

Abstract
Androgen refractory cancer of the prostate (ARCaP) cells contain androgen receptor (AR) and synthesize and secrete prostate specific antigen (PSA). We isolated epithelia-like ARCaPE from parental ARCaP cells and induced them to undergo epithelial–mesenchymal transition (EMT) by exposing these cells to soluble factors including TGFβ1 plus EGF, IGF-1, β2-microglobulin (β2-m), or a bone microenvironment. The molecular and behavioral characteristics of the resultant ARCaPM were characterized extensively in comparison to the parental ARCaPE cells. In addition to expressing mesenchymal biomarkers, ARCaPM gained 100% incidence of bone metastasis. ARCaPM cells express receptor activator of NF-κB ligand (RANKL), which was shown to increase tartrate-resistant acid phosphatase (TRAP)-positive osteoclasts in culture, and when metastatic to bone in vivo. We provide evidence that RANKL expression was promoted by increased cell signaling mediated by the activation of Stat3-Snail-LIV-1. RANKL expressed by ARCaPM cells is functional both in vitro and in vivo. The lesson we learned from the ARCaP model of EMT is that activation of a specific cell signaling pathway by soluble factors can lead to increased bone turnover, mediated by enhanced RANKL expression by tumor cells, which is implicated in the high incidence of prostate cancer bone colonization. The AR-CaP EMT model is highly attractive for developing new therapeutic agents to treat prostate cancer bone metastasis.
Keywords: EMT, Cell signaling, RANKL, Osteoclastogenesis, LIV-1, Bone metastasis, Prostate, Breast, Lung, Kidney, TGFβ1, EGF
Introduction
Two thirds of the cancer-related deaths in the US involve bone metastasis [1, 2]. ARCaP cells, isolated from the ascites fluid of a patient with bone metastasis, express a host of human prostate cancer biomarkers, have high propensity for rapid and predictable bone and soft tissue growth/metastases through orthotopic, intracardic and intraosseous injections [3–5], and undergo EMT on exposure to soluble factors or host bone microenvironment [4, 6]. The ARCaP EMT confers increased cell migratory, invasive and metastatic potential to soft tissue and bone [4, 6]. Here we used this unique model of prostate carcinoma progression to study soluble growth factor-induced EMT and intracellular signaling pathways that lead to RANKL expression, which activates osteoclastogenesis. We suggest that soluble factor-induced RANKL expression in the ARCaP EMT model accounts for subsequent prostate cancer cell bone colonization. We showed the plasticity of cancer cells, which upon induction by these soluble factors, switch to express markers commonly found in osteoblasts (termed “osteomimicry”), such as non-collagenous bone matrix proteins, osteocalcin (OC), osteopontin (OPN) and bone sialoprotein (BSP), and RANKL, collectively allowing cancer cells to survive and thrive in the bone microenvironment [7–9].
Previous studies of other solid tumors support the association of increased EMT with the ability of cancer cells to migrate, invade and metastasize [10–13]. Several studies have characterized EMT during embryonic development and related it to cancer metastasis. These studies pave the way for novel gene discovery and biomarker validation for cancer progression. In this communication, we have determined the roles of LIV-1, a zinc transporter, in the control of EMT, RANKL and Stat3-Snail-LIV-1 signaling during prostate cancer progression. Below we discuss the plasticity of prostate cancer cells, which are capable of undergoing EMT. In addition, it has been shown by others that prostate cancer cells can also revert via mesenchymal–epithelial transition (MET) [14–16], mesenchymal–epithelial-reverting transition (MErT) [16] and mesenchymal–mesenchymal transition (MMT) [17] to control their malignant and invasive potentials.
Materials and methods
Cell culture, reagents and animal studies
ARCaPE and ARCaPM cells were maintained in T medium (Invitrogen, Carlsbad, CA) and 5% fetal bovine serum (FBS) at 37°C supplemented with 5% CO2 in a humidified incubator. ARCaP cells were established from the ascites fluid of a patient with prostate cancer bone metastasis [5]. ARCaPE and ARCaPM are sublines of ARCaP that were generated in our laboratory by single cell dilution cloning [4]. To study the effect of growth factor treatment, ARCaP cells were treated with 4 ng/ml TGF β1 and 50 ng/ml EGF either alone or in combination. Data represent triplicate samples from two independent experiments. Recombinant-human TGF-β1, -mouse RANKL, -mouse M-CSF-1 and -human osteoprotegerin (OPG) were from R&D Systems, Inc. (Minneapolis, MN). FBS, recombinant human EGF, G418, anti-flag M2 monoclonal antibody and mouse monoclonal anti-human actin antibody were from Sigma–Aldrich, Inc. (St. Louis, MO). IGF-1 treatment was reported in a separate manuscript [18].
Animal studies were conducted by previously published procedures [19, 20].
Gene and protein expression
Gene expression was analyzed by RT-PCR (see [4]). In brief, cells were plated on 6-well dishes at 3 × 105 cells per well and grown to 70% confluence, gently washed with PBS and used for RNA isolation. Total RNA was isolated from cells using the RNeasy Mini kit (Qiagen, Valencia, CA) and subjected to reverse transcription using the Superscript first-strand cDNA Synthesis kit (Invitrogen, Carlsbad, CA). The primers used for PCR analysis were: E-cadherin-F-5′-TCCATTTCTTGGTCTACGCC-3′ R-5′-CACCTTCAGCCAACCTGTTT-3′; N-cadherin-F-5′-GTGCCATTAGCCAAGGAATTCAGC-3′ R-5′-GCGTTCCTGTTCCACTCATAGGAGG-3′; Vimentin-F5′-TGGCACGTCTTGACCTTGAA-3′ R-5′-GGTCATCGTGATGCTGAGAA-3′; LIV-1-F-5′-GCAATGGCGAGGAAGTTATCT-3′ R-5′-CTATTG TCTCTAGAAAGTGAG-3′; Snail-F-5′-CGAAAGGCCTTC AACTGCAAAT-3′; R-5′-ACTGGTACTTCTTGACATC TG-3′; RANKL-F-5′-CAGCACATCAGAGCAGAGAA AG-3′ R-5′-TGTTGGCATACAGGTAATAAAAGC-3′ and Stat3-F 5′-AACTCTTGGGACCTGGTGTG-3′ R-5′-TAGGCGCCTCAGTCGTATCT-3′. The thermal profiles for E-cadherin, N-cadherin, vimentin, and Stat3 were 32 cycles with denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 1 min. LIV-1, Snail and RANKL cDNA amplification were 30, 33 and 35 cycles starting with denaturation for 30 s at 94°C, followed by 1 min of annealing at 43°C (for LIV-1), 55°C (for Snail) and 55°C (for RANKL) and 1 min of extension at 72°C. RT-PCR products were analyzed by agarose gel electrophoresis.
Protein expression was determined by immunohistochemical (IHC) and western blot analyses. For IHC, antibodies and their sources are: monoclonal antibodies against cytokeratin 18/19 (CK18/19), vimentin (VM) (Dako Corp., Ltd., Carpinteria, CA); polyclonal antibodies to E-cadherin and N-cadherin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); RANKL (CHEMICON International, Inc., Temecula, CA). Cells and deparaffinized, rehydrated tissues after pressure-cooking antigen retrieval at 125°C and 20 p.s.i. for 30 s were subjected to 10-min double endogenous enzyme block, 30-min primary antibody reaction and 30-min DakoCytomation EnVision + HRP reagent incubation. Signals were detected by adding substrate hydrogen peroxide using diaminobenzidine as a chromogen, counter-stained by hematoxylin. All detection reagents were obtained from Dako Corporation (Carpinteria, CA). Matched IgG, or diluted rabbit serum was used as a negative control. Antibodies for western blot are: Snail (SN9H2, Rat mAb from Cell Signaling Technology, Inc. Beverly, MA), RANKL (sc-9073, Santa Cruz Biotechnology, Inc.), Stat3 and phosphor-Stat3 (Ser 727; Cell Signaling Technology), and LIV-1 (see below). Total cell lysates were prepared using a lysis buffer [50 mM Tris (pH 8), 150 mM NaCl, 0.02% NaN3, 0.1% SDS, 1% NP40 and 0.5% sodium deoxycholate] containing 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Protein concentration was determined by Bradford assay using Coomassie Plus Protein Reagent (Pierce, Rockford, IL). Western blot was performed using the Novex system (Invitrogen) as described previously [9, 21] Protein bands were detected by Enhanced Chemiluminescence Western Blotting Detection Reagents (Amersham-Pharmacia Biotech, Piscataway, NJ).
Snail transfection and induction of RANKL expression
A constitutively active Snail, the Flag-tagged Snail-6SA [22] kindly provide by Dr. M. C. Hung (MD Anderson Cancer Center, Houston, TX) was used to transfect ARCaPE cells. An aliquot of 4 μg plasmid DNA was used in 6-well plate with GenePORTER (Genlantis, San Diego, CA) following manufacturer recommended protocol. A total of 24 single transfected clones were selected subsequent to further selection with G418 (400 μg/ml) and limited dilution.
Over-expression of the Flag-Snail-6SA was confirmed by western blot with antibody (sc-807, Santa Cruz Biotechnology) against the Flag epitope. ARCaPE cells transfected with empty vector served as controls. EMT of the transfected clones was confirmed both morphologically and biochemically and these results will be reported elsewhere.
In vitro osteoclastogenesis assay
Osteoclastogenesis assay was performed as described previously with minor modifications [23]. Briefly, 1 × 103 cancer cells were seeded with 2 × 105 haematopoietic pre-osteoclasts harvested from adult mouse spleen by disaggregating through a wire sieve. The co-cultures were performed in a 48-well plate containing 500 μl of α-MEM media supplemented with 10% FBS, and 1 ng/ml recombinant mouse M-CSF (R&D Systems, Inc., Minneapolis, MN). As a positive control, 100 ng/ml recombinant mouse RANKL (R&D Systems) was added to 2 × 105 pre-osteoclastic cells. A total of 50 ng/ml recombinant human osteoprotegerin (OPG, R&D Systems) was added to certain wells to block RANKL-mediated osteoclastogenesis. The cells were fed twice weekly by replacing half of the cultured media with fresh media containing M-CSF (and RANKL, for the positive control). After 14 days, the cells were analyzed by TRAP staining (Sigma–Aldrich, Inc., St. Louis, MO), and the TRAP+ multinucleated cells in the entire well were counted as mature osteoclasts.
Cell signaling studies and LIV-1 transfection
Human LIV-1 plasmid DNA (2 μg cloned from human breast cancer cells, characterized and sequenced by Dr. Binhua P. Zhou), cloned into pcDNA 3.1/V5-His vector (Invitrogen), was transfected into ARCaPE cells using Lipofectamine 2000 Reagent (Invitrogen). The effects of LIV-1 on EMT gene expression were assayed 3 days after transfection. ARCaPE cells transfected with empty vector served as controls. LIV-1 expression in ARCaPM cells was silenced transiently by LIV-1 siRNA duplex (0.7 μg of 5′–3′-AUAAAGGACAGCCUGCUUAACGGUC and 5′–3′-GACCGUUAAGCAGGCUGUCCUUUAU interference RNA) using Lipofectamine 2000 Reagent (Invitrogen). EMT marker expression in ARCaPM cells with LIV-1 knockdown was assessed 3 days post-transfection. LIV-1 mRNA expression was assayed by RT-PCR and western blot as described above.
LIV-1 polyclonal antibody was produced in rabbits immunized with KLH-conjugated peptide, CPDHDSDSS GKDPRNS. A total of five injections were performed, with each injection consisting of 300–500 μg peptide mixed with complete or incomplete adjuvant. After the fourth injection, blood was collected and centrifuged. For western blotting, primary antibody was used at 1:2,000 dilution and anti-rabbit secondary antibody was used at 1:5,000 dilution. Protein bands were detected by western blot as described above.
Results and discussion
A novel human prostate cancer ARCaP EMT model for the study of prostate cancer progression and metastasis: effects of soluble growth factors
Unlike other animal models bearing human prostate cancer cells, the ARCaP model represents a continuum of prostate cancer progression closely mimicking the pathophysiology of advanced and lethal clinical human prostate cancer bone metastasis. Epithelium-like ARCaPE cells had a relatively low propensity for bone metastasis (1/8) after intracardiac (IC) injection but gained 100% metastasis to bone and 33% metastasis to adrenal gland upon recovering from mouse bone [4]. While the derivative ARCaP cells from mouse bone still expressed distinct epithelial cell markers, they also switched their morphology and gene expression profile to mesenchymal cells and thus were designated as ARCaPM. Figure 1 depicted that ARCaPM cells had decreased expression of E-cadherin (E-cad) and cytokeratins 18 and 19 but increased expression of N-cadherin (N-cad) and two novel EMT associated genes; RANKL, a novel mesenchymal marker in ARCaPM cells, and IL-13 receptor α2 (IL-13R α2), a decoy IL-13 receptor capable of blocking IL-13Rα1 signaling and enhancing tumor growth [24, 25]. In addition to the observed induction of EMT by the bone microenvironment, we also derived mesenchymal-like ARCaPM cells in vitro from parental ARCaPE cells, based on cell morphology, gene expression (see below) and behavioral characteristics. We noted that the transition of ARCaPE cells to ARCaPM cells can be promoted by a host of growth factors, such as TGFβ1 plus EGF (see below), IGF-1 [18] or β2-microglobulin (β2-m, [8]). Figure 2 shows an example in which TGFβ1 plus EGF induced the transition of ARCaPE to ARCaPM cells. We observed that this transition was transient upon growth factor induction since this induction was reversible based on the expression of EMT markers, such as downregulation of E-cad, and upregulation of vimentin and RANKL upon growth factor removal. These results, when compared with permanent induction of EMT by the inoculation of ARCaPE cells in mouse tibia [4] suggest that other factors in bone and/or prolonged exposure to bone milieu play a critical role. These results raised the possibility that ARCaPE cells could “reawaken” from their dormancy to undergo permanent transition and become more aggressive ARCaPM cells.
Fig. 1.

ARCaP EMT model in which selected growth factors or bone microenvironment drives ARCaPE cells to undergo mesenchymal-like transition to ARCaPM. Upon this morphologic transition (a), ARCaPM cells expressed both classic EMT markers (decreased expression of E-cadherin and cytokeratins 18/19 and increased expression of vimentin and N-cadherin) and novel markers (increased expression of RANKL and IL13Rα2) as analyzed by RT-PCR (b), western blot (c) and IHC (d)
Fig. 2.

TGFβ1 and EGF promote ARCaP prostate cancer EMT as evidenced by morphology and gene expression analyses. (a) ARCaPE cells have a typical cobblestone-like epithelial morphology, which could be altered to mesenchymal-like morphology by simultaneous treatment of TGFβ1 and EGF for 3–37 days. (b) The treatment inhibited the epithelial E-cad marker, but increased the mesenchymal vimentin and RANKL markers. (c) Effect of TGFβ1 and EGF treatment was transient, as termination of the treatment led to re-expression of the E-cad with decreased vimentin expression, 9 days after removal of the growth factors
RANKL, a novel EMT marker in ARCaP model, increased upon induction by soluble growth factors and functionally induced increased bone resorption
The induction of RANKL on the cell surface of prostate cancer cells is of particular interest to us since we observed a threefold increase of tartrate-resistant acid phosphatase (TRAP)-positive staining osteoclasts in mouse bone upon the introduction of ARCaPM compared to ARCaPE cells (33 ± 9 vs. 9 ± 1 TRAP-positive multinucleated osteoclasts/well in ARCaPM and ARCaPE co-cultured with hematopoetic pre-osteoclasts). Further, we found that RANKL expressed by ARCaPM or RANKL expression induced by Snail transfection into ARCaPE cells is functional in vitro, with increased activation of osteoclasts noted when incubated with mouse macrophages and this induction can be blocked by osteoprotegerin (OPG), a RANK decoy receptor (Fig. 3). Snail, a known TGF-β target gene, suppresses E-cadherin expression through the activation of a series of nuclear transcription factors. These results, in aggregate, suggest that the RANKL expression induced in ARCaPM cells is functional and promotes osteoclastogenesis, bone turnover and release of growth factors deposited in bone, conditions which favor prostate cancer cell survival and bone colonization [26, 27]. Our results from the ARCaPE transition to ARCaPM, suggest that increased bone turnover in prostate cancer patients could result from both osteoblast-derived as well as prostate cancer cell-derived RANKL. Targeting RANKL–RANK interaction seems to be a rational approach to reduce bone turnover and subsequent prostate cancer growth and colonization in bone. It should be emphasized that the RANKL–RANK interaction can be both paracrine (such as prostate cancer–osteoclast, osteoblast–osteoclast, or intercellular communication among prostate cancer cells) and autocrine in nature (soluble RANKL–RANK interaction within a cancer cell or RANKL–RANK interaction between cancer cells). This interaction could be affected by the levels of OPG, a decoy RANK receptor [28]. Numerous previous studies demonstrated that a paracrine interaction with RANKL derived from osteoblasts, in response to tumor-derived soluble factors such as PTHrP [26, 29, 30], contributes to increased bone turnover through interaction with RANK, a cell surface receptor of osteoclasts. However, our results show that functional RANKL can also be derived directly from prostate cancer cells along with their osteomimetic properties, and thus contribute to increased bone turnover and cancer cell growth, survival and migration in bone [8]. There remain some discrepancies regarding to the expression of RANKL by prostate cancer cells, since previous reports by others have shown that bone metastatic cancer cell lines lack RANKL expression [31–33], consistent with the view that in osteolytic bone metastasis RANKL is osteoblast-derived and is induced by cancer cell-derived PTHrP [30, 32]. However, we have recently conducted multiplexed quantum dot analysis of RANKL expression in clinical human prostate cancer bone metastatic specimens. In this study, we confirmed, at the single cell level, that RANKL was expressed by human prostate cancer epithelial cells (Ep-CAM+ cells), human prostate epithelial cells that had undergone mesenchymal transition (Ep-CAM+ and vimentin+ cells) and cancer cells expressed only mesenchymal marker (Ep-CAM− but vimentin+ cells), (unpublished results presented and discussed in “The 2008 Frontiers of Cancer Nanotechnology Symposium”). In our study, we found the fixation protocol used in the preservation of prostate cancer specimens is critical for determining the quality and quantity of the RANKL expression. Our results are in agreement with Mori et al. [26], who showed that RANKL–RANK autocrine signaling in human prostate cancer PC-3 and DU-145 cells could drive the migration and survival of these cancer cells and confer their ability to invade and metastasize, the well-known characteristic features of EMT. Because of the potential downstream cell signaling convergence between RANKL–RANK interaction and EMT, we suggest a dual targeting strategy where both the paracrine/autocrine RANKL–RANK interaction and EMT could be interrupted to reduce the overall bone colonization and EMT associated with prostate cancer cell growth, survival and migration. The section below describes our results of the involvement of a developmental gene, LIV-1, in EMT during human prostate cancer progression and bone metastasis.
Fig. 3.

Increased osteoclastogenesis in vitro after co-culturing mouse haematopoietic pre-osteoclasts with ARCaPM or ARCaPE-Snail transfectants. (a) Top panels: RANKL induced osteoclastogenesis in haematopoietic pre-osteoclastic precursor cells was blocked by OPG. Bottom panels: ARCaPM cells that overexpress RANKL when co-cultured with mouse haematopoietic pre-osteoclasts induced more TRAP positive staining cells, an indication of inducing osteoclast maturation, as seen under light microscopy, compared to ARCaPE co-culture. This increased osteoclastogenesis was effectively antagonized by osteoprotegerin (OPG), suggesting that the RANKL overexpressed by ARCaPM cells is functional (200×). Inset, multi-nucleated osteoclast at 400×. (b) Multinucleated TRAP positive cells from ARCaPE, ARCaPM, or ARCaPE-Snail 12 clone co-culture were counted and quantified. Overexpression of Snail induced marked TRAP positive cells indicative of functional RANKL which was blocked by OPG. Data represent triplicates obtained from 2 independent experiments
Involvement of Stat3, Snail and LIV-1 signaling in EMT and RANKL expression by ARCaP cells
To understand how EMT and RANKL may be regulated by soluble factors in cancer cells, we sought to examine the effects of β2-m, a soluble factor that has been shown to contribute to osteomimicry by prostate cancer cells [9, 21], in a number of human cancer models including human breast (MCF-7), lung (H358) and renal (SN12C) cells. Figure 4 shows that upon the selection of stably β2-m-transfected cancer cells (with either intermediate or high expression), these cell clones consistently had activated Stat3, Snail, LIV-1 and RANKL with all of the transfected cells expressing markers indicative of EMT and a morphologic transition to fibroblastic (data not shown). We further conducted studies to show a direct link between Snail and RANKL by overexpressing a Flag-tagged Snail and observed increased expression of RANKL only in Snail stably-transfected cells, but not in neo control clones (Fig. 5). These data further supported a direct link between Snail and RANKL expression.
Fig. 4.

Stable β2-m expression in human breast (MCF7, a), lung (H358, b) and renal (SN12C, c) cancer cells increased the expression of RANKL protein and the signaling mediated by Stat3 (increased both basal level and pStat3), Snail and LIV1 (a gene drives EMT in Zebrafish). β2-m-1 and β2-m-2 represent, respectively, the intermediate and high β2-m stably expressing clones. Note that the levels of activation of cell signaling correlated with levels of β2-m expression. P: parental cells. Neo: neo-transfected cells. A represents western blots of RANKL and β-actin, B represents the RT-PCR data of stat3, LIV-1 and Snail with GAPDH as a control gene, and C represents the western blots of p-stat3, stat3 with β-actin as the loading control
Fig. 5.

Combination treatment of ARCaPE.cells with TGFβ1 and EGF induced the protein expression of Snail and RANKL (left panels). Cells were treated with EGF (50 ng/ml) plus TGFβ1 (4 ng/ml) for 3 days (E + T). Note that ARCaPM cells expressed higher levels of endogenous Snail and RANKL than ARCaPE (lanes denoted with C). Right panels: over-expression of Snail induced RANKL expression in the ARCaPE cells. An expression vector containing a constitutively active Snail (Flag-Snail-6SA) was used. EV1–3: vector controls; clones 1–8: stable Snail transfected clones
To test if there is a link between LIV-1 and EMT in the ARCaP model, we overexpressed LIV-1 transiently in ARCaPE cells, which contain low levels of LIV-1, and found that ARCaPE cells underwent EMT by the expression of mesenchymal markers, increased Snail, vimentin, and RANKL but decreased E-cad expression (Fig. 6). This observation linking LIV-1 and EMT is further supported by a follow up experiment whereby the knockdown of LIV-1, which is expressed abundantly in ARCaPM cells, with LIV-1 siRNA resulted in the expression of epithelial markers (increased expression of E-cad, and decreased expression of N-cad, vimentin and RANKL, Fig. 6). These studies led to the conclusion that LIV-1, a zinc transporter protein known to promote EMT in Zebrafish gastrula organizer [34] and associated with human breast cancer [13], plays a central role in ARCaPE transition to ARCaPM and is possibly correlated with the progression of human prostate cancer. We have confirmed this concept in localized and bone metastatic human prostate cancer tissue arrays where LIV-1 protein expression level had statistically significant association with prostate cancer progression from normal, benign, PIN, primary cancer to bone metastasis [35].
Fig. 6.
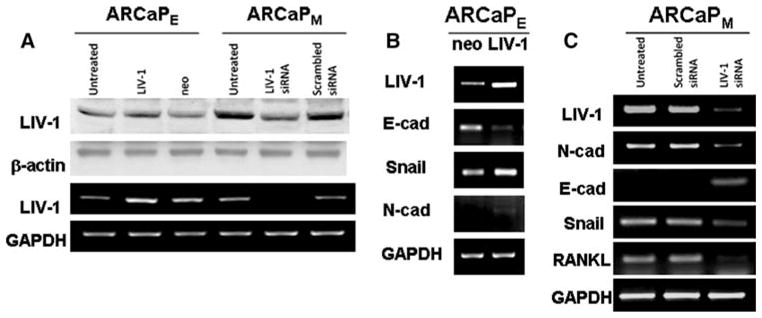
LIV-1 plays an important role in ARCaP EMT. (a) LIV-1 antibody recognized a single band at 85 kDa and was expressed at higher levels in ARCaPM than in ARCaPE cells and is sensitive to siRNA targeting LIV-1 at both RNA and protein levels. Since this was a transient knockdown study, we observed only a slight change of LIV-1 protein level possibly due to the longer t1/2 of this protein in comparison to LIV-1 mRNA. (b) LIV-1 transfected ARCaPE cell clones expressed increased Snail and decreased E-cad, an indication of mesenchymal-like transition. (c) LIV-1 siRNA treated ARCaPM cells expressed decreased levels of LIV-1, Snail, N-cad, vimentin and RANKL, but increased E-cad, indicative of epithelial differentiation in comparison to untreated or scrambled LIV-1 siRNA-treated ARCaPM cells
EMT, MET, MErT and MMT: dynamics of the ARCaP model
EMT is a dynamic process which may occur at primary as well as metastatic sites where reversion of EMT toward an epithelial phenotype via MET takes place [16, 36, 37]. The plasticity of ARCaP cells was supported by the experimental observation that parental epithelial-like ARCaPE cells, exposed to bone or cultured in vitro in the presence of TGFβ1 plus EGF, underwent mesenchymal transition. Because the importance of the bone microenvironment, which is rich in growth factors that can drive ARCaP EMT, we suggest that although the homing of prostate cancer cells to bone is important, it is even more critical that soluble factors released during bone turnover play a key role in the promotion of EMT and subsequent prostate cancer cell growth, survival and bone colonization. This idea is also supported by clinical observations, where patients with serum prostate-specific antigen (PSA) < 0.1 ng/ml were found to harbor prostate cancer cells in bone marrow. Since only a fraction of these patients will develop bone metastasis in their lifetime, this supports the idea that while the presence of prostate cancer cells in bone is common, they are mostly “dormant” and only if they are responsive to soluble factors eliciting the expression of bone-like properties, such as the expression of OC, BSP and RANKL, they gain the ability to undergo EMT, with increased expression of RANKL which promotes bone turnover, cancer cell survival and subsequent colonization in bone.
Mesenchymal to epithelial reversing transition or MErT occurred when a human prostate cancer cell line, DU-145, was co-cultured with human hepatocytes under three-dimensional (3-D) conditions [16]. In analogous conditions, ARCaPM cells cultured by 3-D Rotary Wall Vessel (RWV) underwent MET (data not included). Such rapid transition of ARCaPM to ARCaPE under 3-D conditions showed how ARCaPM cells could “settle” comfortably in the skeleton as a part of the metastastic cascade, since re-expressing E-cad provides ARCaPM cells with strong intercellular adhesion and facilitates their colonization in bone. This, however, did not seem to be the case with ARCaPM cells isolated from the bone environment, which retained mesenchymal features despite establishing stable bone metastatic potential and exhibiting osteolytic response when residing in bone. The derivation of a mesenchymal subline from he bone marrow environment is consistent with the findings of vimentin positivity in 15 of 15 breast cancer cell lines developed by Pantel and co-workers from micrometastases in this environment [38].
MMT, mesenchymal to mesenchymal transition, occurs in situ and during embryonic development, such as with the induction of “reactive” stroma [39]. The plasticity of cancer cells and their surrounding fibromuscular stromal cells involves a highly dynamic reverting program (or trans-differentiation) of cells toward certain phenotypes through selective activation of cell signaling pathways [14, 40, 41]. The underlying biologic basis of these transitions is found in embryonic development and apparently persists in adult cells and contributes to neoplastic progression. Further analysis of the ARCaP model system may reveal the molecular mechanisms underlying such dynamic transitions.
Conclusions
We demonstrated that the host bone microenvironment and soluble growth factors are crucial in facilitating EMT and RANKL expression, which subsequently promote prostate cancer cell migration, invasion and metastasis to the skeleton and soft tissues. Our data demonstrated a close link between the ARCaP EMT model and human prostate cancer bone metastasis. This model, therefore, is well suited to investigate the biology and targeting of the lethal progression of human prostate cancer to bone.
We confirmed the plasticity of prostate cancer cells in the ARCaP EMT model and their ability to express the embryonic EMT-associated LIV-1 gene. Our results suggest that through proper cell signaling switches mimicking embryonic development, invasive ARCaP cells armed with mesenchymal gene expression profiles gained invasive and migratory phenotypes and can then re-acquire epithelial and embryonic phenotypes and grow and flourish in the metastatic niche. We suggest that tumor cell-host microenvironment interaction is critical in determining the temporal and spatial transition of epithelial and mesenchymal cells. The ARCaP model provides an excellent tool to dissect the molecular links in heterogeneous cancer cells between EMT, RANKL and bone turnover, fundamental to understanding why many solid tumors, including prostate cancer, metastasize to bone.
Acknowledgments
We thank Gary Mawyer for editing. The helpful discussion and support from Guodong Zhu, Weiping Qian, Daqing Wu and Clayton Yates are greatly appreciated. The authors are particularly indebted to the helpful suggestions made by the editors, Drs. Rik Thompson and Elizabeth Williams, during the revision of this manuscript. This study was supported in part by CA082739 (HYEZ), U54 CA119338, CA098912 and CA766201 (LWKC). We are grateful to the generous gift from Frances and Clarence Wilkins in support of this study.
Abbreviations
- AR
Androgen receptor
- ARCaP
Androgen-refractory human prostate cancer cell model from a patient with prostate cancer bone metastasis
- ARCaPE
ARCaP clone with epithelial phenotype
- ARCaPM
ARCaP clone with mesenchymal phenotype
- β2-m
β2-Microglobulin
- BSP
Bone sialoprotein
- C4-2
Lineage derivative cells from LNCaP
- C4-2B
C4-2 cells metastasized to bone
- CK18/19
Cytokeratin 18/19
- CM
Conditioned medium
- CREB
cAMP-responsive element-binding protein
- E-cad
E-cadherin
- EGF
Epidermal growth factor
- EMT
Epithelial–mesenchymal transition
- FBS
Fetal bovine serum
- IC
Intracardiac
- IGF-1
Insulin-like growth factor 1
- IHC
Immunohistochemistry
- IL13Rα2
Interleukin13 receptor α2
- LNCaP
Prostate cancer cells metastasized to lymph node
- MET
Mesenchymal–epithelial transition
- MErT
Mesenchymal–epithelial-reverting transition
- MMT
Mesenchymal–mesenchymal transition
- N-cad
N-cadherin
- NFκB
Nuclear factor kappa B
- OC
Osteocalcin
- OPG
Osteoprotegerin
- OPN
Osteopontin
- RANKL
Receptor activator of NFκB ligand
- RT-PCR
Reverse transcriptase-polymerase chain reaction
- SCID
Severe combined immunodeficiency
- siRNA
Small interfering RNA
- Stat
Signal transducer and activator of transcription
- TGFβ1
Transforming growth factor β1
- TRAP
Tartrate-resistant acid phosphatase
- VM
Vimentin
Contributor Information
Haiyen E. Zhau, Email: hzhau@emory.edu, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA
Valerie Odero-Marah, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA.
Hui-Wen Lue, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA. Department of Biology, Georgia State University, Atlanta, GA 30302, USA.
Takeo Nomura, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA.
Ruoxiang Wang, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA.
Gina Chu, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA. Department of Biology, Georgia State University, Atlanta, GA 30302, USA.
Zhi-Ren Liu, Department of Biology, Georgia State University, Atlanta, GA 30302, USA.
Binhua P. Zhou, Sealy Center for Cancer Cell Biology, The University of Texas Medical Branch, Galveston, TX 77555, USA
Wen-Chin Huang, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA.
Leland W. K. Chung, Email: lwchung@emory.edu, Molecular Urology & Therapeutics Program, Department of Urology, Emory University School of Medicine, 1365B Clifton Road, Suite 5107, Atlanta, GA 30322, USA
References
- 1.Robinson VL, Kauffman EC, Sokoloff MH, et al. The basic biology of metastasis. Cancer Treat Res. 2004;118:1–21. doi: 10.1007/978-1-4419-9129-4_1. [DOI] [PubMed] [Google Scholar]
- 2.Tu SM, Lin SH. Clinical aspects of bone metastases in prostate cancer. Cancer Treat Res. 2004;118:23–46. doi: 10.1007/978-1-4419-9129-4_2. [DOI] [PubMed] [Google Scholar]
- 3.Wu TT, Sikes RA, Cui Q, et al. Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int J Cancer. 1998;77(6):887–894. doi: 10.1002/(SICI) 1097-0215(19980911). 77:6<887::AID-IJC15>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Xu J, Wang R, Xie ZH, et al. Prostate cancer metastasis: role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate. 2006;66(15):1664–1673. doi: 10.1002/pros.20488. [DOI] [PubMed] [Google Scholar]
- 5.Zhau HY, Chang SM, Chen BQ, et al. Androgen-repressed phenotype in human prostate cancer. Proc Natl Acad Sci USA. 1996;93(26):15152–15157. doi: 10.1073/pnas.93.26.15152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odero-Marah V, Shi C, Zhau HE, et al. Dual role of Snail transcription factor in epithelial to mesenchymal transition and neuroendocrine differentiation in human prostate cancer cells. AACR Proc. 2007;48:247. [Google Scholar]
- 7.Chung LW, Baseman A, Assikis V, et al. Molecular insights into prostate cancer progression: the missing link of tumor microenvironment. J Urol. 2005;173(1):10–20. doi: 10.1097/01.ju.0000141582.15218.10. [DOI] [PubMed] [Google Scholar]
- 8.Chung LW, Huang WC, Sung SY, et al. Stromal–epithelial interaction in prostate cancer progression. Clin Genitourin Cancer. 2006;5(2):162–170. doi: 10.3816/CGC.2006.n.034. [DOI] [PubMed] [Google Scholar]
- 9.Huang WC, Xie Z, Konaka H, et al. Human osteocalcin and bone sialoprotein mediating osteomimicry of prostate cancer cells: role of cAMP-dependent protein kinase A signaling pathway. Cancer Res. 2005;65(6):2303–2313. doi: 10.1158/0008-5472 CAN-04-3448. [DOI] [PubMed] [Google Scholar]
- 10.Gotzmann J, Mikula M, Eger A, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566(1):9–20. doi: 10.1016/S1383-5742(03)00033-4. [DOI] [PubMed] [Google Scholar]
- 11.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17(5):548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Brabletz T, Hlubek F, Spaderna S, et al. Invasion and metastasis in colorectal cancer: epithelial–mesenchymal transition, mesenchymal–epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005;179(1–2):56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- 13.Taylor KM, Hiscox S, Nicholson RI. Zinc transporter LIV-1: a link between cellular development and cancer progression. Trends Endocrinol Metab. 2004;15(10):461–463. doi: 10.1016/j.tem. 2004.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Larue L, Bellacosa A. Epithelial–mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene. 2005;24(50):7443–7454. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- 15.Thiery JP. Epithelial–mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15(6):740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Yates C, Shepard CR, Papworth G, et al. Novel three-dimensional organotypic liver bioreactor to directly visualize early events in metastatic progression. Adv Cancer Res. 2007;97:225–246. doi: 10.1016/S0065-230X(06)97010-9. [DOI] [PubMed] [Google Scholar]
- 17.Albalate M, de la Piedra C, Fernandez C, et al. Association between phosphate removal and markers of bone turnover in haemodialysis patients. Nephrology Dialysis Transplantation. 2006;21(6):1626–1632. doi: 10.1093/ndt/gfl034. [DOI] [PubMed] [Google Scholar]
- 18.Graham TRZHE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW, O’Regan RM. IGF-1-dependent upregulation of ZEB1 expression drives EMT in human prostate cancer cells in vitro. Cancer Res. 2008;68(7):2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 19.Gleave M, Hsieh JT, Gao CA, et al. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Res. 1991;51(14):3753–3761. [PubMed] [Google Scholar]
- 20.Thalmann GN, Sikes RA, Wu TT, et al. LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. Prostate. 2000;44(2):91–103. doi: 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 21.Huang WC, Wu D, Xie Z, et al. beta2-microglobulin is a signaling and growth-promoting factor for human prostate cancer bone metastasis. Cancer Res. 2006;66(18):9108–9116. doi: 10.1158/0008-5472.CAN-06-1996. [DOI] [PubMed] [Google Scholar]
- 22.Zhou BP, Deng J, Xia W, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat Cell Biol. 2004;6(10):931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi N, Akatsu T, Udagawa N, et al. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988;123(5):2600–2602. doi: 10.1210/endo-123-5-2600. [DOI] [PubMed] [Google Scholar]
- 24.Fichtner-Feigl S, Strober W, Kawakami K, et al. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12(1):99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 25.Kawakami K. Cancer gene therapy utilizing interleukin-13 receptor alpha2 chain. Curr Gene Ther. 2005;5(2):213–223. doi: 10.2174/1566523053544227. [DOI] [PubMed] [Google Scholar]
- 26.Mori K, Le Goff B, Charrier C, et al. DU145 human prostate cancer cells express functional receptor activator of NFkappaB: new insights in the prostate cancer bone metastasis process. Bone. 2007;40(4):981–990. doi: 10.1016/j.bone.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Vessella RL, Corey E. Targeting factors involved in bone remodeling as treatment strategies in prostate cancer bone metastasis. Clin Cancer Res. 2006;12(20 Pt 2):6285s–6290s. doi: 10.1158/1078-0432.CCR-06-0813. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong AP, Miller RE, Jones JC, et al. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate. 2008;68(1):92–104. doi: 10.1002/pros.20678. [DOI] [PubMed] [Google Scholar]
- 29.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2(8):584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 30.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350(16):1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 31.Chamulitrat W, Schmidt R, Chunglok W, et al. Epithelium and fibroblast-like phenotypes derived from HPV16 E6/E7-immortalized human gingival keratinocytes following chronic ethanol treatment. Eur J Cell Biol. 2003;82(6):313–322. doi: 10.1078/0171-9335-00317. [DOI] [PubMed] [Google Scholar]
- 32.Thomas RJ, Guise TA, Yin JJ, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999;140(10):4451–4458. doi: 10.1210/en.140.10.4451. [DOI] [PubMed] [Google Scholar]
- 33.Schwaninger R, Rentsch CA, Wetterwald A, et al. Lack of noggin expression by cancer cells is a determinant of the osteoblast response in bone metastases. Am J Pathol. 2007;170(1):160–175. doi: 10.2353/ajpath.2007.051276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamashita S, Miyagi C, Fukada T, et al. Zinc transporter LIVI controls epithelial–mesenchymal transition in zebrafish gastrula organizer. Nature. 2004;429(6989):298–302. doi: 10.1038/nature02545. [DOI] [PubMed] [Google Scholar]
- 35.Arnold RCL, Farah-Carson MC, et al. Prostate cancer bone metastasis: reactive oxygen species, growth factors and heparan sulfate proteoglycans provide a signaling triad that supports progression. J Urol. 2008;179(Suppl 4):192. [Google Scholar]
- 36.Chaffer CL, Brennan JP, Slavin JL, et al. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66(23):11271–11278. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- 37.Vincan E, Brabletz T, Faux MC, et al. A human three-dimensional cell line model allows the study of dynamic and reversible epithelial–mesenchymal and mesenchymal–epithelial transition that underpins colorectal carcinogenesis. Cells Tissues Organs. 2007;185(1–3):20–28. doi: 10.1159/000101299. [DOI] [PubMed] [Google Scholar]
- 38.Willipinski-Stapelfeldt B, Riethdorf S, Assmann V, et al. Changes in cytoskeletal protein composition indicative of an epithelial–mesenchymal transition in human micrometastatic and primary breast carcinoma cells. Clin Cancer Res. 2005;11(22):8006–8014. doi: 10.1158/1078-0432.CCR-05-0632. [DOI] [PubMed] [Google Scholar]
- 39.Cat B, Stuhlmann D, Steinbrenner H, et al. Enhancement of tumor invasion depends on transdifferentiation of skin fibroblasts mediated by reactive oxygen species. J Cell Sci. 2006;119(Pt 13):2727–2738. doi: 10.1242/jcs.03011. [DOI] [PubMed] [Google Scholar]
- 40.Lee JM, Dedhar S, Kalluri R, et al. The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172(7):973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]