma protein leading to stability of the complex formed with E2F [24,25], (iii) inhibition of erbB1 activation with reduced ligand binding to this receptor as demonstrated in prostate cancer DU145 cell line [26], (iv) inhibition of the activation of transcription factor NF-κB, an important component in cell survival and chemotherapy resistance [27,28], and (v) increasing the expression of IGFBP3 in human prostate carcinoma DU145 and PC-3 tumor xenografts in athymic nude mice [14,29,30]. The pleiotropic effects of silibinin have also been extended to several efficacious chemo-combination studies. Silibinin along with doxorubicin showed strong synergism in the growth inhibition of DU145 cells [15]. Another combination study showed that silibinin sensitizes the androgen-independent DU145 cells to cisplatin- and carboplatin-induced cell growth inhibition and apoptotic death [16]. A combination of silibinin and docetaxel exhibited little or no synergy in growth inhibitory and apoptotic effects in PC-3, DU145, and LNCaP cells, however, silibinin in combination with mitoxantrone decreased cell viability of all three cell lines and was able to overcome the relative resistance of PC-3 cells to mitoxantrone-induced apoptosis [31]. Furthermore, silibinin in combination with paclitaxel has been proposed to be a beneficial chemotherapeutic strategy, especially in patients with tumors refractory to paclitaxel alone [32].
With respect to lung cancer, silibinin has been implicated to cause significant growth inhibition and apoptosis, in both SCLC (SHP-77 cells) and NSCLC (A549 cells), together with some alterations in cell cycle checkpoints [17]. In another in vitro study, using A549 cells, silibinin was shown to reduce MMP-2 and u-PA levels, through the suppression of ERK1/2 (MAPK) and by enhancing the TIMP-2 expression [18,19]. Inhibition of multiple cytokine-induced signaling pathways regulating iNOS expression, in A549 cells, has been demonstrated using silibinin [20]. Extensive studies in vivo, using silibinin along with doxorubicin has showed synergistic suppression of human lung A549 xenograft growth in athymic nude mice [33]. Furthermore, dietary silibinin (0.1–1%) was reported to inhibit tumor cell proliferation and angiogenesis in a chemically-induced lung tumorigenesis A/J mouse model [34]. Though these encouraging findings have established silibinin as a promising agent in chemoprevention of lung cancer, its role in non-small cell lung cancer is an unexplored area of interest. Hence, the focus of current study was to evaluate the anti-neoplastic potency of silibinin in non-small cell lung cancer cell lines, and to characterize its mechanism of action.
MATERIALS AND METHODS
Cell Lines and Reagents
Silibinin (MW = 482.4), Propidium iodide (PI) and β-Actin were obtained from Sigma Aldrich Chemical Co. (St. Louis, MO). RPMI 1640 media and other cell culture materials were from Invitrogen Corporation (Gaithersburg, MD). Human NSCLC H1299 (null p53, wt Rb) and H460 (wt p53, wt Rb) cells were obtained from American Type Culture Collection (Manassas, VA) and H322 (mutated p53, wt Rb) cells were a kind gift from Dr. Daniel Chan (Department of Medicine, Division of Medical Oncology, University of Colorado Denver). Antibodies for CDK4, CDK6, CDK2, cyclin D1, cyclin D3 and cyclin E, and RB-GST fusion protein were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody for p21/Cip1 was from Millipore Corporation (Temecula, CA) and p27/Kip1 was from Neomarkers (Fremont, CA). ECL detection system and anti-mouse HRP-conjugated secondary antibody were from GE Healthcare (Piscataway, NJ) and anti-rabbit peroxidase-conjugated secondary antibody was from Cell Signaling Technology (Danvers, MA).
Cell Culture and Silibinin Treatments
Human NSCLC H1299 cells (passage 3–45), H460 cells (passage 3–34), and H322 cells (passage 3–30) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin G and 100 μg/ml streptomycin sulfate at 37°C in a humidified 5% CO2 incubator. After plating, 40–50% confluent cells were treated with varying doses of silibinin (10–75 μM in medium), which was originally dissolved in dimethyl sulfoxide (DMSO), for different time periods (24–72 h) under serum condition. Apart from the control, an equal amount of DMSO was present in each treatment; though its concentration did not exceed 0.1% (v/v) in any treatment group.
Cell Growth and Cell Death Assays
Five thousand cells/cm2 were plated in 60-mm dishes under standard conditions and left overnight. Cells were subsequently treated with DMSO control alone or with varying doses of silibinin (10–75 μM) as mentioned above. At the end of respective treatment time points (24–72 h), both adherent and non-adherent cells were harvested by brief trypsinization, washed with phosphate-buffered saline (PBS) and collected in separate tubes. Each sample was counted in duplicate using a hemocytometer and an inverted microscope and total cell number was determined. Each treatment group at different time points had 3–4 independent plates. Trypan blue dye exclusion method was used to differentiate between live and dead cells.
Quantitative Detection of Apoptosis
To quantify silibinin-induced apoptotic death of NSCLC cells, annexin V and propidium iodide staining was performed followed by flow cytometry, as described recently (8). Briefly, cells were plated in 60 mm dishes, and at 50% confluency, cells were treated with different concentrations of silibinin. After these treatments, cells were collected by brief trypsinization and washed with phosphate-buffered saline twice, and subjected to annexin V and propidium iodide staining using Vybrant Apoptosis Assay Kit2 following the step-by-step protocol provided by the manufacturer. After the staining, fluorescence-activated cell sorter analysis was performed for the quantification of the percent apoptotic cells.
Cell Cycle Distribution Analysis using Fluorescence-activated Cell Sorting
As mentioned earlier, after 24, 48 or 72 h of treatment time, cells were harvested by brief trypsinization, washed with phosphate-buffered saline and collected by centrifugation. Cell pellets were washed twice with ice-cold PBS, and approximately 0.5 × 106 cells were suspended in 500 μl of saponin/PI solution [0.3% saponin (w/v), 25 μg/ml PI (w/v), 0.1 mM ethylenediaminetetraacetic acid and 10 μg/ml RNase A (w/v) in PBS] and incubated at 4°C for 24 h in the dark. Stained cells were then subjected to flow cytometry at fluorescence-activated cell sorting analysis core facility at the University of Colorado Cancer Center to analyze the cell cycle distribution.
Western Blotting
At 60–65% confluency, cells were treated as described earlier and cell lysates were prepared in non-denaturing lysis buffer [10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% triton X-100, 1 mM EDTA, 1 mM EGTA, 0.3mM phenyl methyl sulfonyl fluoride, 0.2 mM sodium orthovanadate, 0.5% NP40, 5U/ml aprotinin]. Briefly, media was aspirated from the culture plates followed by two washings with ice-cold PBS. Subsequently, cells were incubated in lysis buffer for 10 min on ice, collected by scraping and kept on ice for 30 min. Finally, after freeze and thaw, cell lysates were centrifuged at 4°C for 30 min at 14,000 rpm. Protein concentrations in lysates were determined by using Bio-Rad DC protein assay kit (Bio Rad, Hercules, CA). Fifty-eighty μg of protein lysate per sample was denatured in 2X sample buffer and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 8, 12 or 16% Tris-glycine gel. The separated proteins were transferred on to nitrocellulose membrane followed by blocking with 5% non-fat milk powder (w/v) in Tris-buffered saline [10 mM Tris-HCl (pH 7.5), 100 mM NaCl, 0.1% Tween 20] for 1h at room temperature. Specific primary antibodies were used to probe the protein levels of the different desired molecules, following which appropriate peroxidase-conjugated secondary antibodies were used and visualized by ECL. Each membrane was stripped and reprobed with anti β-actin antibody, to ensure equal protein loading.
Kinase Assays
As reported earlier [9], to determine CDKs (4 and 2) associated kinase activity, 200–300 μg of protein lysates from each sample were pre-cleared with protein A/G-plus agarose beads and CDK4 or CDK2 protein was immunoprecipitated using specific antibody and protein A/G plus agarose beads. After overnight incubation at 4°C, beads conjugated with antibody and protein were washed 3 times with Rb-lysis buffer [50 mM HEPES-KOH (pH 7.5), 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 1 mM DTT, 80 mM β-glycerophosphate, 1 mM NaF, 0.1 mM sodium orthovanadate, 0.1% Tween 20, 10% glycerol, 1 mM PMSF and 10 μg/ml aprotinin and leupeptin] and 2 times with Rb-kinase assay buffer [50 mM HEPES-KOH (pH 7.5), 2.5 mM EGTA, 1 mM DTT, 10 mM β-glycerophosphate, 10 mM MgCl2, 1 mM NaF, 1 mM DTT and 10 mM sodium orthovanadate]. Phosphorylation of the specific substrate (Rb-GST for CDK4 and Histone H1 for CDK2) was measured by incubating the beads with 30 μl of hot kinase solution (2.5 μg of Rb-GST or histone H1, 0.5μl of γ-32P ATP, 0.5μl of 0.1 mM ATP and 28.75 μl of kinase buffer) for 30 min at 37° C. The reaction was stopped by boiling the samples in 5X SDS sample buffer for 5 min. Samples were resolved by SDS-PAGE and the gel was dried and subjected to autoradiography. For in vitro kinase assay, total cell lysate from untreated human NSCLC H1299 cells was used and both CDK4 and CDK2 immunocomplexes were obtained as described above, and incubated with specific doses of silibinin (0–75 μM) along with the substrate for 30 min following the detailed kinase assay as described above.
Densitometry and Statistical Analyses
Using Adobe Photoshop 6.0 (Adobe systems, San Jose, CA), bands were scanned and their mean density was analyzed by Scion Image program (National Institutes of Health, Bethesda, MD). Densitometry data which is represented below the bands are the ‘fold change’ as compared with respective DMSO control, after normalization with respective loading controls (β-actin). Statistical analysis was done using Sigma Stat 3.5 software (Jandel Scientific, San Rafael, CA) and the statistical significance for the differences between the control and treated groups was determined by one way ANOVA, followed by Bonferroni’s t-test for multiple comparisons. p<0.05 was considered statistically significant.
RESULTS
Effect of Silibinin on the Growth of NSCLC Cells
First, we assessed the effects of silibinin on NSCLC cell proliferation (H1299, H460 and H322 cells). Cells were treated with varying concentrations of the drug (10–75 μM) dissolved in DMSO for 24, 48 and 72 h. At the end of each time point, the total cell number, live cell number and cell death were determined. H1299 cells showed cell growth inhibitory effects in a dose- and time- dependent manner. The range of decrease in total cell number was 22% (p<0.05) to 43% (p<0.001), and 21% (p<0.05) to 72% (p<0.001) following 48 and 72 h treatments, respectively, using the specified doses of silibinin (Figure 1B). In H460 cells, total cell number decreased by 33% (p<0.001) to 53% (p<0.001) following 48h and 19% (p<0.05) to 65% (p<0.001) following 72 h of similar silibinin treatments (Figure 1B). In H322 cells, the same silibinin concentrations were effective in decreasing the total cell number by 28% (p<0.05) to 53% (p<0.001), 17% (p<0.05) to 43% (p<0.001) and 24% (p<0.05) to 62% (p<0.001), following 24, 48 and 72 h treatments, respectively (Figure 1B). The rank order of potency for growth inhibition with 75μM silibinin concentration at 72 h was H1299 (72%, p<0.001) > H460 (65%, p<0.001) > H322 (62%, p<0.001). We also calculated the total live and dead cell numbers to further address their involvement in the growth inhibitory effect of silibinin in all the three cell lines. As shown in Figure 1C, silibinin treatment (10–75 μM) for 24–72 h resulted in a decrease in live cells in both dose- and time-dependent manner in all the three cell lines. Importantly, the patterns of silibinin growth inhibitory effects in terms of total versus live cell numbers in all three cell lines were very close, suggesting that silibinin has potent anti-proliferative efficacy in all the three NSCLC cell lines studied at physiologically achievable concentrations.
Effect of Silibinin on NSCLC Cell Death
A linear increase in percent of dead cells was observed with increasing concentrations of silibinin at 24, 48 and 72 h time points in H1299 and H322 cells; which was moderate but significant at higher doses (Figure 1D). In contrast, a similar trend of significant increase in cell death was not observed in H460 cells after 24 and 48 h of treatment (Figure 1D). However, none of the silibinin concentrations and treatment times induced profound cell death, as the maximum effect observed was upto 9% (p<.001) in H1299, 9% (p<.05) in H460 and 8% (p<.001) in H332 cells compared with 4%, 5% and 3% in vehicle controls, respectively at the highest dose of silibinin for 72 h except in H460 cell line, where maximum effect was observed at 50 μM dose of silibinin for 48 h (Figure 1D). Consistent with a lack of profound cell death effect by silibinin in all three NSCLC cell lines in Trypan blue assay, there was also no significant apoptotic cell death observed in the three cell lines when treated with specified doses (50–75 μM) of silibinin (Figure 1E). Together, these results convincingly show that the major biological effect of silibinin in NSCLC cells is growth inhibition, and that cell death is not the prime reason for the reduction in cell number.
Effect of Silibinin on Cell Cycle Progression of NSCLC Cells
We next asked the question whether growth inhibitory effect of silibinin is associated with an alteration in cell cycle progression of NSCLC cells, and found that indeed silibinin induces a prominent G1 arrest in the cell cycle progression of H1299, H460 and H322 cells. Compared to vehicle-treated control H1299 cells which showed 30–38% G1 population, silibinin treatment (25–75 μM) increased cells in G1 phase in the range of 35 (p<0.05)-60% (p<0.001), 38 (p<0.05)-56% (p<0.001) and 44–56% (p<0.001) at 24, 48 and 72 h, respectively (Figure 2A). In control H460 cells, 45–60% cells were in G1 phase as compared to the silibinin treatment (25–75μM) where 50% (p<0.001), 53 (p<0.05)-58% (p<0.001) and 66% (p<0.05) of cells were in G1 phase at 24, 48 and 72 h, respectively (Figure 2B). Similar silibinin treatments of H322 cells caused 47 (p<0.001)-56% [(p<0.001), 24h], 46 (p<0.05)-74% [(p<0.001), 48 h] and 82–84% [(p<0.001), 72 h] cells in G1 phase as compared to DMSO treated controls showing 39, 42 and 72% G1 population at 24, 48 and 72h, respectively (Figure 2C). The observed increase in G1 phase cells by silibinin was accompanied by a decrease in both S and G2/M phase cell population, except in H460 cells, which also showed an increased G2/M population at 72 h.
Figure 2.

Effect of silibinin on cell cycle distribution in human NSCLC cells. H1299 (A), H460 (B) and H322 (C) cells were treated with either DMSO control or various doses of silibinin (10–75 μM) for 24, 48 and 72 h. At the end of these treatments, adherent and non-adherent cells were collected and incubated overnight with saponin/PI solution at 4°C as detailed in Materials and methods. The percentage of cells in the different phases of the cell cycle was determined by flow cytometry. Quantitative cell cycle distribution data for silibinin in (A) H1299, (B) H460 and (C) H322 cells after 24, 48, and 72 h of treatment are shown. The data shown are mean ± SE of three samples for each treatment. ‡, p<0.01; *, p<0.05; #, p<0.001 for differences with control group. Sb; silibinin.
Effect of Silibinin on the Protein Expression of Cyclins, CDKs and CDK Inhibitors
Amongst the many pathways altered in lung cancer malignancy, the most critical one involves the disruption of the normal cell cycle regulation [35]. Next, we analyzed the different cell cycle regulatory molecules which could be involved in the strong G1 arrest caused by silibinin. Western blotting analysis showed that 50 and 75 μM doses of silibinin after 12, 24 and 48 h of treatment caused a decrease in the expression of protein levels of G1 regulatory cyclins and CDKs in H1299, H460 and H322 cells that included CDK2, 4 and 6 and cyclin D1, D3 and E (Figs. 3A, B and C). Apart from positive regulators such as CDKs and cyclins, there are specific CDK inhibitors (INK4 and Cip/Kip family) which control CDK activity, thereby regulating cell cycle through various phases [36]. Various genetic or epigenetic events have been accounted for the loss of p16/INK4A protein in Rb-positive lung cancer cell lines [37]. Our results showed that p18/INK4C levels are increased by silibinin at 12h in H1299 and H460 cells, and at 24 and 48 h in H322 cells (Figure 4). Regarding Cip/Kip family molecules, in H1299 cells, silibinin caused an increase in the protein levels of both p21 and p27 at earlier time points of 12 and 24h; importantly, 48h treatment did not show any effect on p21 levels and strongly reduced p27 levels (Figure 4A). In H460 and H322 cells, silibinin increased the protein levels of p21 at all time points and p27 at 12and 24h time points (Figure 4B and C); a strong decrease in p27 levels by 48h silibinin treatments was also observed in both these cell lines. The Cip/Kip proteins are up-regulated by several known pathways, both at the transcriptional and post-transcriptional levels and are known to be extremely short-lived. Their metabolic stability and degradation through proteolytic pathways are determining factors that decide the fate of the cells, through the cell cycle [13]. In the past, several studies have shown that the ubiquitin-proteosome proteolysis system is a major pathway that regulates p27 levels. Phosphorylation of p27 (Thr187) by CDK2 causes p27 protein to bind with ubiquitin ligase (SCF-SKP2) that leads to 26S proteosome degradation [13]. Accordingly, there is a possibility that silibinin treatment (48 h) induced the p27 binding with SKP2 for the proteosomal degradation; however, further studies are needed in future to address this issue. Nevertheless, our results suggest that the G1 phase arrest by silibinin at 48 h is associated at least in part with the accumulation of p21, if not p27, in all the three NSCLC cell lines.
Figure 3.
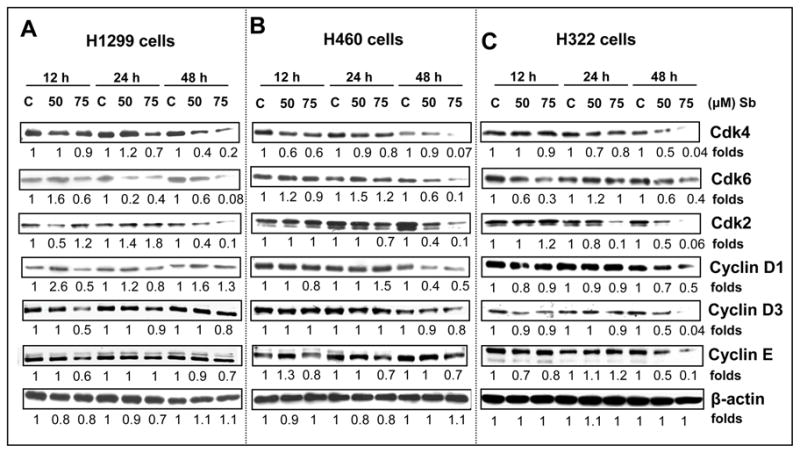
Silibinin modulates the protein levels of G1 phase cell cycle regulatory molecules in human NSCLC cells. H1299 (A), H460 (B) and H322 (C) cells were treated with either DMSO control or various doses of silibinin (50–75 μM) for 12, 24 and 48 h. At the end of each treatment time, cell lysates were prepared in non-denaturing lysis buffer as mentioned in Materials and Methods. For each sample, 50–80 μg of protein lysate was used for SDS-PAGE and western immunoblotting, and membranes were probed for CDK4, CDK6, CDK2, cyclin D1, cyclin D3 and cyclin E. Membranes were stripped and reprobed with anti-β-actin antibody for protein loading correction. Blots shown are representative of at least two-three independent experiments. The densitometry data presented below the bands are ‘fold change’ as compared with respective DMSO control, after normalization with respective loading control (β-actin). C, control; Sb; silibinin.
Figure 4.

Effect of silibinin on the expression of G1 phase-related cyclin-dependent kinase inhibitors in human NSCLC cells. H1299 (A), H460 (B) and H322 (C) cells were treated with either DMSO control or various doses of silibinin (50–75 μM) for 12, 24 and 48 h. At the end of each treatment time, cell lysates were prepared in non-denaturing lysis buffer as mentioned in Materials and methods. For each sample, 50–60μg of protein lysate was used for SDS-PAGE and western immunoblotting, and membranes were probed for p18, p21 and p27. Membranes were stripped and reprobed with anti-β-actin antibody for protein loading correction. Blots shown are representative of at least two-three independent experiments. The densitometry data presented below the bands are ‘fold change’ as compared with DMSO control, after normalization with respective loading control (β-actin). C, control; Sb; silibinin.
Effect of Silibinin on the Kinase Activity of CDKs
Sequential activation of CDK4 and CDK2 in early and mid/late G1 phase takes place, respectively, which can be determined by the kinase activity of these serine-threonine kinases [38]. Cells were treated with desired concentrations of silibinin (50–75 μM) and cell lysates were prepared, and subjected to immunoprecipitation with CDK2 and CDK4 antibodies, following which kinase assays were performed. Silibinin showed inhibition of CDK4 kinase activity in both H1299 and H322 cell lines represented by reduced phosphorylation of Rb-GST, which was used as its substrate (Figure 5A and 5C, upper panel). Similarly, in both these cell lines, silibinin also decreased the kinase activity of CDK2, shown by the marked reduction in phosphorylation levels of its specific substrate, histone H1 (Figure 5A and 5C, lower panel). In H460 cells, the CDK4 kinase activity remained unchanged (Figure 5B, upper panel) whereas the kinase activity of CDK2 significantly decreased by silibinin (Figure 5B, lower panel). Furthermore, we also performed in vitro kinase activity assays for both CDK4 and CDK2 employing total cell lysates from untreated control H1299 cells, to address the issue whether the observed inhibitory effect of silibinin on CDK4 and CDK2 kinase activity was a direct response or due the decreased total protein levels of CDK4 and CDK2 after silibinin treatment. The in vitro addition of silibinin in the kinase assay comprising of CDK4 or CDK2 immunocomplex from untreated cells along with the substrate for 30 min showed that it does not inhibit the direct kinase activity of CDK4 and CDK2 (Figure 5D). These results suggested that the inhibitory effect of silibinin on CDK4 and CDK2 kinase activity is not a direct response; rather it involves a possible decrease in the protein levels of CDK4 and CDK2 by silibinin and/or an increased inhibitory activity of CDKIs following an increase in their levels in silibinin-treated samples.
Figure 5.

Effect of silibinin on the kinase activity of CDK4 and CDK2 in human NSCLC cells. H1299 (A), H460 (B), and H322 (C) cells were treated with either DMSO or 50–75 μM doses of silibinin for 24 h. Kinase activity of CDK4 and CDK2 in the cell extracts were analyzed using Rb-GST and histone H1 as their respective substrates. In-bead kinase assays were performed after immunoprecipitation of the specific protein as described in Materials and methods. (D) Employing equal amount of protein extracted from control H1299 cell, CDK4 and CDK2 were immunoprecipitated and then incubated with indicated doses of silibinin followed by kinase activity assay. Blots shown are representative of at least two independent experiments. C, control; Sb, silibinin; IP, immunoprecipitation.
Effect of Silibinin on Total and Phosphorylated Levels of Retinoblastoma Protein
Eukaryotic cell growth is governed by cell cycle progression where a growth signal “turns-on” an interaction between CDKs and cyclins, activating the CDKs, which phosphorylates retinoblastoma (Rb) and other Rb related proteins (e.g. p107, p130), to release the E2F transcription factors, in order to regulate the expression of various genes required for G1-S phase transition [24,36]. Considering the three cell lines chosen for study, where functional Rb protein is expressed, we asked the question whether the up-regulation of protein levels of CDK inhibitors and decrease in CDK (4 & 2) kinase activities was accompanied by the decrease in Rb phosphorylation, contributing to the growth inhibitory effect of silibinin. Western blot analysis showed a strong decrease in the phosphorylation of Rb at ser807/811 and ser780 sites, in the 24 h silibinin-treated samples of H1299 (50–75 μM), H460 (50–75 μM), and H322 cells (at 75 μM) compared to the respective controls (Figure 6A, B and C). The total Rb levels remained almost unchanged in H1299 and H322 cells, whereas in H460 cells a reduction in the total levels of Rb was observed.
Figure 6.

Silibinin induces hypophosphorylation of Rb in human NSCLC cells. H1299 (A), H460 (B) and H322 (C) cells were treated with either DMSO control or 50 –75 μM doses of silibinin for 24h. At the end of these treatments, total cell lysates were prepared, and subjected to SDS-PAGE on 6% gels followed by western immunoblotting as described in Materials and methods. Membranes were probed for phospho-Rb (Ser 807/ 811), phospho-Rb (Ser 780) and Total Rb. Membranes were stripped and reprobed with anti-β-actin antibody for protein loading correction. Blots shown are representative of at least two independent experiments. The densitometry data presented below the bands are ‘fold change’ as compared with DMSO control, after normalization with respective loading control (β-actin). C, control; Sb; silibinin.
DISCUSSION
Prevention and therapeutic intervention of cancer by non-toxic phytochemicals are newer dimensions that remain to be investigated further. The advantages of the non-toxic physiologically available phytochemicals over the conventional treatment modalities like chemotherapy and radiation therapy, qualifies these agents as ideal candidates in chemoprevention strategies [39]. The anticancer potential of these phytotherapeutics has been extensively studied in various epithelial cancers including prostate, skin, colon, breast, lung, liver etc [31,40]. The relationship between diet and lung cancer has also been explored in many ecologic and case-control studies [41], listing the protective effects of vegetables and fruits intake rich in flavonoids and antioxidants [42,43]. In this regard, a large clinical study has suggested the presence of an inverse association between flavonoid intake and subsequent lung cancer incidences [20].
Accordingly, the findings in the present study showed that silibinin, a well known polyphenolic flavonoid, exhibits strong anti-proliferative effect against human NSCLC (H1299, H460 and H322) cells. These include alterations of the G1-associated cell cycle molecules, explaining its direct relevance to the G1 phase arrest, observed in all the three cell lines, by this agent. Approximately 80–85% of lung tumors are classified as NSCLC [44] of which, adenocarcinoma is the most commonly occurring form accounting for 50% of the cases, followed by squamous cell (20%) and large cell carcinoma (10%). The three cell lines (H1299, H460 and H322 cells) selected for studies, not only differ in their histological grades but also possess different genetic backgrounds, differing in their sensitivities to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors and p53 status. EGFR is overexpressed in the majority of cancers like colorectal, head and neck, breast, NSCLC etc. and has been a major target for new therapies [45]. Currently, two molecular-targeted approaches are used for inhibiting EGFR signaling in advanced chemorefractory NSCLC that includes monoclonal antibodies and EGFR TK-inhibitors [44]. One major limitation of this treatment option is the resistance or sensitivity of cancer cells to these inhibitors, the molecular mechanisms of which are not clearly defined. For example, both H1299 and H460 cells are resistant to EGFR tyrosine kinase inhibitors whereas H322 cells are sensitive. With regard to this aspect, in the present study silibinin was found to exert its chemopreventive effects by inhibiting cell growth, most likely via inducing cell cycle arrest at G1-S transition, in the three cell lines irrespective of their differences, as mentioned.
Cell growth and proliferation in mammalian cells are coordinated events that are regulated through cell cycle progression which constitutes the G0, G1, S, G2 and M phases. The aberrations at the G1-S checkpoint are associated with deregulated cell cycle progression in most human cancers. This check point is usually controlled by CDK4, 6 and 2 in association with cyclin D and cyclin E [42]. Our results showed that silibinin treatment caused a decrease in the protein levels of CDK4, 6 and 2, along with a decrease in cyclin D1, and D3 protein levels, explaining the plausible role of these cell cycle regulatory proteins, in the silibinin-induced G1 arrest observed in H1299, H460 and H322 cell lines. CDKs are activated by phosphorylation/ dephosphorylation events and binding to cyclins, the regulatory subunits, to form heterodimeric complexes. The activities of CDKs are constrained by the CDK inhibitors which fall into two categories namely INK4 family of proteins (p16/INK4A, p15/INK4B, p18/INK4C, p19/INK4D) and the Cip/Kip family consisting of p21/Cip1, p27/Kip1 and p57/Kip2 [21,46–49]. The INK4 proteins specifically inhibit the catalytic subunits of CDK4 and CDK6; however, the Cip/Kip family members are the negative regulators of cyclin E- and cyclin A-CDK2 and cyclin B-CDK1 holoenzymes. However, p27/Kip1 is phosphorylated by cyclin E-CDK2 complex and subsequently targeted for degradation [21,46–49]. Cell cycle inhibition by p16 has been associated with the post-transcriptional induction of p21 and strong inhibition of cyclin E–CDK2 kinase activity; however, some other studies have reported that Cip/Kip family members promote the association of D-type cyclins with CDKs by counteracting the effects of p16 molecules. This type of functional cooperation between different cell cycle inhibitory proteins has been proposed to constitute another level of regulation in cell growth control and tumor suppression [50]. Our results revealed that silibinin up-regulated p21 and p27 protein levels in H460 and H322 cells at early time points (12 and 24 h) whereas in H1299 cells, silibinin decreased the protein levels of p21 at 12 and 24 h. Further studies are needed to clarify the role of differential regulation of p21 and p27 in silibinin-mediated G1 arrest in H1299 cells.
CDK4 and 6 are substrate specific and phosphorylate only Rb, whereas cyclin E-CDK2 has a broader range of specificity to phosphorylate histone H1, Rb, p27/Kip1 and possibly other substrates that help trigger the firing of replication origins, centrosome duplication and histone biosynthesis [48,51,52]. In accordance with the previous observations, silibinin treatment reduced the kinase activity of CDK4 and 2 in both H1299 and H322 cells, suggesting their role in silibinin-mediated G1 arrest. In the mid-G1 phase, CDK4 and 6 initiate the phosphorylation of Rb, followed by the sequential activation of cyclin E-CDK2 complex to phosphorylate Rb on the additional sites. The hyper-phosphorylated state of Rb is maintained throughout the cell cycle (by the cyclin A- and B-dependent kinases) till the cell divides and exits mitosis [48]. Rb is known to directly bind to the activation domain of activator E2Fs and block its activity [53]. Upon phosphorylation, Rb and related proteins release the tethered E2F transcription factors that in complex with DP1 and DP2, either activate (E2F1, 2 and 3) or repress (E2F4 and 5) gene transcription essential for the G1-S transition and commitment to mitosis [54]. Reduction in the Rb phosphorylation at ser807/811 and ser780 in silibinin treated samples of H1299, H460 and H322 cells further clarifies the role of CDK-cyclin-Rb axis in silibinin-caused inhibition of G1-S transition.
A recently conducted phase I clinical trial with silibinin in advanced prostate cancer patients showed that the upto 100 μmol/L plasma level of free silibinin was achievable without any associated major toxicity [55]. Thus, the range of silibinin doses used in this cell culture study (10–75 μM) could be physiologically achievable in vivo in humans and therefore further signify the biological relevance of the findings to both prevention and intervention of various human malignancies including lung cancer. To summarize the current findings, silibinin was found to (i) cause inhibition of cell proliferation, (ii) induce G1 arrest, (iii) decrease the protein levels of G1 phase associated CDKs and their corresponding cyclins, (iv) increase the CDKIs protein levels, (v) inhibit the kinase activity of CDKs (4 and 2), and (vi) decrease the phosphorylation status of Rb in NSCLC cells. Collectively, these findings suggest that silibinin exerts strong anti-proliferative effect against human non-small cell lung carcinoma H1299, H460 and H322 cells. Furthermore, this is the first detailed mechanistic study of silibinin and its cell cycle effects in human NSCLC cells of varying clinical and histopathological profiles. Overall, this study identifies the potential key molecular targets of silibinin with relevance to its efficacy in NSCLC cells. These results might provide a valuable insight to use silibinin in combination with chemotherapeutic agents in NSCLC, however, that needs to be investigated. Further, in vitro and in vivo studies are needed to unravel the potential clinical utility of this naturally occurring chemopreventive agent against lung cancer.
Acknowledgments
Grant support: This work was supported by NCI RO1 grant CA113876.
Abbreviations
- NSCLC
non-small cell lung cancer
- SCLC
small cell lung cancer
- PI
propidium iodide
- CDK
cyclin-dependent kinase
- CDKI
cyclin-dependent kinase inhibitor
- FACS
fluorescence-activated cell sorting
- Rb
retinoblastoma
- HRP
horseradish peroxidase
REFRENCES
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics, 2009. CA Cancer J Clin. 2009 doi: 10.3322/caac.20006. (In press) [DOI] [PubMed] [Google Scholar]
- 3.Stearman RS, Dwyer-Nield L, Grady MC, Malkinson AM, Geraci MW. A macrophage gene expression signature defines a field effect in the lung tumor microenvironment. Cancer Res. 2008;68:34–43. doi: 10.1158/0008-5472.CAN-07-0988. [DOI] [PubMed] [Google Scholar]
- 4.Sun S, Schiller JH, Spinola M, Minna JD. New molecularly targeted therapies for lung cancer. J Clin Invest. 2007;117:2740–2750. doi: 10.1172/JCI31809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006;66:10100–10111. doi: 10.1158/0008-5472.CAN-06-1684. [DOI] [PubMed] [Google Scholar]
- 6.Deep G, Singh RP, Agarwal C, Kroll DJ, Agarwal R. Silymarin and silibinin cause G1 and G2-M cell cycle arrest via distinct circuitries in human prostate cancer PC3 cells: a comparison of flavanone silibinin with flavanolignan mixture silymarin. Oncogene. 2006;25:1053–1069. doi: 10.1038/sj.onc.1209146. [DOI] [PubMed] [Google Scholar]
- 7.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Identifying the differential effects of silymarin constituents on cell growth and cell cycle regulatory molecules in human prostate cancer cells. Int J Cancer. 2008;123:41–50. doi: 10.1002/ijc.23485. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal C, Singh RP, Dhanalakshmi S, et al. Silibinin upregulates the expression of cyclin-dependent kinase inhibitors and causes cell cycle arrest and apoptosis in human colon carcinoma HT-29 cells. Oncogene. 2003;22:8271–8282. doi: 10.1038/sj.onc.1207158. [DOI] [PubMed] [Google Scholar]
- 9.Tyagi A, Agarwal C, Harrison G, Glode LM, Agarwal R. Silibinin causes cell cycle arrest and apoptosis in human bladder transitional cell carcinoma cells by regulating CDKI-CDK-cyclin cascade, and caspase 3 and PARP cleavages. Carcinogenesis. 2004;25:1711–1720. doi: 10.1093/carcin/bgh180. [DOI] [PubMed] [Google Scholar]
- 10.Kim NC, Graf TN, Sparacino CM, Wani MC, Wall ME. Complete isolation and characterization of silybins and isosilybins from milk thistle (Silybum marianum) Org Biomol Chem. 2003;1:1684–1689. doi: 10.1039/b300099k. [DOI] [PubMed] [Google Scholar]
- 11.Mohan S, Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Silibinin modulates UVB-induced apoptosis via mitochondrial proteins, caspases activation, and mitogen-activated protein kinase signaling in human epidermoid carcinoma A431 cells. Biochem Biophys Res Commun. 2004;320:183–189. doi: 10.1016/j.bbrc.2004.05.153. [DOI] [PubMed] [Google Scholar]
- 12.Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Dual efficacy of silibinin in protecting or enhancing ultraviolet B radiation-caused apoptosis in HaCaT human immortalized keratinocytes. Carcinogenesis. 2004;25:99–106. doi: 10.1093/carcin/bgg188. [DOI] [PubMed] [Google Scholar]
- 13.Roy S, Kaur M, Agarwal C, Tecklenburg M, Sclafani RA, Agarwal R. p21 and p27 induction by silibinin is essential for its cell cycle arrest effect in prostate carcinoma cells. Mol Cancer Ther. 2007;6:2696–2707. doi: 10.1158/1535-7163.MCT-07-0104. [DOI] [PubMed] [Google Scholar]
- 14.Flaig TW, Su LJ, Harrison G, Agarwal R, Glode LM. Silibinin synergizes with mitoxantrone to inhibit cell growth and induce apoptosis in human prostate cancer cells. Int J Cancer. 2007;120:2028–2033. doi: 10.1002/ijc.22465. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi AK, Singh RP, Agarwal C, Chan DC, Agarwal R. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth Inhibition, G2-M arrest, and apoptosis. Clin Cancer Res. 2002;8:3512–3519. [PubMed] [Google Scholar]
- 16.Dhanalakshmi S, Agarwal P, Glode LM, Agarwal R. Silibinin sensitizes human prostate carcinoma DU145 cells to cisplatin- and carboplatin-induced growth inhibition and apoptotic death. Int J Cancer. 2003;106:699–705. doi: 10.1002/ijc.11299. [DOI] [PubMed] [Google Scholar]
- 17.Sharma G, Singh RP, Chan DC, Agarwal R. Silibinin induces growth inhibition and apoptotic cell death in human lung carcinoma cells. Anticancer Res. 2003;23:2649–2655. [PubMed] [Google Scholar]
- 18.Chu SC, Chiou HL, Chen PN, Yang SF, Hsieh YS. Silibinin inhibits the invasion of human lung cancer cells via decreased productions of urokinase-plasminogen activator and matrix metalloproteinase-2. Mol Carcinog. 2004;40:143–149. doi: 10.1002/mc.20018. [DOI] [PubMed] [Google Scholar]
- 19.Chen PN, Hsieh YS, Chiou HL, Chu SC. Silibinin inhibits cell invasion through inactivation of both PI3K-Akt and MAPK signaling pathways. Chem Biol Interact. 2005;156:141–150. doi: 10.1016/j.cbi.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Chittezhath M, Deep G, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits cytokine-induced signaling cascades and down-regulates inducible nitric oxide synthase in human lung carcinoma A549 cells. Mol Cancer Ther. 2008;7:1817–1826. doi: 10.1158/1535-7163.MCT-08-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Isosilybin B and isosilybin A inhibit growth, induce G1 arrest and cause apoptosis in human prostate cancer LNCaP and 22Rv1 cells. Carcinogenesis. 2007;28:1533–1542. doi: 10.1093/carcin/bgm069. [DOI] [PubMed] [Google Scholar]
- 22.Zi X, Agarwal R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest, leading to differentiation of prostate carcinoma cells: implications for prostate cancer intervention. Proc Natl Acad Sci U S A. 1999;96:7490–7495. doi: 10.1073/pnas.96.13.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zi X, Feyes DK, Agarwal R. Anticarcinogenic effect of a flavonoid antioxidant, silymarin, in human breast cancer cells MDA-MB 468: induction of G1 arrest through an increase in Cip1/p21 concomitant with a decrease in kinase activity of cyclin-dependent kinases and associated cyclins. Clin Cancer Res. 1998;4:1055–1064. [PubMed] [Google Scholar]
- 24.Tyagi A, Agarwal C, Agarwal R. The cancer preventive flavonoid silibinin causes hypophosphorylation of Rb/p107 and Rb2/p130 via modulation of cell cycle regulators in human prostate carcinoma DU145 cells. Cell Cycle. 2002;1:137–142. [PubMed] [Google Scholar]
- 25.Tyagi A, Agarwal C, Agarwal R. Inhibition of retinoblastoma protein (Rb) phosphorylation at serine sites and an increase in Rb-E2F complex formation by silibinin in androgen-dependent human prostate carcinoma LNCaP cells: role in prostate cancer prevention. Mol Cancer Ther. 2002;1:525–532. [PubMed] [Google Scholar]
- 26.Zi X, Grasso AW, Kung HJ, Agarwal R. A flavonoid antioxidant, silymarin, inhibits activation of erbB1 signaling and induces cyclin-dependent kinase inhibitors, G1 arrest, and anticarcinogenic effects in human prostate carcinoma DU145 cells. Cancer Res. 1998;58:1920–1929. [PubMed] [Google Scholar]
- 27.Saliou C, Rihn B, Cillard J, Okamoto T, Packer L. Selective inhibition of NF-kappaB activation by the flavonoid hepatoprotector silymarin in HepG2. Evidence for different activating pathways. FEBS Lett. 1998;440:8–12. doi: 10.1016/s0014-5793(98)01409-4. [DOI] [PubMed] [Google Scholar]
- 28.Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits constitutive and TNFalpha-induced activation of NF-kappaB and sensitizes human prostate carcinoma DU145 cells to TNFalpha-induced apoptosis. Oncogene. 2002;21:1759–1767. doi: 10.1038/sj.onc.1205240. [DOI] [PubMed] [Google Scholar]
- 29.Singh RP, Deep G, Blouin MJ, Pollak MN, Agarwal R. Silibinin suppresses in vivo growth of human prostate carcinoma PC-3 tumor xenograft. Carcinogenesis. 2007;28:2567–2574. doi: 10.1093/carcin/bgm218. [DOI] [PubMed] [Google Scholar]
- 30.Singh RP, Dhanalakshmi S, Tyagi AK, Chan DC, Agarwal C, Agarwal R. Dietary feeding of silibinin inhibits advance human prostate carcinoma growth in athymic nude mice and increases plasma insulin-like growth factor-binding protein-3 levels. Cancer Res. 2002;62:3063–3069. [PubMed] [Google Scholar]
- 31.Raina K, Agarwal R. Combinatorial strategies for cancer eradication by silibinin and cytotoxic agents: efficacy and mechanisms. Acta Pharmacol Sin. 2007;28:1466–1475. doi: 10.1111/j.1745-7254.2007.00691.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhou L, Liu P, Chen B, et al. Silibinin restores paclitaxel sensitivity to paclitaxel-resistant human ovarian carcinoma cells. Anticancer Res. 2008;28:1119–1127. [PubMed] [Google Scholar]
- 33.Singh RP, Mallikarjuna GU, Sharma G, et al. Oral silibinin inhibits lung tumor growth in athymic nude mice and forms a novel chemocombination with doxorubicin targeting nuclear factor kappaB-mediated inducible chemoresistance. Clin Cancer Res. 2004;10:8641–8647. doi: 10.1158/1078-0432.CCR-04-1435. [DOI] [PubMed] [Google Scholar]
- 34.Singh RP, Deep G, Chittezhath M, et al. Effect of silibinin on the growth and progression of primary lung tumors in mice. J Natl Cancer Inst. 2006;98:846–855. doi: 10.1093/jnci/djj231. [DOI] [PubMed] [Google Scholar]
- 35.Caputi M, Russo G, Esposito V, Mancini A, Giordano A. Role of cell-cycle regulators in lung cancer. J Cell Physiol. 2005;205:319–327. doi: 10.1002/jcp.20424. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23:9408–9421. doi: 10.1200/JCO.2005.01.5594. [DOI] [PubMed] [Google Scholar]
- 37.Shapiro GI, Park JE, Edwards CD, et al. Multiple mechanisms of p16INK4A inactivation in non-small cell lung cancer cell lines. Cancer Res. 1995;55:6200–6209. [PubMed] [Google Scholar]
- 38.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 39.Deep G, Agarwal R. Chemopreventive efficacy of silymarin in skin and prostate cancer. Integr Cancer Ther. 2007;6:130–145. doi: 10.1177/1534735407301441. [DOI] [PubMed] [Google Scholar]
- 40.Khan N, Afaq F, Mukhtar H. Cancer chemoprevention through dietary antioxidants: progress and promise. Antioxid Redox Signal. 2008;10:475–510. doi: 10.1089/ars.2007.1740. [DOI] [PubMed] [Google Scholar]
- 41.van Zandwijk N. Chemoprevention in non-small cell bronchial cancers. Rev Pneumol Clin. 2002;58:3S51–53. [PubMed] [Google Scholar]
- 42.Singh RP, Agarwal R. Natural flavonoids targeting deregulated cell cycle progression in cancer cells. Curr Drug Targets. 2006;7:345–354. doi: 10.2174/138945006776055004. [DOI] [PubMed] [Google Scholar]
- 43.Singh RP, Dhanalakshmi S, Agarwal R. Phytochemicals as cell cycle modulators--a less toxic approach in halting human cancers. Cell Cycle. 2002;1:156–161. [PubMed] [Google Scholar]
- 44.Silvestri GA, Rivera MP. Targeted therapy for the treatment of advanced non-small cell lung cancer: a review of the epidermal growth factor receptor antagonists. Chest. 2005;128:3975–3984. doi: 10.1378/chest.128.6.3975. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Chang A. Molecular predictors of EGFR-TKI sensitivity in advanced non-small cell lung cancer. Int J Med Sci. 2008;5:209–217. doi: 10.7150/ijms.5.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins I, Garrett MD. Targeting the cell division cycle in cancer: CDK and cell cycle checkpoint kinase inhibitors. Curr Opin Pharmacol. 2005;5:366–373. doi: 10.1016/j.coph.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Canepa ET, Scassa ME, Ceruti JM, et al. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007;59:419–426. doi: 10.1080/15216540701488358. [DOI] [PubMed] [Google Scholar]
- 48.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 49.Fischer PM. The use of CDK inhibitors in oncology: a pharmaceutical perspective. Cell Cycle. 2004;3:742–746. [PubMed] [Google Scholar]
- 50.Esposito V, Baldi A, Tonini G, et al. Analysis of cell cycle regulator proteins in non-small cell lung cancer. J Clin Pathol. 2004;57:58–63. doi: 10.1136/jcp.57.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24:2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 52.Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287:821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 53.Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- 54.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 55.Flaig TW, Gustafson DL, Su LJ, et al. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs. 2007;25:139–146. doi: 10.1007/s10637-006-9019-2. [DOI] [PubMed] [Google Scholar]