

Abstract
The congenital myasthenic syndromes have now been traced to an array of molecular targets at the neuromuscular junction encoded by no fewer than 11 disease genes. The disease genes were identified by the candidate gene approach, using clues derived from clinical, electrophysiological, cytochemical, and ultrastructural features. For example, electrophysiologic studies in patients suffering from sudden episodes of apnea pointed to a defect in acetylcholine resynthesis and CHAT as the candidate gene (Ohno et al., Proc Natl Acad Sci USA 98:2017–2022–2001); refractoriness to anticholinesterase medications and partial or complete absence of acetylcholinesterase (AChE) from the endplates (EPs) has pointed to one of the two genes (COLQ and ACHET) encoding AChE, though mutations were observed only in COLQ. After a series of patients carrying mutations in a disease gene have been identified, the emerging genotype–phenotype correlations provided clues for targeted mutation analysis in other patients. Mutations in EP-specific proteins also prompted expression studies that proved pathogenicity, highlighted important functional domains of the abnormal proteins, and pointed to rational therapy.
Keywords: Congenital myasthenic syndromes, Acetylcholine esterase, Choline acetyltransferase, Acetylcholine receptor, Dok-7
Congenital myasthenic syndromes (CMS) were described as early as 1937 (Rothbart 1937) but received little attention until after the autoimmune origin of acquired myasthenia gravis was discovered in the 1970s. In the late 1970s and 1980s, distinct phenotypic features of a number of CMS were described (Engel et al. 1977; Engel et al. 1982; Engel and Lambert 1987; Mora et al. 1987), but their molecular basis remained unsolved until the 1990s. Since then, CMS caused by defects in each subunit of the acetylcholine receptor (AChR), and in ColQ, choline acetyltransferase (ChAT), rapsyn, Nav1.4, MuSK, and Dok-7 have been identified [see reviews by Engel et al. (2003a) and Sine and Engel (2006) and articles by Chevessier et al. (2004) and Beeson et al. (2006)]. These advances had four important consequences: (1) the emerging genotype–phenotype correlations yielded clues for targeted mutation analysis; (2) understanding the diverse molecular basis of the different CMS provided clues for rational therapy; (3) the identified mutations in conjunction with expression studies resulted in precise structure–function correlations; (4) and the novel insights gained from CMS mutations complemented knowledge derived from site-directed mutagenesis studies.
Table 1 shows a classification of the 295 CMS based on patients investigated at the Mayo Clinic to date. In addition to the different CMS listed in the Table 1, a single case of CMS due to a mutation in MuSK had been reported by Chevessier et al. (2004). The table indicates that the postsynaptic CMS are far more frequent than the presynaptic or synaptic space-associated CMS and that most postsynaptic CMS stem from defects in the AChR. The relative frequencies of the presynaptic, synaptic, and postsynaptic CMS observed at other institutions are similar (Beeson et al. 2005). This presentation will focus on the CMS investigated at the Mayo Clinic.
Table 1.
Classification of congenital myasthenic syndrome based on the site of the defect
| Index cases | |
|---|---|
| Presynaptic (8%) | |
| ChAT deficiencya | 16 |
| Paucity of synaptic vesicles and reduced quantal release | 1 |
| Congenital Lambert–Eaton-like | 2 |
| Other presynaptic defects | 2 |
| Synaptic space (15%) | |
| EP AChE deficiencya | 43 |
| Postsynaptic (78%) | |
| Primary kinetic defect ± AChR deficiencya | 52 |
| Primary AChR deficiency ± minor kinetic defecta | 108 |
| Rapsyn deficiencya | 43 |
| Dok-7 myastheniaa | 25 |
| Na channel myastheniaa | 1 |
| Plectin deficiencya | 2 |
| Total | 295 |
Classification was based on a cohort of CMS patients investigated at the Mayo Clinic between 1988 and 2008. One hundred seventeen patients had intercostal biopsies for in vitro electrophysiology and electron microscopy studies. Others were studied using DNA isolated from blood
Gene defect identified
Endplate Acetylcholinesterase Deficiency Due to Mutations in COLQ
With few exceptions, endplate (EP) acetylcholinesterase (AChE) deficiency causes a severe CMS refractory to cholinergic agents. Other clues to the diagnosis are a repetitive compound muscle action potential (CMAP) evoked by a single nerve stimulus and absence of AChE from the EPs. Electron microscopy shows small nerve terminals, often isolated from the synaptic cleft by an intruding Schwann cell process, and degeneration of the junctional folds (Fig. 1c). The small size and encasement by Schwann cells of the nerve terminals restricts evoked release of ACh. The absence of AChE prolongs the lifetime of ACh in the synaptic space; this increases the duration of the synaptic current (Fig. 1b), so that it outlasts the refractory period of the muscle fiber and excites a second CMAP (Fig. 1a). The prolonged EP currents also overload the postsynaptic region with cations and instigate an EP myopathy causing loss of AChR (Fig. 1c). The safety margin of neuromuscular transmission is compromised by decreased quantal release, loss of AChR, and desensitization of AChR at physiologic rates of stimulation (Engel et al. 1977).
Figure 1.

CMS caused by defects in AChE. a Repetitive CMAPs are generated by prolonged synaptic potentials that outlast the absolute refractory period of the muscle fiber. b MEPCs recorded from a normal and an AChE-deficient patient EP. Vertical arrows indicate decay time constants. The patient MEPC decays slowly as ACh repeatedly binds to AChRs before it exits the synaptic space by diffusion; the MEPC amplitude is reduced owing to the associated EP myopathy. c AChE-deficient EP region. AChR is localized with peroxidase-labeled α-bungarotoxin. Numerous junctional folds are degenerating, shedding their terminal expansions into the synaptic space. The nerve terminals are abnormally small and cover only a fraction of the postsynaptic region. d Schematic diagram showing domains of a ColQ strand and components of the A12 species of asymmetric AChE with 24 identified ColQ mutations. e Clockwise from upper left density gradient ultracentrifugation profiles of extracts of COS cells transfected with wild-type AChET, wild-type AChET and wild-type COLQ, wild-type AChET and the P59Q PRAD domain mutant of COLQ, wild-type AChET and the W148X collagen domain mutant of COLQ; wild-type AChET and the 1082delC C-terminal domain mutant of COLQ; and wild-type AChET and the D342E C-terminal domain mutant of COLQ. Note that the asymmetric forms of AChE appear only after transfection with the C-terminal D342E mutant of COLQ. AChE acetylcholinesterase, MEPC miniature EP current, HSPBD heparan sulfate proteoglycan binding domain, PRAD proline-rich attachment domain, Wt wild type, G globular moiety of AChET, A asymmetric moiety of AChE, M mutant peak. (Reproduced by permission from Engel et al. 2003b)
The molecular form of AChE at the EP is an asymmetric enzyme composed of one, two, or three homotetramers of globular catalytic subunits (AChET) attached to a triple-stranded collagenic tail (ColQ) forming A4, A8, and A12 species of AChE (Bon et al. 1997). When wild-type AChET and COLQ are transfected into COS cells, the density gradient sedimentation profile of the cell extract reveals the abundance of the globular and asymmetric species of AChE (Fig. 1e, upper central panel). After human COLQ cDNA was cloned (Donger et al. 1998; Ohno et al. 1998a), the genomic structure of COLQ was determined (Ohno et al. 1998a) and the molecular basis of EP AChE deficiency was traced to recessive mutations in COLQ (Donger et al. 1998; Ohno et al. 1998a). Figure 1d shows the COLQ mutations identified to date. The consequences of some of the mutations are illustrated in density gradient profiles of extracts of COS cells that have been transfected with wild-type AChET and mutant COLQ constructs (see panels in Fig. 1e).
Defects in Choline Acetyltransferase
The phenotype is distinguished by sudden episodes of severe respiratory distress and bulbar weakness culminating in apnea precipitated by infections, fever, excitement, or no identified cause against a background of variable interictal myasthenic symptoms. In some patients, the disease presents at birth with respiratory distress or apnea that gradually improves, but is followed by apneic attacks in later life; other patients are normal at birth and develop myasthenic symptoms and apneic attacks during infancy or childhood. The synaptic response to ACh, reflected by the amplitude of the miniature EP potential (MEPP) and EP potential (EPP), is normal in rested muscle but decreases abnormally during 10 Hz stimulation for 5 min, and then recovers slowly over the next 10 to 15 min while the quantal content of the EPP is essentially unaltered (Mora et al. 1987; Byring et al. 2002).
That the MEPP and EPP amplitudes decline abnormally when neuronal impulse flow is increased and then recover slowly has pointed to a defect in the resynthesis or vesicular packaging of ACh and implicated four candidate genes: the presynaptic high-affinity choline transporter, ChAT, the vesicular ACh transporter, and the vesicular proton pump. Mutation analysis in the first five patients revealed one nonsense and nine missense recessive mutations in CHAT. Kinetic studies of nine bacterially expressed and purified missense mutants revealed that one (E441K) lacked catalytic activity and eight (L210P, P211A, I305T, R420C, R482G, S498L, V506L, and R560H) had catalytic efficiencies for coenzyme A ranging from 1% to 39% of the wild type (Ohno et al. 2001). Because each patient carried two different mutations of different biochemical pathogenicity, no clear genotype–phenotype correlation emerged except that, in the most severely affected patient, both mutations markedly reduced the expression as well as the overall catalytic efficiency of ChAT. Subsequently, eight additional CHAT mutations were detected in our laboratory, and five additional mutations (V194L, I336T, S694L, S694C, and R548X) were reported by other investigators (Maselli et al. 2003; Schmidt et al. 2003; Kraner et al. 2003; Barisic et al. 2005) but were not functionally characterized. Interestingly, no patients harboring CHAT mutations had central or autonomic nervous system findings.
The crystal structure of rat (Fig. 2; Cai et al. 2004) and human (Kim et al. 2006) ChAT reveals a monomer consisting of two domains with an interfacial active site tunnel and a histidine residue at the center of the active site. Six of the identified mutations (V194L, L210P, P211A, E441K, R560H, and S694C) are at the substrate binding site or at, or near to, the catalytic site of the enzyme; the remaining missense mutation likely alter enzyme activity allosterically or change the folding or stability of the enzyme.
Figure 2.

ChAT mutations in CMS patients mapped on stereo image of structural model of rat ChAT (PDB code1Q6X). The active site is in a solvent-accessible tunnel at the interface of the N (upper) and C (lower) domains. Histidine at the active site is shown in stick representation. Residue numbers correspond to the human sequence. The indicated mutations, except those in boxes, were identified in our laboratory
CMS Caused by Defects in AChR
Mutations have now been discovered in the α, β, δ, and ε subunits of adult AChR and in the γ subunit of fetal AChR that harbors the γ instead of the ε subunit. The mutations fall into two major classes: those that reduce expression of AChR and those that alter its kinetic properties.
The low-expressor mutations are predominantly in the AChR ε subunit because persistent expression of the fetal AChR γ subunit can partially compensate for the absence or low expression of the ε subunit and thereby rescue the phenotype (Engel et al. 1996a, 2003a). Most low-expressor mutation on both alleles of the α, β, or δ AChR subunits are likely lethal in the embryos; only few such patients have been identified and they had a severe course with high fatality.
The kinetic mutations divide further into two types: slow-channel mutations that increase the synaptic response to ACh and fast-channel mutations that decrease the synaptic response to ACh. The slow-channel and fast-channel mutations represent physiologic opposites in their phenotypic consequences and steps underlying receptor activation. Some fast-channel mutations also reduce the expression of AChR, which contributes to, or even dominates, the clinical phenotype.
Slow-Channel CMS
The name of the syndrome originates from the abnormally slow decay of synaptic currents caused by abnormally prolonged opening burst of the AChR channel (Fig. 3b, c). The clinical phenotypes vary. Some slow-channel CMS present in early life and cause severe disability by the end of the first decade; others present later in life and progress slowly, resulting in little disability even in the sixth or seventh decade of life. Most patients show selectively severe involvement of cervical and of wrist and finger extensor muscles. Except for severely affected patients, the cranial muscles tend to be spared. Progressive spinal deformities and respiratory embarrassment are common complications during the evolution of the illness.
Figure 3.

Slow-channel syndromes. a Schematic diagram of AChR subunits with slow-channel mutation. b Single-channel currents from wild-type and slow-channel (αV249F) AChRs expressed in HEK cells. c MEPC recorded from EPs of a control subject and a patient harboring the αV249F slow-channel mutation. The slow-channel MEPC decays biexponentially due to expression of both wild-type and mutant AChRs at the EP. d Slow-channel EP. The junctional folds have disintegrated and the synaptic space is filled with debris (black asterisk). The junctional sarcoplasm displays fragmented apoptotic nuclei (white asterisk) and myeloid structures. Bar=1 μm. (a to c are from Engel 2004, by permission)
Since 1995, no fewer than 19 slow-channel mutations have been uncovered (Sine et al. 1995; Gomez et al. 1996, 2002; Engel et al. 1996b; Wang et al. 1997; Milone et al. 1997; Croxen et al. 1997; Ohno et al. 1998b, 2000; Shen et al. 2002, 2006; Hatton et al. 2003). The different mutations occur in different AChR subunits and in different functional domains of the subunits (Fig. 3a).
In the kinships observed to date, mutations in the channel domain caused more severe disease than those at the ACh binding site, but the phenotypic expressivity varied between and within kinships harboring the same mutation. Thus, although suggestive, phenotypic severity is not a fully reliable predictor of mutation site.
Patch clamp studies at the EP, mutation analysis, and expression studies in human embryonic kidney (HEK) cells indicate that the αG153S mutation near the extracellular ACh binding site (Sine et al. 1995) and the αN217K (Wang et al. 1997) as well as εL221F (Hatton et al. 2003) mutations in the N-terminal part of the M1 domain act mainly by enhancing affinity for ACh. This slows dissociation of ACh from the binding site and results in repeated channel reopenings during the prolonged receptor occupancy. Mutations in the M2 domain that line the channel pore, such as βV266M, εL269F, εT264P, and αV249F, promote the open state by affecting channel opening and closing steps (Ohno et al. 1995; Engel et al. 1996b; Milone et al. 1997), especially by slowing the channel closing rate α and variably speeding the channel opening rate β. Some M2 domain mutations also increase affinity for ACh; this is most marked in the case of αV249F (Milone et al. 1997), pronounced with εL269F (Engel et al. 1996b) and εT264P (Ohno et al. 1995), and not apparent in the case of βV266M (Engel et al. 1996b).
The prolonged synaptic currents predispose to cationic overloading of the postsynaptic region. This instigates an EP myopathy associated with the destruction of the junctional folds, degeneration of postsynaptic organelles, and apoptosis of the junctional nuclei (Fig. 3d).
For the normal adult human AChR, 7% of the synaptic current is carried by Ca2+; this is higher than for human fetal AChR or for muscle AChR of other species and predisposes to postsynaptic Ca2+ overloading when the synaptic current is prolonged (Fucile et al. 2006). However, at least two slow-channel mutations in the ε M2 domain of AChR (εT264P and εV259F) increase the Ca2+ permeability 1.5-fold and twofold, respectively (Di Castro et al. 2007) and further enhance the deleterious effects of the prolonged synaptic currents and the intrinsically high Ca2+ permeability of human AChR.
The safety margin of neuromuscular transmission is compromised by the EP myopathy which destroys junctional folds, causes loss of AChR, and alters the EP geometry; by staircase summation of markedly prolonged EPPs that cause a depolarization block at physiologic rates of stimulation; and by an increased propensity of the mutant receptors to become desensitized. The slow-channel syndrome can be effectively treated by long-lived open-channel blockers of AChR, such as quinidine and fluoxetine (Harper and Engel 1998; Fukudome et al. 1998; Harper et al. 2003).
Fast-Channel CMS
Fast-channel CMS are caused by recessive loss-of-function mutations usually accompanied by a low-expressor or null mutation in the second allele, so that the fast-channel mutation determines the phenotype. The name of the syndrome stems from abnormally rapid decay of the synaptic response caused by abnormally brief opening burst of the AChR channel. The underlying mechanisms include a decreased opening rate (β) or an increased closing rate (α) of the AChR channel, decreased agonist affinity, or a combination of these factors. A decrease in β or an increase in α decrease the gating efficiency (θ) determined by β/α. The diagnosis of a fast-channel syndrome depends on in vitro microelectrode recordings because there are no clinical, electromyographic, or anatomic clues to the diagnosis. Accurate diagnosis, however, is important for therapy, for this syndrome can be effectively treated with cholinesterase inhibitors and 3,4-diaminopyridine. A schematic diagram of the fast-channel mutations observed to date is presented in Fig. 4. Two examples of recently observed fast-channel CMS are presented below.
Figure 4.

Fast-channel syndromes. a MEPC recorded from EPs of a control subject and a patient harboring the αV285I fast-channel mutation. Arrows indicate decay time constants. b Single-channel currents from wild-type and fast-channel (αV285I) AChRs expressed in HEK cells. c Schematic diagram of fast-channel mutations in the AChR α, β, and δ subunits. (From Engel 2004, by permission)
An N346del fast-channel mutation was observed in the AChR ε subunit in two unrelated patients whose second ε allele harbored a splice site and a frameshift mutation. Both patients had severe myasthenic symptoms since birth (Shen et al. 2005). The deleted Asn residue is at the C terminus of the long cytoplasmic link between the third (M3) and fourth (M4) transmembrane domains of the ε subunit. The deletion shortens the M3–M4 link and shifts a negatively charged Asp residue against M4. EP studies showed reduced expression of εN346del-AChR and compensatory accumulation of fetal γ-AChR. When expressed in HEK cells, εN346del-AChR decreases the duration of channel opening bursts 2.7-fold compared to the wild type due to a 2.3-fold decrease in gating efficiency and a 2.5-fold decrease in agonist affinity of the diliganded closed state (see Fig. 5, left lower panel). Mutagenesis studies established that the effects of εN346del are not due to juxtaposition of a negative charge against M4 but to the shortening of the M3–M4 linker. Deletion of the C-terminal residue of the M3–M4 link in the β and δ subunits also results in fast-channel kinetics, but in the α subunit it dictates slow-channel kinetics. Thus, M3–M4 linkers contribute in an asymmetric manner to optimizing the activation of AChR through allosteric links to the channel and the agonist binding site (Shen et al. 2005).
Figure 5.
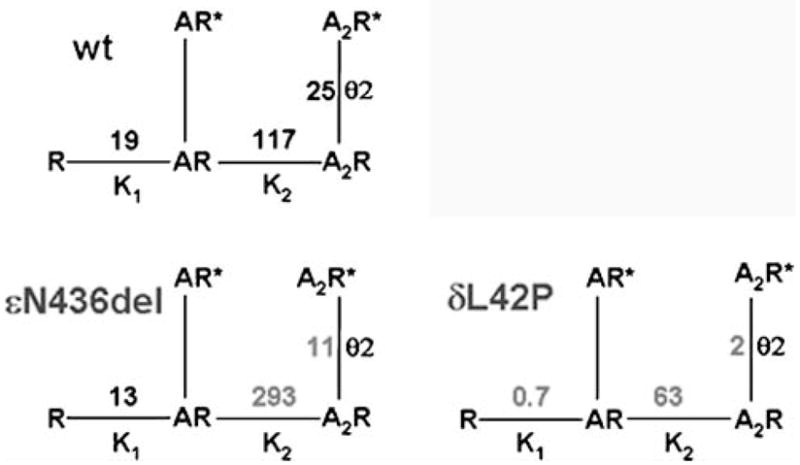
Kinetic scheme for activation of wild-type, εN436del, and δL42P AChR. AChR activation involves reversible binding of two molecules of agonist (A) to the AChR in the resting, closed state (R), followed by reversible formation of the open state (R*). K1 and K2 are equilibrium dissociation constants of the first and second agonist binding steps; θ indicates the gating equilibrium constant. Values indicated in gray differ markedly from corresponding values for the wild type
An L42P fast-channel mutation was identified in the AChR δ subunit in a patient with moderately severe to severe myasthenic symptoms since birth (Shen et al. 2008). The mutated Leu-42 is in a β1 strand at a ligand binding interface with an α subunit and positioned in the transitional zone where β strands of the extracellular domain merge with the α-helices of the transmembrane domain (see Fig. 6a). Expression studies in HEK cells revealed that δL42P decreases gating efficiency 12-fold, has a modest effect on agonist affinity, and shortens the channel burst length to 20% of normal (see right lower panel in Fig. 5). Replacing δ Leu-42 with a small flexible Gly, or charged Lys or Asp, or substitutions of Pro along the β1 strand around δ Leu-42 have similar kinetic consequences. Prosubstitution of the corresponding Leu in the ε subunit also has similar effects, in the β subunit it has no kinetic effect, but in the α subunits it markedly prolongs the channel burst length and results in slow-channel kinetics.
Figure 6.

Structural model of the AChR α and δ subunits and mutant cycle analyses. a An enlarged view of the coupled intersubunit residues αY127 and δN41 in the structural model of the Torpedo AChR (Protein Data Bank code 2BG9). b A mutant cycle for the mutations αY127T, δL42P, and εL40P. Single-channel currents correspond to each AChR elicited by 100 μM ACh. Changes in gating free energy along each limb of the cycle are shown, and the overall coupling free energy (ΔΔGint) in units of kilocalories per mole computed from −RT ln[(θwwθmm)/(θwmθmw)] where the θ(β/α) is the gating equilibrium constant for diliganded receptors for wild-type, single-mutant, or double-mutant AChRs. Horizontal bar indicates 20 ms for wild-type AChRs and 100 ms for the mutant AChRs. Vertical bar indicates 5 pA. (Reproduced by permission from Shen et al. 2008)
Because δ Asn-41 and ε Asn-39 were previously shown to be energetically coupled to α Tyr-127 in effecting rapid and efficient opening of the AChR channel (Mukhtasimova and Sine 2007), we postulated that δL42P and the corresponding substitution in the ε subunit, εL40P, could also hinder interaction with αY127. To examine how mutation of one residue in an AChR subunit affects the functional property of another residue in the same or another subunit, we performed mutant cycle experiments (Horovitz and Fersht 1990). Using −RTlnθ2 to compute the free energy of channel gating of each moiety of AChR, we generated a two-dimensional mutant cycle composed of gating free energies for wild-type, δL42P/εL40P, αY127T, and αY127T/δL42P/εL40P AChRs (see Fig. 6b). These experiments revealed that δL42 and εL40 are energetically coupled to αY127 with a free energy of 3.9 kcal/mol (Shen et al. 2008). This value approaches the 5.8-kcal/mol coupling energy of δN41A and εN39A with αY127T revealed by previous mutant cycle experiments (Mukhtasimova and Sine 2007).
The overall findings indicate that (1) δL42 together with εL40 are functionally coupled to the proximal αY127 and thus essential for efficient gating, (2) the equivalent α L40 residues positioned at nonreceptor binding interfaces of the α subunits also contribute to channel gating but in a novel manner (Shen et al. 2008).
Dok-7 Myasthenia
In 2006, Okada and coworkers identified Dok-7 as a muscle-intrinsic activator of MuSK required for synaptogenesis (Okada et al. 2006). Dok-7 harbors N-terminal pleckstrin homology and phosphotyrosine binding domains and is strongly expressed at the postsynaptic region of the skeletal muscle and in the heart. Subsequently, Beeson et al. (2006) detected recessive DOK7 mutations in CMS patients whose weakness was mostly in a limb–girdle distribution. In six of these patients, Slater et al. (2006) found that the EPs were small relative to muscle fiber size and had few and simple junctional folds, which the authors considered a constitutive feature of the disease. The number of AChRs per EP was also reduced, but this was deemed appropriate for the small size of the EPs. The amplitude of the MEPPs was decreased; this was largely attributed to the decreased input resistance of the simplified postsynaptic architecture. The quantal content (m) and the amplitude of the EPP were decreased but the amplitude of the miniature EP currents (MEPCs) was not.
After Okada’s discovery, we considered DOK7 to be a good candidate gene in patients who had small EPs, or a reduced number of AChRs per EP, or a limb–girdle phenotype and no mutations in AChR or rapsyn. Using these criteria, we identified 25 patients who carry DOK7 mutations and the findings in the first 16 patients were published (Selcen et al. 2008): each patient harbored two heteroallelic pathogenic mutations, 11 detected in genomic DNA, and six in cDNA isolated from intercostal muscle specimens. No consistent phenotype–genotype correlations emerged. Fourteen patients had intercostal muscle biopsies with detailed microelectrode and electron microscopy studies of the EPs (Selcen et al. 2008). All EPs consisted of one to multiple small synaptic contacts. The mean MEPP as well as the MEPC amplitude and the quantal content of the EPP were reduced by ~25%, but for each of these parameters the distribution of patient and control values overlapped. Electron microscopy of 613 regions of 409 EPs revealed disintegrating junctional folds in 33% (Figs. 7a and 8a, b), denuded postsynaptic regions with nearby nerve sprouts in 18% (Fig. 8b), and degenerating subsynaptic organelles in 10% (Fig. 8b). Importantly, however, some EPs in each patient had a normal architecture and the density and distribution of AChR on the crests of the preserved junctional folds were normal (Fig. 7b). These findings indicate that (1) a simplified postsynaptic architecture is due at least in part to the degeneration and regeneration of the junctional folds; (2) although Dok-7 acts in concert with MuSK in activating rapsyn to concentrate AChRs on the junctional folds, the identified defects in Dok-7 do not decrease the concentration of AChR on preserved junctional folds.
Figure 7.
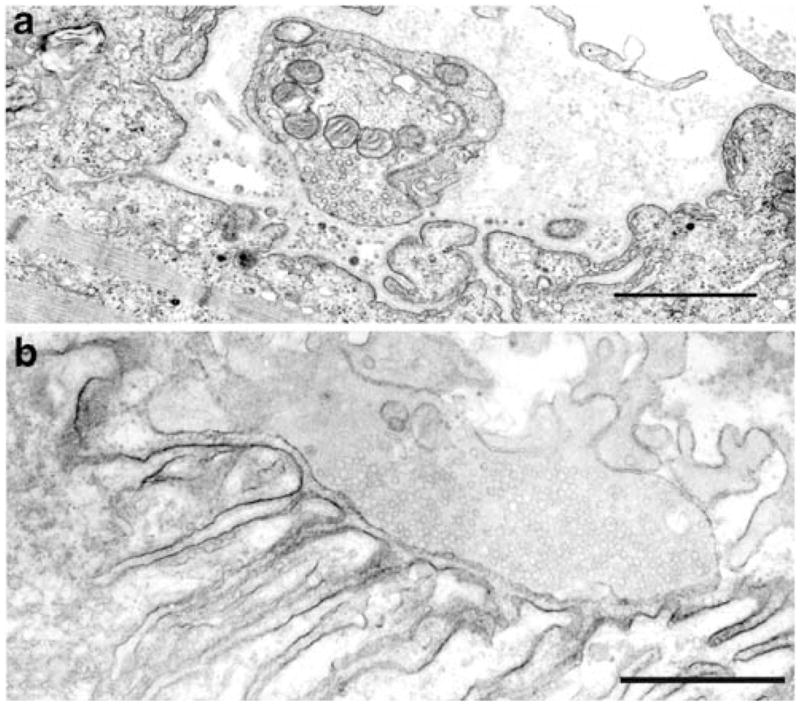
Dok-7 myasthenia. a Degenerating EP region displaying loss or amputation of junctional folds and accumulation of degenerate fold remnants in the widened synaptic space. b Unstained normal EP region reacted for AChR with peroxidase conjugated α-bungarotoxin. The junctional folds are intact and display a normal concentration and distribution of AChR on the junctional fold. Bars=1 μm
Figure 8.
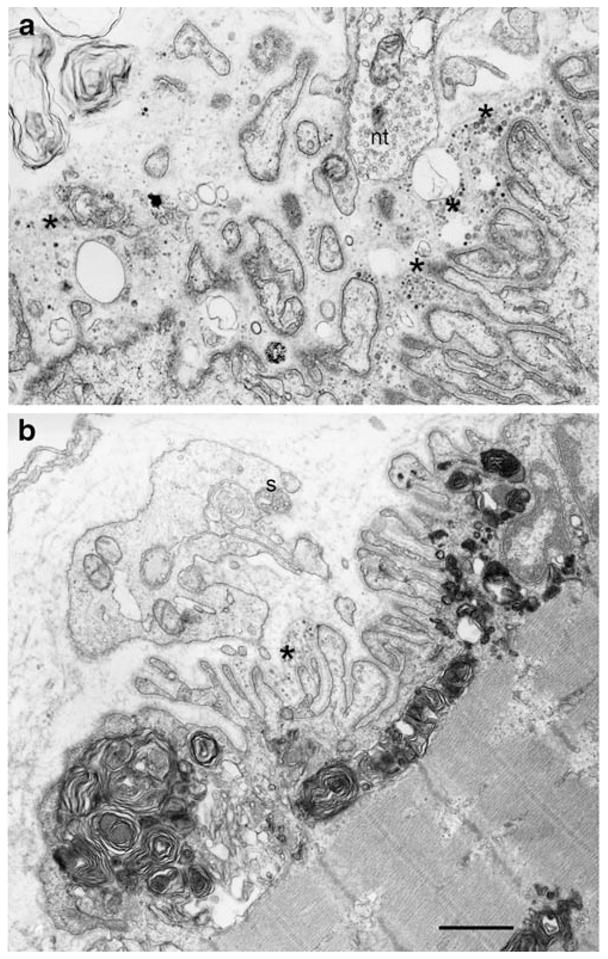
Dok-7 myasthenia; EPs with presynaptic and postsynaptic abnormalities. a A highly abnormal EP showing extensive degeneration of the junctional folds (asterisks) and widening of the synaptic space. b This EP region shows extensive accumulation of myeloid structures in the junctional sarcoplasm. Some junctional folds are degenerating (asterisk). The postsynaptic region is denuded of its nerve terminal. A nerve sprout (s) surrounded by Schwann cell appears above the junction. Bars=1 μm
What Have We Learned from the Congenital Myasthenic Syndromes
Among targets for genetic disorders, the neuromuscular junction is ideal for identifying causality. It is highly localized and accessible, and its anatomy and physiology are well characterized. Neuromuscular transmission is essential for life and has few mechanisms of plasticity or compensation, further restricting the set of candidate genes. Once the genetic cause and underlying molecular process are identified, the potential for treatment is high.
From the CMS targeting ChAT, we have learned that the enzyme is rate limiting for ACh resynthesis and vesicular filling at the neuromuscular junction whereas central and autonomic cholinergic synapses can function normally even if the catalytic efficiency of the enzyme is diminished (Ohno et al. 2001).
From the CMS targeting the ColQ subunit AChE, we have learned that the absence of the enzyme from the synaptic space, though highly disabling, is not lethal; that both the collagenic and globular domains of ColQ are crucial for its insertion into the synaptic space; and that the absence of the enzyme activates negative trophic effects on the size of the nerve terminal and thereby decreases the number of quanta available for release (Engel et al. 1977; Ohno et al. 1998a; Kimbell et al. 2004). We also learned that prolonged synaptic currents can repeatedly excite the muscle fiber action potential and that cationic overloading of the postsynaptic region by the prolonged currents causes an EP myopathy (Engel et al. 1977).
From the low-expressor and null mutations in the AChR ε subunit, we have learned that persistent expression of the fetal γ subunit can rescue the phenotype, though unfortunately no rescue is available for low-expressor or null mutations in the other AChR subunits (Engel et al. 1996a, 2003a).
Perhaps unexpectedly, kinetic mutations of the AChR shed light on biology itself. For many years, it was supposed that the decay phase of the postsynaptic response resulted from the inherent ability of AChR channels to open and close repeatedly before finally closing. The CMS mutation αG153S, by increasing affinity for ACh, increased bursting of the AChR and consequently the decay phase of the response, confirming the postulated mechanism (Sine et al. 1995). Insight into the inherently homogeneous kinetics of single AChRs emerged from study of the mutation εA411P, which causes heterogeneous kinetics of single AChRs; the findings were reconciled by postulating an intrinsically corrugated free energy landscape that was funnel-shaped for the wild-type AChR but flattened for the εA411p mutant (Wang et al. 2000). Many disease-causing mutations have yielded unexpected insights into structure–function relationships of the AChR, including mutations of the ion-conductive pore that enhance channel gating (Engel et al. 1996b, 2003a), mutations of the ACh binding site that reduce affinity of ACh for the open-channel state (Ohno et al. 1996), and mutations at subunit interfaces that mediate intersubunit communication required for rapid and efficient channel opening (Shen et al. 2008).
From pioneering studies by Beeson et al. (2006) and Okada et al. (2006), we learned that Dok-7 is a muscle-intrinsic activator of MuSK and important in synaptogenesis and that mutations in Dok-7 cause a CMS that preferentially affects the limb–girdle and axial rather than cranial muscles. Subsequently, we also learned that Dok-7 is essential for maintaining the structure of the mature junction, so that defects in Dok-7 result in cycles of degeneration and regeneration of the junction (Selcen et al. 2008).
Finally, and importantly, understanding the underlying molecular defect has shown the way to effective therapy. When ACh release is compromised, as in ChAT deficiency, or if the synaptic response to ACh is reduced by low-expressor of fast-channel mutations in AChR or mutations in rapsyn, then therapy aimed at increasing quantal release with 3,4-diaminopyridine and the number of AChRs activated by each quantum with cholinesterase inhibitors is the rational approach. In the slow-channel syndrome, the increased synaptic response to ACh is effectively dampened by long-lived open-channel blockers of the AChR channel. Cholinergic agonists are contraindicated in EP AChE deficiency and Dok-7 myasthenia. Fortunately, in these two diseases, treatment by β-adrenergic agonists is an empiric but nonetheless effective approach (Engel 2007).
Acknowledgments
Work in our laboratories was supported by NIH grants to A.G. Engel (NS-6277) and to S.M. Sine (NS-31744) and by a Muscular Dystrophy Association grant to A.G. Engel.
Contributor Information
Andrew G. Engel, Email: age@mayo.edu, Department of Neurology and Muscle Research Laboratory, Mayo Clinic College of Medicine, Rochester, MN 55905, USA
Xin-Ming Shen, Department of Neurology and Muscle Research Laboratory, Mayo Clinic College of Medicine, Rochester, MN 55905, USA.
Duygu Selcen, Department of Neurology and Muscle Research Laboratory, Mayo Clinic College of Medicine, Rochester, MN 55905, USA.
Steven M. Sine, Department of Physiology and Biomedical Engineering, Mayo Clinic College of Medicine, Rochester, MN 55905, USA
References
- Barisic N, Muller JS, Paucic-Kirincic E, et al. Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. European Journal of Paediatric Neurology. 2005;9:7–12. doi: 10.1016/j.ejpn.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Beeson D, Hantai D, Lochmuller H, Engel AG. Report of the 126th International Workshop: Congenital myasthenic syndromes. Neuromuscular Disorders. 2005;15:498–512. doi: 10.1016/j.nmd.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- Bon S, Coussen F, Massoulié J. Quaternary associations of acetylcholinesterase. II. The polyproline attachment domain of the collagen tail. Journal of Biological Chemistry. 1997;272:3016–3021. doi: 10.1074/jbc.272.5.3016. [DOI] [PubMed] [Google Scholar]
- Byring RF, Pihko H, Shen XM, et al. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscular Disorders. 2002;12:548–553. doi: 10.1016/s0960-8966(01)00336-4. [DOI] [PubMed] [Google Scholar]
- Cai Y, Cronin CN, Engel AG, Ohno K, Hersh LB, Rodgers D. Choline acetyltransferase structure reveals distribution of mutations that cause motor disorders. EMBO Journal. 2004;23:2047–2058. doi: 10.1038/sj.emboj.7600221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevessier F, Faraut B, Ravel-Chapuis A, et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Human Molecular Genetics. 2004;13:3229–3240. doi: 10.1093/hmg/ddh333. [DOI] [PubMed] [Google Scholar]
- Croxen R, Newland C, Beeson D, et al. Mutations in different functional domains of the human muscle acetylcholine receptor α subunit in patients with the slow-channel congenital myasthenic syndrome. Human Molecular Genetics. 1997;6:767–774. doi: 10.1093/hmg/6.5.767. [DOI] [PubMed] [Google Scholar]
- Di Castro A, Martinello K, Grassi F, Eusebi F, Engel AG. Pathogenic point mutations in a transmembrane domain of the ε subunit increase the Ca2+ permeability of the human endplate ACh receptor. Journal of Physiology (London) 2007;579:671–677. doi: 10.1113/jphysiol.2007.127977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donger C, Krejci E, Serradell P, et al. Mutation in the human acetylcholinesterase-associated gene, COLQ, is responsible for congenital myasthenic syndrome with end-plate acetylcholin-esterase deficiency. American Journal of Human Genetics. 1998;63:967–975. doi: 10.1086/302059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG. Congenital myasthenic syndromes. In: Engel AG, Franzini-Armstrong C, editors. Myology. 3. New York: McGraw-Hill; 2004. pp. 1801–1844. [Google Scholar]
- Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics. 2007;4:252–257. doi: 10.1016/j.nurt.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Lambert EH. Congenital myasthenic syndromes. Electroencephalography and Clinical Neurophysiology Supplement. 1987;39:91–102. [PubMed] [Google Scholar]
- Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Annals of Neurology. 1977;1:315–330. doi: 10.1002/ana.410010403. [DOI] [PubMed] [Google Scholar]
- Engel AG, Lambert EH, Mulder DM, et al. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Annals of Neurology. 1982;11:553–569. doi: 10.1002/ana.410110603. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Bouzat C, Sine SM, Griggs RG. End-plate acetylcholine receptor deficiency due to nonsense mutations in the ε subunit. Annals of Neurology. 1996a;40:810–817. doi: 10.1002/ana.410400521. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Milone M, et al. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Human Molecular Genetics. 1996b;5:1217–1227. doi: 10.1093/hmg/5.9.1217. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nature Reviews. Neuroscience. 2003a;4:339–352. doi: 10.1038/nrn1101. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: A diverse array of molecular targets. Journal of Neurocytology. 2003b;32:1017–1037. doi: 10.1023/B:NEUR.0000020639.22895.28. [DOI] [PubMed] [Google Scholar]
- Fucile S, Sucapane A, Grassi A, Eusebi F, Engel AG. The human adult subtype AChR channel has high Ca2+ permeability. Journal of Physiology (London) 2006;573:35–43. doi: 10.1113/jphysiol.2006.108092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukudome T, Ohno K, Brengman JM, Engel AG. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. NeuroReport. 1998;9:1907–1911. doi: 10.1097/00001756-199806010-00044. [DOI] [PubMed] [Google Scholar]
- Gomez CM, Maselli R, Gammack J, et al. A beta-subunit mutation in the acetylcholine receptor gate causes severe slow-channel syndrome. Annals of Neurology. 1996;39:712–723. doi: 10.1002/ana.410390607. [DOI] [PubMed] [Google Scholar]
- Gomez CM, Maselli R, Vohra BPS, et al. Novel delta subunit mutation in slow-channel syndrome causes severe weakness by novel mechanism. Annals of Neurology. 2002;51:102–112. doi: 10.1002/ana.10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CM, Engel AG. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Annals of Neurology. 1998;43:480–484. doi: 10.1002/ana.410430411. [DOI] [PubMed] [Google Scholar]
- Harper CM, Fukudome T, Engel AG. Treatment of slow channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:170–173. doi: 10.1212/01.wnl.0000061483.11417.1b. [DOI] [PubMed] [Google Scholar]
- Hatton CJ, Shelley C, Brydson M, Beeson D, Colquhoun D. Properties of the human muscle nicotinic receptor, and of the slow-channel myasthenic syndrome mutant εL221F, inferred from maximum likelihood fits. Journal of Physiology (London) 2003;547:729–760. doi: 10.1113/jphysiol.2002.034173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz A, Fersht A. Strategy for analyzing the cooperativity of intramolecular interactions in peptides and proteins. Journal of Molecular Biology. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- Kim AR, Rylett RJ, Shilton BH. Substrate binding and catalytic mechanism of human acetylcholinesterase. Biochemistry. 2006;45:14621–14631. doi: 10.1021/bi061536l. [DOI] [PubMed] [Google Scholar]
- Kimbell LM, Ohno K, Engel AG, Rotundo RL. C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. Journal of Biological Chemistry. 2004;279:10997–11005. doi: 10.1074/jbc.M305462200. [DOI] [PubMed] [Google Scholar]
- Kraner S, Lufenberg I, Strassburg HM, Sieb JP, Steinlein OK. Congenital myasthenic syndrome with episodic apnea in patients homozygous for a CHAT missense mutation. Archives of Neurology. 2003;60:761–763. doi: 10.1001/archneur.60.5.761. [DOI] [PubMed] [Google Scholar]
- Maselli RA, Chen D, Mo D, Bowe C, Fenton G, Wollman RL. Choline acetyltransferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle and Nerve. 2003;27:180–187. doi: 10.1002/mus.10300. [DOI] [PubMed] [Google Scholar]
- Milone M, Wang HL, Ohno K, et al. Slow-channel syndrome caused by enhanced activation, desensitization, and agonist binding affinity due to mutation in the M2 domain of the acetylcholine receptor alpha subunit. Journal of Neuroscience. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora M, Lambert EH, Engel AG. Synaptic vesicle abnormality in familial infantile myasthenia. Neurology. 1987;37:206–214. doi: 10.1212/wnl.37.2.206. [DOI] [PubMed] [Google Scholar]
- Mukhtasimova N, Sine SM. An inter-subunit trigger of channel gating in the muscle nicotinic receptor. Journal of the Neurological Sciences. 2007;27:4110–4119. doi: 10.1523/JNEUROSCI.0025-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Hutchinson DO, Milone M, et al. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the ε subunit. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Wang HL, Milone M, et al. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor ε subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- Ohno K, Brengman JM, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proceedings of the National Academy of Sciences of the United States of America. 1998a;95:9654–9659. doi: 10.1073/pnas.95.16.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Milone M, Brengman JM, Lo Monaco M, Evoli A, Tonali P, et al. Slow-channel congenital myasthenic syndrome caused by a novel mutation in the acetylcholine receptor ε subunit. Neurology. 1998b;50:A432. Ref Type: Abstract. [Google Scholar]
- Ohno K, Wang HL, Shen XM, et al. Slow-channel mutations in the center of the M1 transmembrane domain of the acetylcholine receptor α subunit. Neurology. 2000;54(Suppl 3):A183. [Google Scholar]
- Ohno K, Tsujino A, Brengman JM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2017–2022. doi: 10.1073/pnas.98.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Inoue A, Okada M, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- Rothbart HB. Myasthenia gravis. Familial occurrence. JAMA. 1937;108:715–717. [Google Scholar]
- Schmidt C, Abicht A, Krampfl K, et al. Congenital myasthenic syndrome due to a novel missense mutation in the gene encoding choline acetyltransferase. Neuromuscular Disorders. 2003;13:245–251. doi: 10.1016/s0960-8966(02)00273-0. [DOI] [PubMed] [Google Scholar]
- Selcen D, Milone M, Shen XM, et al. Dok-7 myasthenia: Phenotypic and molecular genetic studies in 16 patients. Annals of Neurology. 2008;64:71–87. doi: 10.1002/ana.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XM, Ohno K, Adams C, Engel AG. Slow-channel congenital myasthenic syndrome caused by a novel epsilon subunit mutation in the second AChR transmembrane domain. Journal of the Neurological Sciences. 2002;199(Suppl 1):S96. [Google Scholar]
- Shen XM, Ohno K, Sine SM, Engel AG. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle AChR M3–M4 linker. Brain. 2005;128:345–355. doi: 10.1093/brain/awh364. [DOI] [PubMed] [Google Scholar]
- Shen XM, Deymeer F, Sine SM, Engel AG. Slow-channel mutation in AChR αM4 domain and its efficient knockdown. Annals of Neurology. 2006;60:128–136. doi: 10.1002/ana.20861. [DOI] [PubMed] [Google Scholar]
- Shen XM, Fukuda T, Ohno K, Sine SM, Engel AG. Congenital myasthenia-related AChR ä subunit mutation interferes with intersubunit communication essential for channel gating. Journal of Clinical Investigation. 2008;118:1867–1876. doi: 10.1172/JCI34527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2006;440:448–455. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- Sine SM, Ohno K, Bouzat C, et al. Mutation of the acetylcholine receptor α subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Slater CR, Fawcett PRW, Walls TJ, et al. Pre- and postsynaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain. 2006;127:2061–2076. doi: 10.1093/brain/awl200. [DOI] [PubMed] [Google Scholar]
- Wang HL, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptor alpha subunit decreases the rate of agonist dissociation. Journal of General Physiology. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Ohno K, Milone M, et al. Fundamental gating mechanism of nicotinic receptor channel revealed by mutation causing a congenital myasthenic syndrome. Journal of General Physiology. 2000;116:449–460. doi: 10.1085/jgp.116.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]