

Abstract
Aerosolized hyaluronan (HA) has been previously shown to prevent cigarette smoke-induced airspace enlargement and elastic fiber injury in mice when given concurrently with smoke. In the present study, a more stringent test of the therapeutic potential of HA was performed by delaying treatment with this agent for 1 month. After treatment with cigarette smoke for 3 h per day for 5 days per week for 1 month, mice (DBA/2J) began receiving aerosolized HA (0.1%) for 1 h prior to smoke exposure (controls were given aerosolized water). The results indicate that much of the damage to the lung elastic fibers occurred within the first several months of smoke exposure, as measured by levels of desmosine and isodesmosine (DID) in bronchoalveolar lavage fluid (BALF). In contrast to previously published studies, where concurrent administration of aerosolized HA significantly reduced BALF DID levels within 3 months of smoke exposure, the same effect was not seen until 6 months when HA treatment was delayed. However, despite the prolonged breakdown of elastic fibers in the current study, a significant reduction in airspace enlargement was observed after only 2 months of HA treatment. These findings provide further support for testing this agent in patients with preexisting chronic obstructive pulmonary disease.
Keywords: Hyaluronan, Lung, Desmosine, Cigarette Smoke, COPD
Introduction
Although a number of different mechanisms may be responsible for the loss of alveoli in pulmonary emphysema, damage to elastic fibers is a significant factor in the pathogenesis of this disease [1, 2]. These fibers are responsible for the mechanical recoil that facilitates the expiration of air from the lungs, and their breakdown can lead to alveolar distention and rupture. Due to the importance of elastic fiber injury in pulmonary emphysema, a number of therapeutic approaches have focused on protecting this extracellular matrix component from degradation by elastases and other injurious agents [3, 4]. However, determining the effectiveness of such treatment is often difficult because of the lack of sensitive, real-time indicators of successful therapeutic intervention. Both pulmonary function tests and high-resolution computerized tomography (HRCT) require prolonged time intervals to assess the potential benefits of a therapy, while more sensitive markers such as proinflammatory cytokines lack the necessary specificity to determine efficacy [5–9].
One possible solution to this problem is measurement of elastic fiber breakdown products themselves. Recent advances in detecting the elastin-specific amino acid crosslinks desmosine and isodesmosine (DID) have greatly increased the sensitivity and specificity of this test procedure [10–14]. Levels of DID in urine and plasma have been shown to correlate with physiological and radiological measures of chronic obstructive pulmonary disease (COPD) [12].
In the current study, we used this biomarker to determine both the progression of elastic fiber damage in a mouse model of smoke-induced pulmonary emphysema and the potential therapeutic effects of aerosolized hyaluronan (HA) on smoke-induced injury. This agent has previously been shown to significantly reduce smoke-induced airspace enlargement and prevent elastic fiber injury when given concurrently with smoke [15]. The current investigation modifies the original experimental protocol by delaying therapeutic intervention for 1 month following initiation of smoke exposure, thereby providing a more clinically relevant approach to evaluating this form of treatment. The ability of HA to limit airspace enlargement and prevent elastic fiber injury, despite pre-existing smoke-induced lung injury, would support clinical testing of this agent in patients who already have significant evidence of COPD.
Methods
Experimental Plan
Eight-week-old female DBA/2J mice (The Jackson Laboratory, Bar Harbor, ME) were divided into two treatment groups as follows: Group 1 was treated with aerosolized HA, beginning 1 month following initiation of smoke exposure; Group 2 was treated with aerosolized water, beginning 1 month following initiation of smoke exposure. Groups 1 and 2 were exposed to smoke for 3 h per day, 5 days/week, for a period of 10 months. Group 1 was treated with a 0.1% solution of HA in water for 1 h prior to each smoking session. Group 2 received aerosolized water for a similar interval. At various intervals following initial smoke exposure, animals were euthanized to determine (1) DID content in bronchoalveolar lavage fluid (BALF) and whole lungs at 2, 4, 6, 8, and 10 months; (2) lung histopathology at 3, 6, and 10 months; and (3) airspace enlargement as measured by the mean linear intercept (MLI) at 3 months. To obtain baseline values, additional 8-week-old animals, not exposed to either smoke or HA, were assayed for DID content in both BALF and whole lungs.
Exposure to Cigarette Smoke
Following administration of either aerosolized HA or water, the nebulizer was disconnected and the smoking machine (Model TE-10, Teague Enterprises, Davis, CA) was attached to the exposure chamber. Both treatment groups were exposed to cigarette smoke for a period of 3 h/day, 5 days/week. The smoking machine simultaneously burned two filtered research-grade cigarettes (type 2R4F, University of Kentucky). Each cigarette was puffed once per minute for 2 s at a flow rate of 1.05 L/min, yielding 35 cc of smoke. This cycle was repeated nine times before ejecting the cigarette and loading a new one. Proper flow rate was maintained by a vacuum pump that established negative pressure at the exhaust port.
Exposure to HA Aerosol
Beginning 1 month following initial smoke exposure, Group 1 was administered a 0.1% solution of low-molecular-weight (150 kDa) streptococcal HA in water (Bayer, Shawnee, KS), using a Misty-Ox nebulizer (Vital Signs, Totowa, NJ). Group 2 received aerosolized water alone. The nebulizer was connected to a heavy-duty air compressor that delivered a constant pressure of 30 psi. The aerosol entered the exposure chamber through an inflow port attached to the roof and was drawn through the chamber by negative pressure created by a vacuum pump connected to an exhaust port on the side wall. The chamber was large enough (28 × 19 × 15 in.) to permit the mice to remain in their cages while inhaling the aerosol, thereby minimizing direct handling of the animals.
Light Microscopic Studies
At 3, 6, and 10 months following initiation of smoke exposure, mice were asphyxiated with CO2 and their lungs were inflated in situ with 10% neutral-buffered formalin at a constant pressure of 20 cmH2O. After 2 h, the chest contents were removed and fixed for several days in formalin. The extrapulmonary structures were then dissected off and the lung tissues were randomly cut and entirely submitted for histological processing. Slide sections stained with hematoxylin and eosin were examined with the light microscope to determine histological changes and to quantify airspace diameter by the mean linear intercept method [16]. Additional sections were treated with the Verhoeff-Van Gieson stain to identify elastic fibers.
Measurement of DID
The levels of the elastin-specific crosslinking amino acids, DID, were measured in both BALF and whole-lung tissues. Animals were asphyxiated with CO2 and their lungs were lavaged three times with 1-ml aliquots of Hanks' solution. Both cell-free lavage fluids and homogenized lung tissues were then hydrolyzed in 6 N HCl at 110°C for 24 h, and the hydrolysates were filtered and evaporated to remove acid. DID were quantified by high-performance liquid chromatography and electrospray ionization mass spectrometry according to previously published procedures [11].
Data Analysis
All data were expressed as mean ± standard error of the mean (SEM). Both the two-sample t test and the Newman-Keuls multiple comparison test were used to determine statistically significant differences (p < 0.05) between treatment groups.
Results
BALF DID
As shown in Fig. 1, the amount of BALF DID in both the HA/Smoke (Group 1) and Smoke-Only (Group 2) groups dropped precipitously after 4 months of smoke exposure. In the HA/Smoke group, there was a decrease from 0.8 ng/ml to less than 1 pg/ml during this interval. While differences in BALF DID between the groups were not statistically significant at 2 and 4 months, subsequent measurements showed significantly lower levels of these amino acids in the HA-treated animals. Levels of DID were undetectable (<1 pg/mg protein) in 8-week-old animals that were not exposed to either HA or smoke.
Fig. 1.

The levels of BALF DID in both treatment groups showed a marked decrease after 4 months of smoke exposure. Although differences between the groups were not statistically significant at 2 and 4 months, subsequent measurements in the HA-treated animals were significantly lower (n ≥ 3 for each bar). Levels of DID were undetectable (<1 pg/mg protein) in 8-week-old animals that were not exposed to either HA or smoke (not shown; n = 4)
Lung DID
The amount of DID in the lungs was also measured at bimonthly intervals, beginning 2 months after the smoking regimen began. Both the HA/Smoke and Smoke-Only groups showed an increase in DID during the first 2 months, followed by a decline over the next 4 months and a second increase between 6 and 10 months (Fig. 2). Differences between the groups were not statistically significant over the entire course of the study.
Fig. 2.

An increase in lung DID was seen in both treatment groups during the first 2 months of smoke exposure, followed by a decrease over the next 4 months and a second increase between 6 and 10 months (n ≥ 3 for each bar). Differences between the groups were not statistically significant over the 10-month period following initial smoke exposure
Lung Histopathology
Exposure to tobacco smoke for 3 months resulted in significant airspace enlargement in animals receiving smoke alone, whereas only minimal alveolar changes were seen in those treated with HA (Fig. 3). The MLI of animals treated with HA was 42.6 μm compared to 58.4 μm for those receiving smoke alone (p < 0.01; Fig. 4). There was no significant difference between the MLI of the HA-treated group and that of 8-week-old control animals from our previous study (40.5 ± 0.6 μm), which were not exposed to either smoke or HA [15]. However, animals receiving only smoke had a significantly higher MLI than this control group (p < 0.01). Similar measurements were not performed at later time intervals because there was little progression of airspace enlargement in either group, which is consistent with previous morphological findings [15].
Fig. 3.
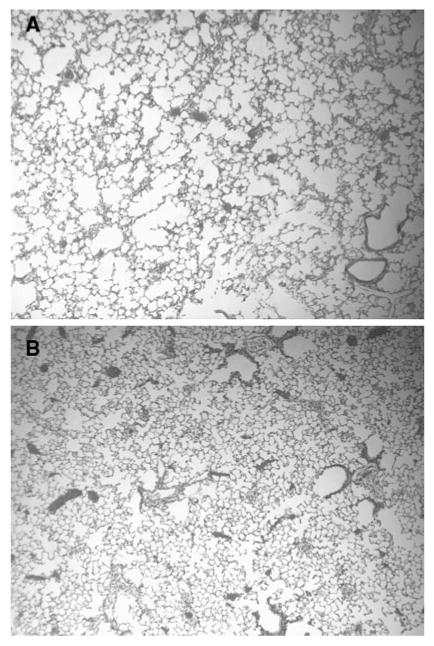
a Lungs exposed to smoke for 3 months without HA treatment showed a moderate degree of airspace enlargement. b Lungs exposed to smoke for 3 months and treated with HA for 2 months showed only minimal airspace enlargement. Original magnification of both figures: 40 × (hematoxylin and eosin)
Fig. 4.

After 3 months of smoke exposure, animals treated with HA showed a significant reduction in airspace enlargement compared to those receiving smoke alone (n = 5 for each bar)
Both groups showed inflammation of the larger airways at 3 months, including prominent papillary hyperplasia of bronchial epithelium, and accumulation of elastic fibers within the connective tissue stalks associated with the papillary epithelium (Fig. 5). The airway inflammation persisted at later time intervals (6 and 10 months), but there was no evidence of alveolitis or interstitial fibrosis in either group, despite the increase in total lung DID between these two time points. While several studies have shown that low-molecular-weight HA may enhance the expression of a variety of cytokines [17, 18], we observed no evidence of an inflammatory response in the HA-treated animals beyond that induced by smoke exposure.
Fig. 5.
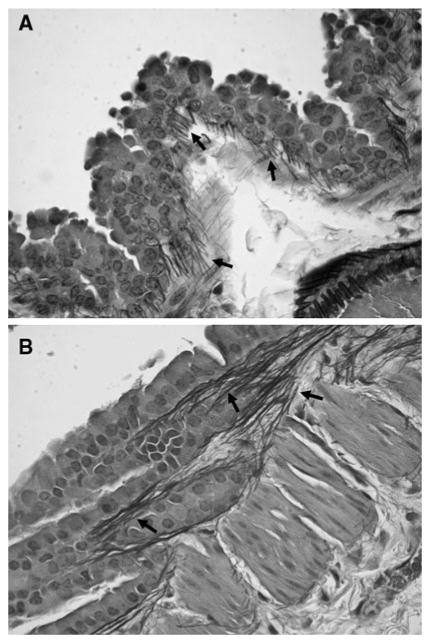
a Photomicrograph of a mouse lung treated with HA and exposed to cigarette smoke for 3 months showing papillary hyperplasia of bronchial epithelium and proliferation of elastic fibers (arrows). b Similar histological changes were seen in lungs receiving smoke alone for the same period of time. Original magnification of both figures: 400 × (Verhoef-Van Gieson)
Discussion
The concept of using nebulized HA to prevent elastic fiber injury is based on a series of experiments designed to determine the potential role of agents other than elastases in the pathogenesis of pulmonary emphysema. Previous studies from this laboratory indicated that intratracheal instillation of a nonelastolytic enzyme, hyaluronidase, induced pulmonary airspace enlargement in hamsters when administered in conjunction with 60% oxygen [19]. Damage to elastic fibers occurred only when both agents were given concomitantly, suggesting the possibility that hyaluronidase may facilitate the breakdown of these fibers by increasing their susceptibility to injury by other injurious agents such as elastases or oxidants. This concept was supported by subsequent work demonstrating that pretreatment of the lung with hyaluronidase enhances airspace enlargement induced by intratracheal administration of elastase [20, 21].
Studies were then undertaken to examine the effect of HA itself on this model of emphysema. Animals were exposed to an aerosol composed of 0.1% HA in water for 50 min prior to intratracheal instillation of elastase. Compared to controls treated with aerosolized water and elastase, those that received HA had significantly less airspace enlargement [22, 23].
Although the precise mechanism by which HA prevents lung injury is not yet well understood, our laboratory has shown that HA does not directly inhibit elastases but instead appears to bind to elastic fibers and prevent elastases from attacking them [20, 22, 24]. Studies using aerosolized fluorescein-labeled HA demonstrated preferential adherence of the polysaccharide to lung elastic fibers [22, 24]. This finding was complemented by additional experiments in which the binding of HA to elastic fibers in vitro prevented elastolysis by several different types of elastase, including human metalloproteinase 12, an enzyme that may be responsible for emphysematous changes associated with cigarette smoking [22].
Interactions between HA and elastic fibers may involve formation of electrostatic or hydrogen bonds. The binding sites may not be situated on the elastin protein itself but may instead be located in the surrounding matrix composed of microfibrils or other glycoproteins. Alternatively, the exogenously administered HA could combine with native HA in close proximity to elastic fibers by a process of self-aggregation [25, 26]. The resulting molecular complexes of HA may provide a protective barrier against both free elastases and the cells that secrete them.
In contrast to earlier studies in which HA was administered concomitantly with cigarette smoke, the current investigation allowed elastic fiber breakdown to proceed unimpeded for the first month, thus providing a more clinically relevant test of the therapeutic potential of HA. While concurrent administration of aerosolized HA significantly reduced BALF DID levels within 3 months of smoke exposure [15], the same effect was not seen until 6 months in the present study. Nevertheless, the delay in administering HA did not affect its ability to prevent emphysematous changes in the lung.
In the current study, the lack of airspace enlargement in the HA-treated group, despite significant elastic fiber breakdown, may possibly be explained by the fact that airway injury is an early feature of this model of smoke-induced lung injury. Although the precise contribution of airway inflammation to BALF DID levels remains uncertain, it may be speculated that the high levels of BALF DID at 2 and 4 months following initiation of smoke exposure are a consequence of elastin turnover in the walls of the larger airways rather than the distal lung.
With regard to total-lung DID, there were no significant differences between the two groups at any time point, suggesting that this parameter is not a sensitive measure of elastic fiber degradation but rather reflects the balance between elastic fiber injury and repair. Rapid resynthesis of these fibers could mask any differences with regard to their rate of breakdown.
The development of airway inflammation within the first 2 months of smoke exposure may explain why HA was initially ineffective in reducing BALF DID levels. The release of enzymes and oxidants by inflammatory cells may cause the exogenous HA to undergo breakdown, thereby impairing its ability to form larger, protective complexes in proximity to airway elastic fibers. While such a process remains hypothetical, this laboratory has previously shown that the same preparation of aerosolized HA used in the current study is effective in preventing acute lung injury only when given prior to intratracheal instillation of endotoxin [27].
Whether a similar pattern of elastic fiber breakdown and proliferation occurs in human lungs in response to smoking remains unclear. However, there is some experimental evidence which suggests that both forms of injury have certain features in common. In one study, DID levels in plasma and urine were significantly elevated in COPD patients, with and without emphysema, indicating that elastic fiber injury occurs in both airways and lung parenchyma [12]. Other investigators have also reported a reduction in urinary desmosine levels as the disease progresses, although their findings were attributed to a loss of lung elastic fiber mass rather than a specific decrease in the rate of elastin breakdown [28].
The leveling off of airspace enlargement in smoke-exposed mice after several months is consistent with an adaptive response to chronic injury. A number of studies suggest that enhanced synthesis of endogenous antioxidants may limit the damaging effects of tobacco smoke and other oxidants [29–31]. Furthermore, changes in the interstitial extracellular matrix resulting from continued injury and repair could decrease the likelihood of alveolar wall rupture due to elastase activity or mechanical stress. Regarding this possibility, an increase in lung collagen content has been reported after prolonged exposure to cigarette smoke, suggesting a transition from a degradative to a proliferative process, similar to that observed in the current study [32].
Notwithstanding these limitations, experimental models of smoke-induced lung injury provide a means of evaluating the usefulness of potential therapeutic agents. In the current study, the ability of HA to mitigate both airspace enlargement and elastic fiber injury, despite a 1-month delay in treatment, provides added support for testing this agent in patients with pre-existing COPD. The gradual progression of this disease suggests that even a small decrease in the rate of elastic fiber injury could have a significant impact on the decline of lung function.
Acknowledgments
This work was supported by NHLBI HL68383, the Alpha-1 Foundation, the Ned Doyle Foundation, the Charles A. Mastronardi Fund, the Franklyn Bracken Fund, and the James P. Mara Center for Lung Disease at the St. Luke's-Roosevelt Hospital Center.
Contributor Information
Jerome O. Cantor, Email: jocantor@pol.net, St Luke's Roosevelt Hospital Center, New York, NY, USA; St John's University, Queens, NY, USA; School of Pharmacy and Allied Health Sciences, 8000 Utopia Pkwy, Queens, NY 11439, USA.
Joseph M. Cerreta, St John's University, Queens, NY, USA
Marcos Ochoa, St Luke's Roosevelt Hospital Center, New York, NY, USA.
Shuren Ma, St Luke's Roosevelt Hospital Center, New York, NY, USA.
Ming Liu, Brookdale Hospital, Brooklyn, NY, USA.
Gerard M. Turino, St Luke's Roosevelt Hospital Center, New York, NY, USA
References
- 1.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–678. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro SD. Proteinases in chronic obstructive pulmonary disease. Biochem Soc Trans. 2002;30:98–102. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 3.Owen CA. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3:253–268. doi: 10.2147/copd.s2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Djekic UV, Gaggar A, Weathington NM. Attacking the multi-tiered proteolytic pathology of COPD: new insights from basic and translational studies. Pharmacol Ther. 2009;121:132–146. doi: 10.1016/j.pharmthera.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Celli BR. The importance of spirometry in COPD and asthma: effect on approach to management. Chest. 2000;117(2 Suppl):15S–19S. doi: 10.1378/chest.117.2_suppl.15s. [DOI] [PubMed] [Google Scholar]
- 6.Newell JD, Hogg JC, Snider GL. Report of a workshop: quantitative computed tomography scanning in longitudinal studies of emphysema. Eur Respir J. 2004;23:769–775. doi: 10.1183/09031936.04.00026504. [DOI] [PubMed] [Google Scholar]
- 7.Nakano Y, Muller NL, King GG, Niimi A, Kalloger SE, Mishima M, Pare PD. Quantitative assessment of airway remodeling using high-resolution CT. Chest. 2002;122:271S–275S. [PubMed] [Google Scholar]
- 8.Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56:515–548. doi: 10.1124/pr.56.4.2. [DOI] [PubMed] [Google Scholar]
- 9.Tzortzaki EG, Lambiri I, Vlachaki E, Siafakas NM. Biomarkers in COPD. Curr Med Chem. 2007;14:1037–1048. doi: 10.2174/092986707780362943. [DOI] [PubMed] [Google Scholar]
- 10.Ma S, Lin YY, Tartell L, Turino GM. The effect of tiotropium therapy on markers of elastin degradation in COPD. Respir Res. 2009;10:12. doi: 10.1186/1465-9921-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma S, Lieberman S, Turino GM, Lin YY. The detection and quantitation of free DID in human urine and their peptide-bound forms in sputum. Proc Natl Acad Sci USA. 2003;100:12941–12943. doi: 10.1073/pnas.2235344100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boschetto P, Quintavalle S, Zeni E, Leprotti S, Potena A, Ballerin L, Papi A, Palladini G, Luisetti M, Annovazzi L, Iadorola P, De Rosa E, Fabbri LM, Mapp CE. Association between markers of emphysema and more severe chronic obstructive pulmonary disease. Thorax. 2006;61:1037–1042. doi: 10.1136/thx.2006.058321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiorenza D, Viglio S, Lupi A, Baccheschi J, Tinelli C, Trisolini R, Iadarola R, Luisetti M, Snider GL. Urinary desmosine excretion in acute exacerbations of COPD: a preliminary report. Respir Med. 2002;96:110–114. doi: 10.1053/rmed.2001.1224. [DOI] [PubMed] [Google Scholar]
- 14.Viglio S, Iadorola P, Lupi A, Trisolini R, Tinelli C, Balbi B, Grassi V, Worlitzsch D, Doring G, Meloni F, Meyer KC, Dowson L, Hill SL, Stockley RA, Luisetti M. MEKC of DID in urine of chronic destructive lung disease patients. Eur Respir J. 2000;15:1039–1045. doi: 10.1034/j.1399-3003.2000.01511.x. [DOI] [PubMed] [Google Scholar]
- 15.Cantor JO, Cerreta JM, Ochoa M, Ma S, Chow T, Grunig G, Turino GM. Aerosolized hyaluronan limits airspace enlargement in a mouse model of cigarette smoke-induced pulmonary emphysema. Exp Lung Res. 2005;31:417–430. doi: 10.1080/01902140590918669. [DOI] [PubMed] [Google Scholar]
- 16.Dunnill MS. Quantitative methods in the study of pulmonary pathology. Thorax. 1962;17:320–328. doi: 10.1136/thx.17.4.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mascarenhas MM, Day RM, Ochoa CD, Choi WI, Yu L, Ouyang B, Garg HG, Hales CA, Quinn DA. Low molecular weight hyaluronan from stretched lung enhances interleukin-8 expression. Am J Respir Cell Mol Biol. 2004;30:51–60. doi: 10.1165/rcmb.2002-0167OC. [DOI] [PubMed] [Google Scholar]
- 18.McKee CM, Penno MB, Cowman M, Burdick MD, Streiter RM, Bao C, Noble PW. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J Clin Invest. 1996;98:2403–2413. doi: 10.1172/JCI119054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantor JO, Cerreta JM, Armand G, Keller S, Turino GM. Pulmonary air-space enlargement induced by intratracheal instillment of hyaluronidase and concomitant exposure to 60% oxygen. Exp Lung Res. 1993;19:177–192. doi: 10.3109/01902149309031718. [DOI] [PubMed] [Google Scholar]
- 20.Cantor JO, Cerreta JM, Keller S, Turino GM. Modulation of airspace enlargement in elastase-induced emphysema by intratracheal instillment of hyaluronidase and hyaluronic acid. Exp Lung Res. 1995;21:423–436. doi: 10.3109/01902149509023717. [DOI] [PubMed] [Google Scholar]
- 21.Murakami H, Yoshida M, Aritomi T, Shiraishi M, Ishibashi M, Watanabe K. Effects of hyaluronidase on porcine pancreatic elastase-induced lung injury. J Jpn Respir Soc. 1998;36:577–584. [PubMed] [Google Scholar]
- 22.Cantor JO, Shteyngart B, Cerreta JM, Armand G, Liu M, Turino GM. The effect of hyaluronan on elastic fiber injury in vitro and elastase-induced airspace enlargement in vivo. Proc Soc Exp Biol Med. 2000;225:65–71. doi: 10.1046/j.1525-1373.2000.22508.x. [DOI] [PubMed] [Google Scholar]
- 23.Cantor JO, Cerreta JM, Armand G, Turino GM. Aerosolized hyaluronic acid decreases alveolar injury induced by human neutrophil elastase. Proc Soc Exp Biol Med. 1998;217:471–475. doi: 10.3181/00379727-217-44260. [DOI] [PubMed] [Google Scholar]
- 24.Cantor JO, Cerreta JM, Armand G, Turino GM. Further investigation of the use of intratracheally administered hyaluronic acid to ameliorate elastase-induced emphysema. Exp Lung Res. 1997;23(3):229–244. doi: 10.3109/01902149709087369. [DOI] [PubMed] [Google Scholar]
- 25.Baccarani-Contri M, Vincenzi D, Cicchetti F, Mori G, Pasquali-Ronchetti I. Immunocytochemical localization of proteoglycans within normal elastin fibers. Eur J Cell Biol. 1990;53:305–312. [PubMed] [Google Scholar]
- 26.Scott JE, Cummings C, Brass A, Chen Y. Secondary and tertiary structures of hyaluronan in aqueous solution, investigated by rotary shadowing-electron microscopy and computer simulation. Hyaluronan is a very efficient network-forming polymer. Biochem J. 1991;274(Pt 3):699–705. doi: 10.1042/bj2740699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nadkarni PP, Kulkarni GS, Cerreta JM, Ma S, Cantor JO. Dichotomous effect of aerosolized hyaluronan in a hamster model of endotoxin-induced lung injury. Exp Lung Res. 2005;31:807–818. doi: 10.1080/01902140600574942. [DOI] [PubMed] [Google Scholar]
- 28.Cocci F, Miniati M, Monti S, Cavarra E, Gambelli F, Battolla L, Lucattelli M, Lungarella G. Urinary desmosine excretion is inversely correlated with the extent of emphysema in patients with chronic obstructive pulmonary disease. Int J Biochem Cell Biol. 2002;34:594–604. doi: 10.1016/s1357-2725(02)00015-8. [DOI] [PubMed] [Google Scholar]
- 29.Comhair SA, Erzurum SC. Antioxidant responses to oxidant-mediated lung diseases. Am J Physiol Lung Cell Mol Physiol. 2002;283:L246–L255. doi: 10.1152/ajplung.00491.2001. [DOI] [PubMed] [Google Scholar]
- 30.Baskaran S, Lakshmi S, Prasad PR. Effect of cigarette smoke on lipid peroxidation and antioxidant enzymes in albino rat. Indian J Exp Biol. 1999;37:1196–2000. [PubMed] [Google Scholar]
- 31.Wurzel H, Yeh CC, Gairola C, Chow CK. Oxidative damage and antioxidant status in the lungs and bronchoalveolar lavage fluid of rats exposed chronically to cigarette smoke. J Biochem Toxicol. 1996;10:11–17. [PubMed] [Google Scholar]
- 32.Wright JL, Churg A. Smoke-induced emphysema in guinea pigs is associated with morphometric evidence of collagen breakdown and repair. Am J Physiol Lung Cell Mol Physiol. 1995;12:17–20. doi: 10.1152/ajplung.1995.268.1.L17. [DOI] [PubMed] [Google Scholar]