

Abstract
Background
A large energy gap between the native state and the non-native folded states is required for folding into a unique three-dimensional structure. The features that define this energy gap are not well understood, but can be addressed using de novo protein design. Previously, α2D, a dimeric four-helix bundle, was designed and shown to adopt a native-like conformation. The high-resolution solution structure revealed that this protein adopted a bisecting U motif. Glu7, a solvent-exposed residue that adopts many conformations in solution, might be involved in defining the unique three-dimensional structure of α2D.
Results
A variety of hydrophobic and polar residues were substituted for Glu7 and the dynamic and thermodynamic properties of the resulting proteins were characterized by analytical ultracentrifugation, circular dichroism spectroscopy, and nuclear magnetic resonance spectroscopy. The majority of substitutions at this solvent-exposed position had little affect on the ability to fold into a dimeric four-helix bundle. The ability to adopt a unique conformation, however, was profoundly modulated by the residue at this position despite the similar free energies of folding of each variant.
Conclusions
Although Glu7 is not involved directly in stabilizing the native state of α2D, it is involved indirectly in specifying the observed fold by modulating the energy gap between the native state and the non-native folded states. These results provide experimental support for hypothetical models arising from lattice simulations of protein folding, and underscore the importance of polar interfacial residues in defining the native conformations of proteins.
Keywords: conformational specificity, de novo protein design, dimeric proteins, four-helix bundle, protein folding
Introduction
How the sequence of a protein specifies its structure has challenged biochemists for decades. The role of sidechains in the hydrophobic core in defining the native state has been studied thoroughly [1-10]. The importance of sidechains in solvent-exposed positions, however, is less clear. Recently, Cordes and Sauer reported that changes to exterior residues in arc repressor modulated the overall thermodynamic stability of the native state [11]. Here we examine how a solvent-exposed residue defines conformational specificity.
Conformational specificity refers to the ability of a sequence to specify a unique fold with a well-packed interior. Conformational specificity requires not only a stable native state but also a large energy gap (Δ) between the native state and the non-native folded states (molten globule ensemble, Figure 1) [12-19]. Natural proteins have evolved to maximize this energy gap because insufficient conformational specificity can have severe physiological consequences. In human lysozyme, for example, the loss of conformational specificity can lead to amyloid fibril formation, which is associated with disease [20]. It is often difficult, however, to define the attributes that define conformational specificity because they are typically distributed throughout the protein. Thus, conformational specificity reflects the sum of many contributions throughout the chain. Furthermore, it is difficult to ascertain these determinants because the energy gap is large relative to the available thermal energy, kBT. Thus, mutations that result in changes on the order of 1–3 kBT (~1–2 kcal mol−1 at 25°C) are insufficient to give rise to an observable population of non-native folded states.
Figure 1.

Schematic diagram depicting the hypothesized energy levels for a natural protein. Each line denotes a unique conformation and therefore the unfolded states are represented by an astronomically large number of these lines. The non-native folded states, or molten globule ensemble, are a collection of nearly isoenergetic conformations as shown. The number of molten globule states depicted is arbitrary.
Partially solvent-exposed residues, which might play a relatively small role in stabilizing the native state, could help to determine the size of Δ. Although these positions are solvent-exposed in the native states, in non-native folded states they might be more buried. Hence, the sidechain at this position could exert a strong effect on the stability of misfolded forms [21]. For example, a hydrophobic residue would lower the energy of the misfolded states without affecting the energy level of the native state, thereby decreasing the value of Δ.
Designed proteins might be ideal for defining the features responsible for conformational specificity [22-35]. They tend to be small, which limits the number of possible contributions to Δ. Also, their sequences have not been subjected to evolutionary pressure to maximize the magnitude of Δ in contrast to natural proteins that have undergone more than a billion years of evolution. Indeed, Imperiali and co-workers have shown that a single residue change in the β turn of a 23-residue ββα motif prevents this peptide from adopting a unique conformation [36,37].
Here, we explore the origins of conformational specificity in α2D, a de novo designed, dimeric four-helix bundle. This protein is like a coiled coil in that it is unfolded as a monomer but folded as a dimer [38-40]. Unlike a coiled coil, however, this dimeric four-helix bundle is comprised of shorter helices with lengths that are similar to those found in natural proteins. Thus, they form globular proteins rather than the highly elongated rod-like structures that are formed by coiled coils. An advantage to using a homo-oligomeric system for examining conformational specificity is that a given mutation will occur multiply in an equivalent structural context in the oligomer. This duplication amplifies the effects of a single change, and simplifies the interpretation of results.
α2D is the third generation of a series of dimeric four-helix bundles [24,41]. The initial members of this series adopted dynamic, molten-globule-like conformations. Subsequent refinements to the design led to α2D, which adopted a unique, well-defined conformation [21,42]. The structure of α2D in solution has been solved to high-resolution (root mean square deviation [rmsd] 0.28 Å) and features the bisecting U motif (Figure 2) [43]. The sequence of α2D differs from its molten globule-like predecessor, α2C, at three positions, residues 7, 26 and 30. In α2D, residues 26 and 30 are histidine sidechains that engage in an intermonomer hydrogen-bonded cluster. Previous experimental studies indicated that this interaction serves as a conformational lock that contributes to native-like behavior of the dimer [44].
Figure 2.

The structure of α2D, Ac-GEVEELEKKFKELWK-GPRRG-EIEELHKKFHELIKG-NH2 (where the three residues in italics indicate the changes made to α2C in the design of α2D). (a) Topology diagram of α2D showing the topology of the dimeric four-helix bundle that adopts the bisecting U motif. (b) Superposition of the backbone from the 15 lowest energy structures of α2D calculated from NMR-derived data (PDB code 1QP6). (c) A cutaway view of a superposition of the 15 lowest energy NMR structures showing that Glu7 is not directly involved in stabilizing the native state of α2D. Glu7 (shown in green and red) adopts several different conformations in solution, as opposed to the hydrophobic core residues (shown in gray).
The sequence of α2D also differs from that of α2C at the seventh position in which a hydrophobic leucine residue was replaced with a glutamic acid residue. This sidechain is largely exposed to solvent and adopts multiple rotamers in the ensemble of nuclear magnetic resonance (NMR)-derived structures (Figure 2c). The location and mobility of Glu7 suggests that it is not directly involved in defining the thermodynamic stability of the native state relative to the fully unfolded ensemble. Indeed, no change in the structure of α2D upon addition of 1 M NaCl is observed using circular dichroism (CD) spectroscopy. Glu7, however, might aid in specifying a distinct conformation by increasing Δ. To explore this possibility Glu7 was changed to a variety of hydrophobic and polar residues. These variants were initially characterized by thermal denaturation to determine the energetic differences between their folded dimeric state and their fully unfolded, monomeric state. Secondly, the presence of molten-globule-like conformations was assessed by examining the NMR spectrum and the change in specific heat capacity at constant pressure (ΔCp) for folding. The results indicate that an interfacial, polar residue can affect the native behavior of α2D by modulating Δ.
Results
Design and synthesis of E7X variants
A series of sidechains were evaluated in place of Glu7 of α2D. Alanine was initially investigated to confirm that Glu7 was not involved in any stabilizing interactions necessary for native behavior. Glu7 was then replaced with polar (E7D, E7H, E7S and E7Y; mutations written using single-letter amino acid code), aromatic (E7F, E7H and E7Y), and hydrophobic (E7F, E7L and E7V) sidechains. All peptides were synthesized by solid phase methods, purified to homogeneity using high performance liquid chromatography (HPLC), and assessed for proper molecular weight using matrix-associated laser desorption ionisation (MALDI) mass spectrometry.
α2D variants form stable helical dimers
The variants of α2D all form helical dimers as assessed using CD spectroscopy and analytical ultracentrifugation (data not shown). For example, an equilibrium sedimentation experiment for E7A (see Supplementary material) shows that this protein sediments as a single homogenous species with an apparent molecular weight of 8469 Da, which agrees well with the expected molecular weight for the dimer (8459 Da). E7F and E7Y were also fully dimeric over this concentration range. Some of the proteins form less stable homodimers (E7D, E7S, E7H and E7V) and their equilibrium sedimentation curves could not be well described by a single monomolecular species at the lowest concentration examined (50 μM). However, the data for these peptides were very well described by a monomer–dimer equilibrium. Alternative oligomerization schemes such as monomer–trimer and monomer–tetramer failed to give adequate fits to the data for these peptides confirming their dimeric nature. Only one peptide, E7L, showed a tendency to form higher order aggregates. The equilibrium sedimentation data for this peptide can be described equally well by monomer–dimer-trimer or monomer–dimer–tetramer equilibria.
Thermodynamic stability
Thermodynamic parameters for folding were measured from the thermal denaturation curves of each protein, monitoring the ellipticity at 222 nm of their CD spectra. Figure 3a illustrates typical unfolding curves for E7D and E7F, relative to α2D. Changing Glu7 to phenylalanine (E7F) results in a protein with increased resistance to thermal unfolding although the transition is less cooperative (occurs over a wider range of temperatures). The mutation of Glu7 to aspartic acid (E7D) results in a protein that is only partially folded at the concentration shown (144 μM), and hence is less stable than α2D. Because this peptide is not fully folded, it is possible to detect denaturation at low temperatures (cold denaturation) as well as at elevated temperatures. This phenomenon is seen more clearly at lower concentrations in Figure 3b, where the fraction of folded peptide is smaller. This cold denaturation, which we observe for all E7X variants (where X is any residue), allows a reasonably accurate determination of the change in heat capacity of unfolding, ΔCp [45].
Figure 3.

Thermal denaturation of α2D and two variants. (a) Temperature dependence of the helical CD signal at 222 nm for α2D (93 μM), E7D (144 μM), and E7F (82 μM) in 50 mM Na2HPO4 pH 7.3. (b) Temperature dependence of the helical CD signal at 222 nm for E7D at 29 μM, 39 μM, 75 μM, and 144 μM. Because of the oligomeric nature of these peptides, the CD signal is concentration-dependent. In both panels, data were fit to the Gibbs-Helmholtz equation and the best fit is shown as a line (see text).
A great advantage of a dimeric system is that it is possible to change the fraction that is folded at a given temperature by simply changing the concentration. Thus, it is possible to obtain excellent estimates of the folded and unfolded base-lines as well as essential thermodynamic parameters by globally fitting data sets at multiple peptide concentrations to the Gibbs-Helmholtz equation (Figure 3b). This contrasts with unimolecularly folded proteins in which the fraction folded at a given temperature is modulated by addition of denaturants — a procedure that introduces uncertainties in the slopes of the baseline, and also requires extrapolation of thermodynamic parameters to physiological conditions.
Table 1 lists the values of the midpoint, Tm; the Gibbs free energy, ΔG298; the van’t Hoff enthalpy, ΔHm; and ΔCp for the folding–unfolding transition at 25°C evaluated at 100 μM standard state and extrapolated to a 1 M standard state. As expected for a surface modification, the differences in the free energies for folding at room temperature are small, and within ± 1.0 kcal mol−1 (or 0.5 kcal mol−1 on a per monomer basis). The fitting error in ΔCp is approximately 100 cal mol−1 K−1 as assessed from a sensitivity analysis of multiple data sets for each of the variants. With the exception of the E7F and E7V substitutions, the value of ΔCp for all variants was constant within experimental error (860 ± 100 cal mol−1 K−1), and hence was fixed in the fitting procedure. By contrast, this value of ΔCp gave rise to unacceptable fits to the data for E7F and E7V, indicating that ΔCp was significantly lower for these proteins.
Table 1.
Thermodynamic parameters for the unfolding of α2D and variants*
| Glu7 variant |
ΔΔG298† (kcal mol−1) (1 M) |
ΔG298† (kcal mol−1) (1 M) |
Tm‡ (°C) (100 μM) |
Tm‡ (K) (1 M) |
ΔHm§ (kcal mol−1) (100 μM) |
ΔHm§ (kcal mol−1) (1 M) |
ΔCp (cal mol−1 K−1) |
|---|---|---|---|---|---|---|---|
| E | - | 7.0 | 51.8 | 369.8 | 30.7 | 69.3 | 860 |
| A | 0.2 | 7.2 | 53.2 | 370.1 | 32.6 | 70.2 | 860 |
| S | −0.8 | 6.2 | 37.7 | 358.5 | 23.4 | 64.3 | 860 |
| D | −1.4 | 5.6 | 27.3 | 354.4 | 14.2 | 60.6 | 860 |
| H | −1.1 | 5.9 | 36.0 | 363.1 | 16.2 | 62.5 | 860 |
| Y | 1.0 | 8.0 | 65.0 | 379.7 | 39.4 | 75.1 | 860 |
| F | 0.5 | 7.5 | 72.7 | 404.2 | 28.5 | 61.0 | 558 |
| V | −0.4 | 6.6 | 50.1 | 405.1 | 7.6 | 55.5 | 514 |
Thermal denaturation data of peptide samples (pH 7.3, 50 mM Na2HPO4) were obtained by monitoring ellipticity at 222 nm. Multiple datasets at various peptide concentrations were globally analyzed using the Gibbs-Helmholtz equation using a 100 μM (or 1 M) standard state as described [60]. The computed values of ΔCp were constant given the precision of these parameters (± 10% based on sensitivity analysis). Therefore, a single value of ΔCp was used for all samples (860 ± 100 cal mol−1 K−1 dimer–1) except E7F and E7V (see the Discussion section).
ΔG298 is the Gibbs free energy for the folding–unfolding transition at 25°C, 1 M standard state; units are kcal mol−1.
Tm is the midpoint of the folding–unfolding transition (at the standard state indicated) and is precise to 0.4°C.
The ΔHm is the extrapolated enthalpy for the folding–unfolding transition at Tm (at the standard state indicated); units are kcal mol−1 and the uncertainty is ± 0.8 kcal mol−1.
The analysis of ΔCp obtained by the use of the Gibbs-Helmholtz equation was confirmed using an independent method [46,47] in which the van’t Hoff enthalpies of the individual mutants (at 100 μM total peptide concentration) at their thermal unfolding midpoints were plotted versus Tm (Figure 4). The data for all the mutants, except for E7V and E7F, fall along a line, of which the slope is in reasonable agreement with the value of ΔCp determined by the Gibbs-Helmholtz analysis (0.7 versus 0.86 kcal mol−1 K−1). E7V and E7F show a very significant negative deviation from the line, clearly demonstrating that they have lower values of ΔCp. ΔCp primarily reflects the difference in solvent exposure for the folded versus the unfolded states of a protein [47,48]. Thus, as compared with the more native variants, E7V and E7F might experience a smaller change in solvation upon unfolding, possibly because their hydrophobic interiors are less well packed. Alternatively, these variants might bury more nonpolar surface area in the unfolded state. This possibility seems less probable, however, because the unfolded states of these peptides is likely to be the same.
Figure 4.
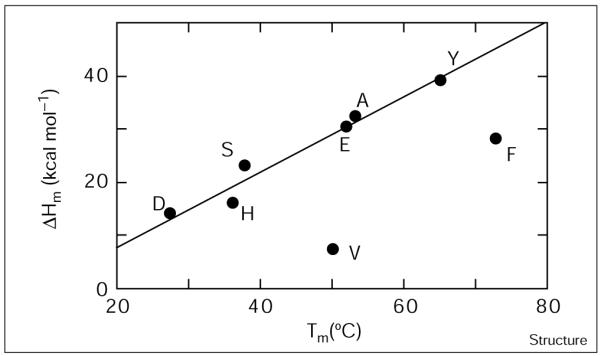
The ΔCp of unfolding is significantly lower for E7F and E7V. The enthalpy of unfolding at the midpoint temperature is plotted as a function of the midpoint temperature for Glu7 variants. The line drawn simply serves to guide the eye.
NMR spectroscopy
The proton 1D NMR spectrum of α2D is typical of native proteins (Figure 5). The excellent packing of its hydrophobic core is apparent from the dispersion of chemical shifts of the methyl region (0–1.25 ppm). E7A and E7S, which have similar thermodynamic stability to α2D, have NMR spectra that are only slightly less well dispersed than α2D. E7D and E7H are significantly less stable than α2D and are only approximately 90% dimeric under conditions for NMR spectroscopy. The chemical shift dispersion for these peptides is similar to that for E7A and E7S. However, the resonances for E7D are broader possibly reflecting conformational exchange between the folded dimeric and unfolded monomeric states.
Figure 5.

Ability of the hydrophobic core of α2D variants to adopt a unique conformation as assessed by 1D 1H-NMR spectroscopy at 500.13 MHz. Proton spectra at 2 mM monomer concentration in 50 mM d11-Tris, pH 7.3 for (a) α2D, E7A, E7S, E7D and E7H, and (b) α2D, E7Y, E7F, E7V and E7L at the temperature of maximum thermal stability. The methyl region of each spectrum is shown and is essentially a fingerprint of the hydrophobic core for each protein.
The introduction of hydrophobic sidechains for Glu7 (E7Y, E7F, E7V and E7L) leads to a deterioration of chemical shift dispersion that correlates with the polarity and flexibility of the residue (Figure 5b). E7Y, the most polar of this group of variants, exhibits reasonable chemical shift dispersion although the resonances are somewhat broadened. In contrast, E7V and E7L show poorly dispersed spectra with broad methyl resonances similar to those observed for molten globule ensembles. E7F shows behavior that is intermediate between E7Y and the aliphatic substituted variants, E7V and E7L. The differences in chemical shift dispersion between native and molten globule states is more clearly illustrated by comparing the methyl regions of the 2D 1H–13C heteronuclear single quantum coherence (HSQC) spectra for each protein. For example, the spectra for α2D and E7V are shown in Figure 6. For α2D, all 14 methyl resonances are observed with chemical shift dispersion typical of natural proteins. However, the resonances E7V are broader and much less well dispersed, which is typical of molten globules.
Figure 6.
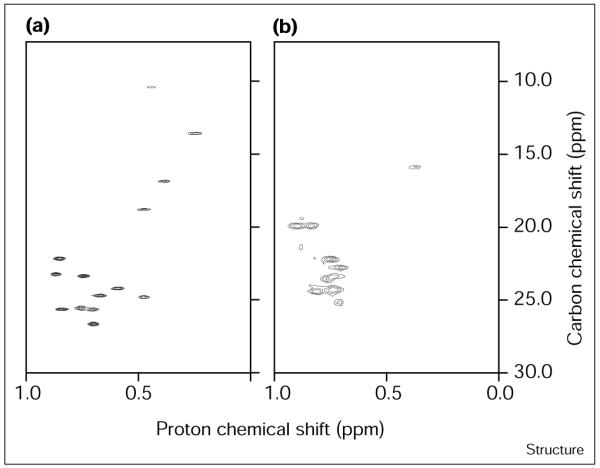
Ability of the hydrophobic core of α2D and E7V to adopt a unique conformation as assessed by 2D 1H–13C NMR spectroscopy at 500.13 MHz. The methyl region of the HSQC spectra is shown at 2 mM monomer concentration in 50 mM H2NaPO4, pH 7.3 for (a) α2D and (b) E7V at the temperature of maximum thermal stability. Note the broadening of resonances and collapse of chemical shift dispersion for E7V compared with α2D. The number of methyl groups is 14 for α2D and 16 for E7V.
The molten globule nature of a protein can be confirmed by the aromatic CD signal in the near-UV region [49,50]. In this region of the CD spectrum, conformational averaging of a molten globule will typically give rise to a decrease in the molar ellipticity. Indeed, the near-UV CD spectrum of α2D shows a double maximum, that is four-fold to fivefold more intense than the most intense band of E7V (Figure 7). Together these data suggest that hydrophobic and particularly aliphatic residues at this position lead to molten globule behavior.
Figure 7.

Ability of the hydrophobic core of α2D and E7V to adopt a unique conformation as assessed by aromatic CD spectroscopy. Near-UV CD spectra were collected at 1 mM monomer concentration in 50 mM H2NaPO4, pH 7.3 for α2D (filled circles) and E7V (open circles) at the temperature of maximum thermal stability. Data were averaged for 10 s every 2 nm.
Discussion
The α2D protein provides an outstanding experimental system for investigating the relationship between thermodynamic stability and conformational specificity. A full description of the conformational properties of a protein requires consideration of the relative energies of the native state, the non-native folded states, and the fully unfolded ensemble. We would expect the fully unfolded states to be similar for α2D and each variant. The folded states for these variants are helical and dimeric in each case, but differ drastically in their dynamic properties. Thus, the nature of a surface-accessible residue must have a profound effect on Δ, as evidenced by the variation in the conformational specificity of these variants. Such dramatic changes in conformational specificity have previously been observed only in coiled coils. Furthermore, these examples involved fully buried hydrogen-bonded polar groups [51-54] rather than the solvent-exposed sidechains studied in this work. Thus, the large effect observed here is particularly striking and supports current theories concerning conformational specificity [13-16,55-57].
As expected from its surface location, changes to the sidechain at position 7 have minimal effects on thermodynamic stability of the dimeric versus monomeric forms of the variants. Expressed on a per monomer basis, the changes in free energy of folding span a range of approximately 0.5 kcal mol−1 and can be explained largely by differences in helical propensities. The expected changes in ΔG298 (Table 1) for folding from helical propensity effects, including interactions between sidechains at position i − i + 3 and i − i + 4, were estimated using parameters described in the computer algorithm AGADIR [58]. A plot of the expected differences in ΔG298, ΔΔGα, versus the measured ΔΔGobs shows a moderate linear correlation (r = 0.72) indicating that much of the variation in ΔΔGobs arises from helix stabilization alone. Interestingly, the aromatic residues (histidine, phenylalanine and tyrosine) tended to be more stable than expected. Examination of a model suggests that a planar aromatic sidechain might provide enhanced stability by lying across the helix–helix interface forming interhelical interactions that are not accounted for in AGADIR (which is parameterized with monomeric α helices). Therefore, we fit the data (Figure 8) to a linear equation in which the dependent variables were ΔΔGα and the number of aromatic residues (narom; 0 or 2 for a dimer). The equation of best fit was:
The marked improvement in the correlation coefficient (r = 0.93) suggests that the aromatic contributions do indeed account for the additional thermodynamic stability observed. The coefficient of determination for ΔΔGa (0.72) is reasonably close to unity and well within the range of slopes observed when different helical propensity scales are compared [59]. The coefficient of determination for narom (0.47) provides the mean energetic contribution (in kcal mol−1) associated with the aromatic sidechains. Hence, aromaticity and helical propensity can describe the stability differences amongst the variants to within approximately 0.4 kcal mol−1.
Figure 8.

Thermodynamic stability of α2D and Glu7 variants. The measured ΔΔGobs is plotted against ΔΔGpred, the contribution to stability differences based on both aromaticity and helical propensity for α2D and Glu7 variants. The data are fit to the linear equation ΔΔGobs =a*ΔΔGα + b*narom + c. The rms deviation of the individual data points from the line is 0.4 kcal mol−1.
The similarity in the free energies of folding for this group of variants is particularly striking given the large variation in their dynamic properties. For example, E7V, which behaves as a molten globule, shows a thermodynamic stability nearly identical to that of the native-like α2D. At 25°C, E7V shows a decrease in the enthalpy of folding compared with α2D, which is offset by a concomitant decrease in the entropy of folding. Thus, the increase in conformational entropy for E7V is completely balanced by a correspondingly unfavorable change in the enthalpic contributions to the free energy.
Although the effects of these substitutions on the overall free energy of folding are small, the corresponding effects on conformational specificity are remarkably large. Thus, changing a glutamic acid to valine residue results in complete abrogation of the ability to define a unique state as evidenced by a loss of aromatic CD signal, a collapse of dispersion in chemical shifts, and a marked lowering of ΔCp. The origins of this behavior can be explained by consideration of how the position 7 variants modulate Δ. A unique conformation is observed only when Δ is sufficiently large (relative to thermal energy) to provide an essentially full population of the native state. Figure 9 illustrates how the variants of α2D affect their energy profiles given the assumption that the unfolded states are essentially identical. Replacement of Glu7 with histidine (E7H) increases the energies of both the native state as well as the misfolded ensemble. Because the increases in energies of both states are roughly similar, E7H retains a significant Δ. Thus, E7H retains its native, dimeric conformation although the stability of the dimer is somewhat attenuated. By contrast, replacement of Glu7 with valine (E7V) causes a selective decrease in the energy of the molten globule ensemble relative to the native state resulting in the elimination of a significant Δ. Thus, E7V adopts an ensemble of nearly isoenergetic, interchanging conformers.
Figure 9.

Schematic diagram depicting the hypothesized energy levels for α2D and variants. Each line denotes a unique conformation as explained in Figure 1.
These results are consistent with theoretical considerations of protein folding that emphasize the need to maximize the energy difference between the native state and ensemble of non-folded states to obtain native behavior [13-16,55-57, 19]. Here, we confirm this prediction in an experimental system. It is interesting to note that the conclusions presented here might have been difficult to obtain from studies on natural proteins. Natural proteins have evolved to maximize this energy gap such that single mutations will not perturb their ability to function. α2D has not yet evolved to the same degree as functional proteins but has allowed us to probe the determinants of conformational specificity.
Biological implications
Natural proteins have evolved a large energy gap, Δ (Figure 1), between the native state and the non-native states to prevent misfolding and aggregation that could have pathological consequences. In addition, a large value of Δ is necessary to withstand the effects of random mutations, many of which would be expected to decrease Δ, resulting in possible deleterious effects. This energy gap can be increased by either specific interactions that stabilize the native state (positive design) or interactions that destabilize the ensemble of misfolded conformers (negative design) [24,26]. Previous studies on natural proteins have focused primarily on the role of buried residues in stabilizing the native state [4,5].
Using α2D, a dimeric four-helix bundle, as a model system, we have identified two distinct contributions to Δ arising from solvent-exposed residues. Previously, it was demonstrated that a hydrogen-bonded network involving an interfacial cluster of histidine residues (in a different region of the protein) provides conformational specificity by specifically stabilizing the native state [44]. Here we show that Glu7 makes a different contribution to this energy gap. Rather than stabilizing the native state, this residue destabilizes alternatively folded states. Thus, residues on the surface of a protein that contribute little to the overall stability of the native state might contribute significantly to Δ and the ability to adopt a unique conformation. This study was possible because α2D has not undergone extensive evolution towards a large Δ. In this sense, our designed protein can be considered a primordial protein and serves as a model system to study the evolution of proteins. We are currently applying these results towards ‘evolving’ our designed proteins into functional proteins.
Materials and methods
Sample preparation
Peptides were synthesized using 9-fluorenylmethyloxycarbonyl-protected amino acids on an Applied Biosystems 433A peptide synthesizer, and purified by preparative reverse phase HPLC using a VYDAC C18 column. The molecular weights of the peptides were confirmed using MALDI mass spectrometry.
Analytical ultracentrifugation
Sedimentation equilibrium data were collected at 25°C on a Beckman XLI Analytical Ultracentrifuge equipped with both absorption and interference optics. Peptide concentration was determined by tryptophan absorbance (ε280 = 5600 M−1cm−1). Protein samples at 100 μM, 200 μM and 500 μM were prepared at pH 7.3 (25 mM Tris). Data were collected at three speeds (30,000, 35,000 and 40,000 rpm) at each peptide concentration and fit globaly using in-house software written in Igor Pro (WaveMetrics, Inc.). The partial specific volume, V, was estimated from amino acid composition using the method of Kharazov [60]. The oligomeric state of each peptide was determined by fitting the sedimentation data to a fixed molecular weight or a monomer-dimer equilibrium as described [61].
Thermal denaturation
Temperature dependence of the helical CD signal at 222 nm was collected for E7X proteins in 50 mM Na2HPO4, pH 7.3. The signal was averaged for 60 sec after a 4 min equilibration period. Data for each protein at three or four different concentrations were globally fit to the Gibbs-Helmholtz equation for a monomer–dimer equilibrium as described [61].
NMR spectroscopy
Proton NMR spectra were collected on E7X proteins at 2 mM monomer concentration in 50 mM d11-Tris, pH 7.3 on a Bruker AMX spectrometer operating at 500.13 MHz proton frequency. Data were collected at the temperature of maximum thermal stability (determined from thermal denaturation studies using circular dichroism spectroscopy). Each spectrum is the average of 256 free induction decays of 4K complex data points and was processed with 4.0 Hz line broadening. The natural abundance 1H–13C HSQC spectra were collected overnight in a non-constant time manner. Each HSQC spectrum is the average of 128 free induction decays; the spectra were collected with 0.04 ppm resolution in 1H and 0.1 ppm resolution in 13C.
Supplementary Material
Acknowledgements
The authors thank Luis Serrano (EMBL-Heidelberg) for providing ΔΔGα values and Jim Lear for software to analyze sedimentation equilibrium data. Walter Englander kindly provided NMR spectrometer time on a Bruker AMX-500. We are grateful to Jim Lear, Chris Summa, Jeff Saven, Giovanna Ghirlanda and Steve Betz for useful discussions. This work was supported by NIH postdoctoral NRSA (to RBH) 1 F32 GM18491-01 and GM54616 (to WFD), as well as the MRSEC program of the NSF.
Footnotes
Supplementary material Supplementary material including a figure of a typical fit to the analytical ultracentrifugation data and a figure showing the ΔCp sensitivity analysis for E7V is available at http://current-biology.com/supmat/supmatin.htm.
References
- 1.Dolphin GT, Brive L, Johansson G, Baltzer L. Use of aromatic amino acid residues to restrict the dynamics in the hydrophobic core of a designed helix–loop–helix dimer. J. Am. Chem. Soc. 1996;118:11297–11298. [Google Scholar]
- 2.Kellis JT, Jr., Nyberg K, Sali D, Fersht AR. Contribution of hydrophobic interactions to protein stability. Nature. 1988;333:784–786. doi: 10.1038/333784a0. [DOI] [PubMed] [Google Scholar]
- 3.Munson M, O’Brien R, Sturtevant JM, Regan L. Redesigning the hydrophobic core of a four-helix bundle protein. Protein Sci. 1994;3:2015–2022. doi: 10.1002/pro.5560031114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews BW. Studies on protein stability with T4 lysozyme. Adv. Protein Chem. 1995;46:249–278. doi: 10.1016/s0065-3233(08)60337-x. [DOI] [PubMed] [Google Scholar]
- 5.Lim WA, Sauer RT. The role of internal packing interaction in determining the structure and stability of a protein. J. Mol. Biol. 1991;219:359–376. doi: 10.1016/0022-2836(91)90570-v. [DOI] [PubMed] [Google Scholar]
- 6.Richards F, Lim W. An analysis of packing in the protein folding problem. Quart. Rev. Biophys. 1993;26:423–498. doi: 10.1017/s0033583500002845. [DOI] [PubMed] [Google Scholar]
- 7.Shortle D. Mutational studies of protein structures and their stabilities. Quart. Rev. Biophys. 1992;25:205–250. doi: 10.1017/s0033583500004674. [DOI] [PubMed] [Google Scholar]
- 8.Cordes MHJ, Davidson AR, Sauer RT. Sequence space, folding and protein design. Curr. Opin. Struct. Biol. 1996;6:3–10. doi: 10.1016/s0959-440x(96)80088-1. [DOI] [PubMed] [Google Scholar]
- 9.Lau K, Dill K. Theory for protein mutability and biogenesis. Proc. Natl Acad. Sci. USA. 1990;87:638–642. doi: 10.1073/pnas.87.2.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perutz M, Lehmann H. Molecular pathology of human haemoglobin. Nature. 1968;219:902–909. doi: 10.1038/219902a0. [DOI] [PubMed] [Google Scholar]
- 11.Cordes MH, Sauer RT. Tolerance of a protein to multiple polar-to-hydrophobic surface substitutions. Protein Sci. 1999;8:318–325. doi: 10.1110/ps.8.2.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handel TM, Williams SA, DeGrado WF. Metal ion-dependent modulation of the dynamics of a designed protein. Science. 1993;261:879–885. doi: 10.1126/science.8346440. [DOI] [PubMed] [Google Scholar]
- 13.Shakhnovich E, Gutin AM. Engineering of stable and fast-folding sequences of model proteins. Proc. Natl Acad. Sci. USA. 1993;90:7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shakhnovich EI, Gutin AM. A new approach to the design of stable proteins. Protein Eng. 1993;6:793–800. doi: 10.1093/protein/6.8.793. [DOI] [PubMed] [Google Scholar]
- 15.Sali A, Shakhnovich E, Karplus M. How does a protein fold? Nature. 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 16.Onuchic JN, Wolynes PG, Luthey-Schulten Z, Socci ND. Towards an outline of the topography of a realistic protein folding funnel. Proc. Natl Acad. Sci. USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Y, Karplus M. Folding of a model three-helix bundle protein: a thermodynamic and kinetic analysis. J. Mol. Biol. 1999;293:917–951. doi: 10.1006/jmbi.1999.2936. [DOI] [PubMed] [Google Scholar]
- 19.Klimov DK, Thirumalia D. Criterion that deterimines the foldability of proteins. Phys. Rev. Lett. 1996;76:4070–4073. doi: 10.1103/PhysRevLett.76.4070. [DOI] [PubMed] [Google Scholar]
- 20.Booth D, et al. Pepys MB. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature. 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 21.Raleigh DP, Betz SF, DeGrado WF. A de novo designed protein mimics the native state of natural proteins. J. Am. Chem. Soc. 1995;117:7558–7559. [Google Scholar]
- 22.Betz SF, Liebman PA, DeGrado WF. De novo design of native proteins: characterization of proteins intended to fold into antiparallel, ROP-like, four-helix bundles. Biochemistry. 1997;36:2450–2458. doi: 10.1021/bi961704h. [DOI] [PubMed] [Google Scholar]
- 23.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. From coiled coils to small globular proteins: design of a native-like three-helix bundle. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryson JW, et al. DeGrado WF. Protein design: a hierarchic approach. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 25.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 26.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. De novo design and structural characterization of proteins and metalloproteins. Annu. Rev. Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 27.Fezoui Y, Connolly PJ, Osterhout JJ. Solution structure of α-t-α, a helical hairpin peptide of de novo design. Protein Sci. 1997;6:1869–1877. doi: 10.1002/pro.5560060907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beasley JR, Hecht MH. Protein design: the choice of de novo sequences. J. Biol. Chem. 1997;272:2031–2034. doi: 10.1074/jbc.272.4.2031. [DOI] [PubMed] [Google Scholar]
- 29.Richardson J, et al. Surles MC. Looking at proteins: representations, folding, packing, and design. Biophysical Society National Lecture. Biophys. J. 1992;63:1186–1209. [PMC free article] [PubMed] [Google Scholar]
- 30.Baltzer L. Functionalization of designed folded polypeptides. Curr. Opin. Struct. Biol. 1998;8:466–470. doi: 10.1016/s0959-440x(98)80124-3. [DOI] [PubMed] [Google Scholar]
- 31.Street A, Mayo S. Computational protein design. Structure. 1999;7:R105–R109. doi: 10.1016/s0969-2126(99)80062-8. [DOI] [PubMed] [Google Scholar]
- 32.Regan L. Proteins to order? Structure. 1998;6:1–4. doi: 10.1016/s0969-2126(98)00001-x. [DOI] [PubMed] [Google Scholar]
- 33.Munoz V, Serrano L. Local versus nonlocal interactions in protein folding and stability. Fold. Des. 1996;1:R71–R77. doi: 10.1016/S1359-0278(96)00036-3. [DOI] [PubMed] [Google Scholar]
- 34.Imperiali B, Ottesen J. Uniquely folded mini-protein motifs. J. Pept. Res. 1999;54:177–184. doi: 10.1034/j.1399-3011.1999.00121.x. [DOI] [PubMed] [Google Scholar]
- 35.Hellinga H. Rational protein design: combining theory and experiment. Proc. Natl Acad. Sci. USA. 1997;94:10015–10017. doi: 10.1073/pnas.94.19.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Struthers M, Ottesen JJ, Imperiali B. Design and NMR analyses of compact, independently folded ββα motifs. Fold. Des. 1998;3:95–103. doi: 10.1016/S1359-0278(98)00015-7. [DOI] [PubMed] [Google Scholar]
- 37.Cheng RP. PhD Thesis. California Institute of Technology; Pasadena: 1998. Design and Synthesis of Metallopeptides: Incorporation of Unnatural Amino Acids and Construction of a Structural Template; p. 170. p170. [Google Scholar]
- 38.O’Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 39.Lumb KJ, Kim PS. Interhelical salt bridges, coiled-coil stability and specificity of dimerization. Science. 1996;271:1137–1138. doi: 10.1126/science.271.5252.1136. [DOI] [PubMed] [Google Scholar]
- 40.Kohn W, Mant C, Hodges R. α-helical protein assembly motifs. J. Biol. Chem. 1997;272:2583–2586. doi: 10.1074/jbc.272.5.2583. [DOI] [PubMed] [Google Scholar]
- 41.Betz SF, Bryson JW, DeGrado WF. Native-like and structurally characterized designed α-helical bundles. Curr. Opin. Struct. Biol. 1995;5:457–463. doi: 10.1016/0959-440x(95)80029-8. [DOI] [PubMed] [Google Scholar]
- 42.Raleigh DP, DeGrado WF. A de novo designed protein shows a thermally induced transition from a native to a molten globule-like state. J. Amer. Chem. Soc. 1992;114:10079–10081. [Google Scholar]
- 43.Hill RB, DeGrado WF. Solution structure of α2D, a native-like de novo designed protein. J. Amer. Chem. Soc. 1998;120:1138–1145. [Google Scholar]
- 44.Hill RB, Hong J-K, DeGrado WF. Hydrogen bonded cluster can specify the native state of a protein. J. Am. Chem. Soc. 2000;122:746–747. [Google Scholar]
- 45.Chen B.-l., Schellman JA. Low-temperature unfolding of a mutant of phage T4 lysozyme. 1. Equilibrium studies. Biochemistry. 1989;28:685–691. doi: 10.1021/bi00428a041. [DOI] [PubMed] [Google Scholar]
- 46.Becktel W, Schellman J. Protein stability curves. Biopolymers. 1987;26:1859–1877. doi: 10.1002/bip.360261104. [DOI] [PubMed] [Google Scholar]
- 47.Cohen DS, Pielak GJ. The stability of yeast iso-1-cytochrome c as a function of pH and temperature. Protein Sci. 1994;3:1253–1260. doi: 10.1002/pro.5560030811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomez J, Hilser V, Xie D, Freire E. The heat capacity of proteins. Proteins. 1995;22:404–412. doi: 10.1002/prot.340220410. [DOI] [PubMed] [Google Scholar]
- 49.Kuwajima K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins. 1989;6:87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- 50.Ptitsyn OB, Pain RH, Semisotnov GV, Zerovnik E, Razgulyaer OL. Evidence for a molten globule state as a general intermediates in protein folding. FEBS Lett. 1990;262:20–24. doi: 10.1016/0014-5793(90)80143-7. [DOI] [PubMed] [Google Scholar]
- 51.Betz S, Fairman R, O’Neil K, Lear J, DeGrado W. Design of two-stranded and three-stranded coiled coil peptides. Proc. of the Royal Society. 1995;348:81–88. doi: 10.1098/rstb.1995.0048. [DOI] [PubMed] [Google Scholar]
- 52.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez L, Jr., Woolfson DN, Alber T. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat. Struct. Biol. 1996;3:1011–1018. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- 54.Oakley M, Kim P. A buried polar interaction can direct the relative orientation of helices in a coiled coil. Biochemistry. 1998;37:12603–12610. doi: 10.1021/bi981269m. [DOI] [PubMed] [Google Scholar]
- 55.Sali A, Shakhnovich E, Karplus M. Kinetics of protein folding, a lattice model study of the requirements for folding to the native state. J. Mol. Biol. 1994;235:1614–1636. doi: 10.1006/jmbi.1994.1110. [DOI] [PubMed] [Google Scholar]
- 56.Yue K, et al. Dill KA. A test of lattice protein folding algorithms. Proc. Natl Acad. Sci. USA. 1995;92:325–329. doi: 10.1073/pnas.92.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou J, Saven J. Statistical theory of combinatorial libraries of folding proteins: energetic discrimination of a target structure. J. Mol. Biol. 2000;296:281–294. doi: 10.1006/jmbi.1999.3426. [DOI] [PubMed] [Google Scholar]
- 58.Petukhov M, Munoz V, Yumoto N, Yoshikawa S, Serrano L. Position dependence of non-polar amino acid intrinsic helical propensities. J. Mol. Biol. 1998;278:279–289. doi: 10.1006/jmbi.1998.1682. [DOI] [PubMed] [Google Scholar]
- 59.Myers JK, Pace CN, Scholtz JM. Helix propensities are identical in proteins and peptides. Biochemistry. 1997;36:10923–10929. doi: 10.1021/bi9707180. [DOI] [PubMed] [Google Scholar]
- 60.Kharakoz D. Partial volumes and compressibilities of extended polypeptide chains in aqueous solution: additivity scheme and implication of protein unfolding at normal and high pressure. J. Mol. Biol. 1997;36:10276–10285. doi: 10.1021/bi961781c. [DOI] [PubMed] [Google Scholar]
- 61.Schneider JP, Lear JD, DeGrado WF. A designed buried salt bridge in a heterodimeric coiled coil. J. Am. Chem. Soc. 1997;119:5742–5743. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.