

Abstract
Neuropilin-1 (NRP-1) is a receptor that plays an essential role in angiogenesis, vascular permeability and nervous system development. Previous studies have shown that peptides with an N-terminal Arg, especially peptides with the four residue consensus sequence R/K/XXR/K bind to NRP-1 cell surfaces. Peptides containing such consensus sequences promote binding and internalization into cells, while blocking the C-terminal Arg (or Lys) prevents the internalization. In this study we use molecular dynamics simulations to model the structural properties of the NRP-1 complex with a prototypic CendR peptide, RPAR. Our simulations show that RPAR binds NRP-1 through specific interactions of the RPAR C-terminus: three hydrogen bonds and a salt bridge anchor the ligand in the receptor pocket. The modeling results were used as the starting point for a systematic computational study of new RPAR analogs based on chemical modifications of its natural amino acids. Comparison of the structural properties of the new peptide - receptor complexes with the original organization suggest that some of the analogs can increase the binding affinity while reducing the natural sensitivity of RXXR to endogenous proteases.
Blood vessels are generally impermeable to macromolecules and supramolecular complexes. Increased permeability of blood vessels is associated with physiological angiogenesis during embryonic development (1). Vascular leakage is also a characteristic of tumor vessels (2). VEGF, originally described as a tumor-secreted vascular permeability factor (3), is at least partially responsible for the leakiness of angiogenic vessels (2,4). VEGF was later independently cloned as an endothelial cell mitogen (5,6). Neuropilin-1 (NRP-1) is a hub receptor with multiple ligands that plays a central role in angiogenesis and cardiovascular development (7,8), and acts as VEGF co-receptor during induction of vascular permeability. We have identified a class of peptides derived from phage display screens that induce cell internalization and tissue penetration via NRP-1 mediation (9). The peptides that promoted such process upon binding to NRP-1 were found to have the R/K/XXR/K motif at their C terminus (known as the C-end rule), a pattern also shared by VEGF-A165 and by certain semaphorins (9). This 4 residue motif contains an Arg or a Lys residue in the first and 4th position and any residue in the 2nd and 3rd positions. The NRP-1 binding is particularly efficient when the RXXR C-end rule motif is present in tandem organization, such as RPARPAR, RGERPPR and RVTPPR (9). Finally, cryptic R/K/XXR/K motifs embedded in a protein can be proteolytically activated by exogenous proteases to trigger vascular leakage and cell internalization (9). CendR peptides are particularly relevant to cancer drug delivery, as NRP-1 is frequently over-expressed in tumor cells. Hence, therapeutic strategies that that include peptides with R/K/XXR/K terminus may help in overcoming problems related to penetration of drugs into the tumor, since binding of such peptides to NRP-1 promotes extravasation and penetration of co-administered drugs to the tumor, as shown recently by Sugahara et al. (10).
Despite the central role of VEGF permeability factor, the structure of NRP-1 complexed with an R/KXXR/K has not yet been elucidated from a crystal structure. However, the structure of the binding domains with a peptide mimic of VEGF terminus was recently reported (11). Such peptide, called tuftsin (an immuno-stimulatory tetrapeptide, TKPR) has a sequence very close to the C-terminus of VEGF-A165, DKPRR, and competed with the VEGF for binding to NRP-1 (12). Both features demonstrated that the NRP-1 – tuftsin complex could represent an accurate structural template for the binding of any active peptide. Based on those findings, in this work we have employed computer simulations to model the RPAR interaction with NRP-1 binding domain, using NRP-1 – tuftsin crystal structure (11) as a template. Figure 1a shows the overall NRP-1 – tuftsin complex while Figure 1b focuses on the binding region. Tuftsin binds to NRP-1 through a network of interactions that mainly clusters in the receptor segments called interaction loops. Thus, the interacting side chains that belong to the receptor are located in the three interactions loops identified in Figure 1b. The strongest and most relevant of such interactions is a salt bridge between tuftsin’s C-terminal Arg and Asp-320 of NRP-1. Other hydrogen bonding interactions involve the Lys backbone of tuftsin and the side chain of Tyr-297 in NRP-1, and the carboxylate oxygen atoms of tuftsin’s C-terminal Arg, which interact simultaneously with the side chains of Ser-346, Thr-349 and Tyr-353 of NRP-1. As we will show in the next sections, most of these interactions are also featured by the RPAR segment and NRP-1.
Figure 1.

(a) Schematic illustration of the NRP-1 – tuftsin complex (PDB: 2ORZ) used to model the NRP-1 – RPAR complex. NRP-1 is shown in purple and tuftsin in green. (b) Focused view of the binding site. NRP-1 interaction loops and the specific residues participating in the binding are labeled for clarity. Specific interactions between loop residues and NRP-1 are drawn: Hydrogen bonds are shown as blue discontinuous lines while salt bridges are marked by yellow discontinuous lines.
To exploit the RPAR potential in targeted drug delivery, it may be advantageous to increase its half-life time by enhancing its resistance to proteases, a goal that can be achieved by introducing single residue chemical modifications. Such modifications should not alter the ability of RPAR to recognize and bind NRP-1. Thus, the interactions pattern featured by the wild type segment bound to the receptor is used as a template that any candidate modification must preserve. In a recent study, we successfully used this strategy on CREKA (13), a linear pentapeptide that selectively binds to tumor vasculature (14), to increase the resistance of the peptide against proteases. In the CREKA study, we found that replacement of a hydrogen atom by a methyl group at selected amide bonds result in peptide analogues that accumulate in tumors more effectively than the wild type peptide (13). We then used the modified CREKA peptides to design nanoparticles that specifically blocked tumor circulation (13). In the present work, we have studied possible chemical modifications that could be introduced to RPAR. In order to discard those that might have higher chances to fail, we have evaluated their potential in silico by assessing their possible interactions, using a simplified model of the NRP-1 binding domain. The modifications introduced into the RPAR peptide have mainly focused on changes in the chemical scaffold of the terminal Arg, since this amino acid plays a key role in binding the receptor. Specifically, backbone and side chain modifications have been examined in detail. We changed as well the directionality of the last peptide bond into a retro-peptide bond (15), and in parallel, we inverted the chiralities of all amino acids in the peptide.
Our results constitute a first step in a comprehensive study of this important peptide – receptor binding, which induces cell internalization and tissue penetration. Computer modeling studies may assist in understanding tissue penetration mechanisms and aid in increasing the efficiency of drug delivery into tumors using R/K/XXR/K motif.
Methods
Molecular Dynamics simulations to model NRP-1 – RPAR interaction
Calculations were performed using the NAMD package (16). All the atoms of the system were considered explicitly, and the energy was calculated using the AMBER ff03 force field (17,18). Water molecules were represented using the TIP3 model (19). Simulations were performed using the NVT ensemble in an orthorhombic simulation box. Periodic boundary conditions were applied using the nearest image convention. The box size was adjusted to fit the complex size, the initial box dimensions being 114.0×93.0×107.0 Å3 to ensure infinite dilution.
The system contained 111486 atoms, of which 5040 were protein atoms and the rest were ions and solvent atoms. The starting molecular structure was modeled based on the tuftsin bound complex (PDB code 2ORZ). The charge of all potential titratable groups was fixed at values corresponding to neutral pH (i.e. all Asp and Lys side chains were represented in their negatively and positively charged forms, respectively). Simulations were performed considering an ionic strength of approximately 0.8% w/w (66 Cl· and 57 Na+ ions) at 298 K. The overall charge of the system was kept neutral for the use of particle mesh Ewald summation to calculate the electrostatic interactions (20) (i.e. counterions were Cl· and Na+).
The protocol for MD simulations was as follows. Before running each simulation, the potential energy of each system was minimized using 5000 conjugate gradient steps. To ensure uniform distribution of the ions in solution, the protein atoms were held fixed while the solvent was heated to a temperature of 500 K. Next, we let the system equilibrate at 298 K with NPT conditions to allow the water box size to shrink to its final dimensions avoiding low water density. After the water box size was stabilized at approximately 112×89×104 Å3 we heated and equilibrated the system. The heating protocol included 15 ps of increasing the temperature of the system from 0 K to the final temperature of 298 K plus 500 ps of equilibration period.
A residue-based cutoff was applied at 14.0 Å, i.e. the interaction was evaluated when two residues had any atoms separated by less than such distance. A numerical integration time step of 1 fs was used for all the simulations. The nonbonded pair list was updated every 5 steps, and the trajectories were saved every 1000 steps (1 ps) for subsequent analysis. During the production runs the system was held in a constant thermal bath of 298 K using thermal coupling. The simulation was run for a period of 40 ns. To ensure that the system reached the equilibrium, the first 4 ns were excluded from the analyses, since during that time the potential energy continued to increase slightly (see next section). Analyses were done using the NAMD program (16), Visual Molecular Dynamics (VMD) software (21) and MatLab.
Targeted modifications of RPAR
A simulation system based upon a simplified receptor
The results provided by the MD simulations of the wild type complex allowed a topological description of the most relevant interactions of the NRP-1 – RPAR binding. This interaction map was used to build a simplified receptor model featuring the most important chemical groups. This model is represented by those receptor residues that enclose the whole binding cavity, including the three interaction loops which are listed in Table 1 (these loops are displayed in Figure 1 for the NRP-1 – tuftsin complex). The use of a reduced model for the NRP-1 receptor allows us to screen for many candidates of RPAR mutants with a significant reduction in computational requirements. At the same time, this strategy allows to overcome to potential drawbacks that other approaches would present. If we were to freeze the less active parts of the receptor, computational resources would not be saved while the receptor would lose conformational flexibility, which is always an important factor for the formation and stabilization of any given ligand – receptor model. The model itself was then constructed by excising small segments of the original protein that included the residues that were in contact directly or indirectly with the RPAR peptide inside the receptor. For each excised segment, their edge residues (i.e. the actual length of each segment) were selected to completely cover the binding pocket space and to assure charge neutrality of the system. Moreover, each peptide segment that defined part of the receptor model was anchored at both edges (N- and C-termini) via a harmonic potential to an inert surface, constituted by van der Waals spheres, similar to the strategy followed in (22). The inert surface was constituted by 100 spheres regularly distributed in a square of 47.5×47.5 Å2. Finally the receptor model bound to each RPAR modified peptide (see below) was solvated using a water cap of radius 23.75 Å with a total number of 851 TIP3 water molecules (19).
Table 1.
NRP-1 sequences used to build a simplified model of the receptor binding pocked. For each peptide segment the original NRP-1 sequence position and the residue single letter code are specified. Residues that are remarked in italic define the three interaction loops cited in text: residues 296–298, 319–321 and 345 –354 constitute loops I, II and III, respectively (11).
| 291 | 292 | 293 | 294 | 295 | 296 | 297 | 298 | 299 | 300 | 301 | |||
| I | T | A | S | S | Q | Y | G | T | D | W | |||
| 312 | 313 | 314 | 315 | 316 | 317 | 318 | 319 | 320 | 321 | 322 | 323 | ||
| E | N | G | W | T | P | G | E | D | S | Y | R | ||
| 343 | 344 | 345 | 346 | 347 | 348 | 349 | 350 | 351 | 352 | 353 | 354 | 355 | 356 |
| G | A | I | S | K | E | T | K | K | K | Y | Y | V | K |
| 411 | 412 | 413 | 414 | 415 | 416 | 417 | |||||||
| W | D | T | G | I | S | M |
RPAR analogs Studied
13 different RPAR analogs have been built by replacing the C-terminal Arg with a synthetic surrogate. The details of each molecular analog and the rational of its design are explained in the Results section (Figure 4).
Figure 4.

Schematic representation of the most relevant chemical modifications introduced on the Arg amino acid. (a) Modifications on the side chain: I coded Arg, II α-dehydro Arg, III β-dehydro Arg, IV γ-dehydro Arg, V Arg in which the side chain has been reduced one methylenic unit (also denoted nor-arginine), and VI Arg in which the side chain has been enlarged one methylenic unit (also denoted homo-Arginine). (b) VII Retro-modification of the main chain to obtain a retroamide bond (retro-Arg).
A chemical modification was considered to be potentially useful for further biomedical applications if it follows three conditions (listed from more to less relevant):
The docking in the binding site must maintain the interactions mapped for the wild type during the simulation.
The binding of the peptide should not cause a conformational rearrangement of the binding pocket (i.e. the RMSD with respect with the initial receptor topology should oscillate around the values observed for the wild type).
The interaction energy between the residues of the receptor interacting with the RPAR analog should be close to the values obtained for the wild type RPAR.
Simulation protocol and energy parameters
For each studied RPAR analog, short MD simulations were performed using the AMBER9 package (23). Before running each simulation, the potential energy of each system was minimized using 5000 conjugate gradient steps. The heating protocol included 15 ps of increasing the temperature of the system from 0 K to the final temperature of 298 K plus 500 ps of thermal equilibration. A residue-based cutoff was applied at 14.0 Å for the nonbonding interactions. A numerical integration time step of 2 fs was used for all the simulations. The nonbonded pair list was updated every 5 steps, and the trajectories were saved every 1000 steps (2 ps) for subsequent analysis. During the production runs the system was held in a constant thermal bath of 298 K using thermal coupling. Each studied system, which includes a RPAR analog, the receptor model and the water cap, was simulated for a period of 5 ns.
All parameters were extrapolated from the AMBER ff03 force field (17) for each proposed synthetic amino acid with the exception of the electrostatic parameters that were explicitly computed. Atomic centered charges were calculated by fitting the (U)HF/6-31G(d) quantum mechanical and the Coulombic Molecular Electrostatic Potentials (MEPs) to a large set of points placed outside the nuclear region. The electrostatic parameters derived at this level of theory are fully compatible with the current parameters of the AMBER force-field (17). For each new amino acid analog, the electrostatic conformation, which usually is the parameters were fitted using as main chain conformation the C7,eq conformation of lowest energy in small model dipeptides.
Results and Discussion
Analysis of the simulated NRP-1 – RPAR complex
Figure 2a shows the temporal evolution of the overall potential energy of the complex through the MD simulation. The energy kept increasing at the beginning of the simulation and stabilized after 4 ns. Such energy increase can be attributed to the initial position of the RPAR peptide in the receptor pocket, which was initially modeled according to the position of tuftsin (11). Accordingly, the first 4ns were excluded from the numerical analyses below to ensure that the complex is properly equilibrated. Figure 2b displays the evolution of the ligand – protein complex RMSD, which was calculated with respect to the minimized structure. As can be seen, the structure of the complex is very stable and the RMSD stays within 1.2–1.25 Å from the initial conformation throughout the simulation. This stability is due to the combination of a good geometric match between NRP-1 and RPAR and a strong network of interactions that holds the complex tightly bound.
Figure 2.

NRP-1 – RPAR trajectory analysis: (a) Temporal evolution of the potential energy for the NRP – RPAR complex during the MD simulation; (b) Temporal evolution of the RMSD of the NRP-1 – RPAR complex during the MD simulation. The RMSD was calculated with respect to the minimized structure and was measured considering the backbone atoms only, to reduce noise; (c) Temporal evolution of the distance between Asp-74 of NRP-1 and the C-terminal Arg of RPAR. The distance is between the charged centers of these two amino acids; (d) The distance between the donor and acceptor centers of the three conserved hydrogen bonds between NRP-1 and RPAR. See inset legend for details. CArg refers to the C-terminal Arg residue of RPAR.
To further validate our results we tested specific interactions that hold the RPAR molecule in its binding pockets. The default criteria of the VMD software (i.e. 3Å distance and 160 degrees) were used to define a hydrogen bond. The interactions we found include:
The C-terminal Arg of RPAR is held by a number of interactions, the strongest of which is a salt bridge with Asp-320 of NRP-1. Figure 2c shows the temporal evolution of the distance between the two charged centers of these two amino acids throughout the whole simulation. As can be seen, the distance fluctuates between 3.4–3.5Å, indicating a strong and stable salt bridge.
A hydrogen bond between the backbone oxygen of the RPAR Pro and the side chain of Tyr-297 of NRP-1. The hydrogen bond is well preserved as is reflected in Figure 2d, which shows that the hydrogen bonding distance remains below 2 Å during the whole trajectory.
A bifurcated hydrogen bond involving the oxygen of the C-terminal Arg of RPAR as hydrogen bonding acceptor and the side chains of Ser-346 and Thr-349 of NRP-1 as hydrogen bonding donors. The two interactions exist in around 98% of the trajectory. The distances between the donor and acceptor centers of this interaction are illustrated in Figure 2d. As seen, the donor-acceptor distances are at and below 2 Å throughout the entire simulation, indicating well preserved hydrogen bonds.
Figure 3 presents the interaction network, showing that the stronger interactions are near the binding pocket. This allows building a molecular template for designing RPAR surrogates based on chemical modifications at specific positions. Table 2 lists the average pair-wise interaction energies between the amino acids participating in the interaction discussed above. Averages were calculated over the entire trajectory and the energy, which was calculated over all the atoms in the participating amino acids and not just in the interacting centers, includes the non-bonded contributions (i.e. van der Waals and electrostatics) with self-energy excluded. The four interactions contribute a highly negative energy indicating strong binding. As expected, the energy of interaction between the C-terminal Arg of RPAR and Asp-320 is the strongest due to the electrostatic nature of this salt bridge, which is formed between the two charged centers. On the other hand, the energy associated with the interaction between the Pro of RPAR and Tyr-297 corresponds to that typically found in conventional hydrogen bonds. This interaction provides further anchoring of RPAR to the binding site outside the binding pocket. Finally, the energy found for the bifurcated interaction between the C-terminal Arg of RPAR and the side chains of Ser-346 and Thr-349 is intermediate between a salt bridge and a conventional hydrogen bond. This is because of the negative charge of the carboxylate of Arg, which enhances the electrostatic nature of the interaction with respect to that of conventional hydrogen bonds.
Figure 3.
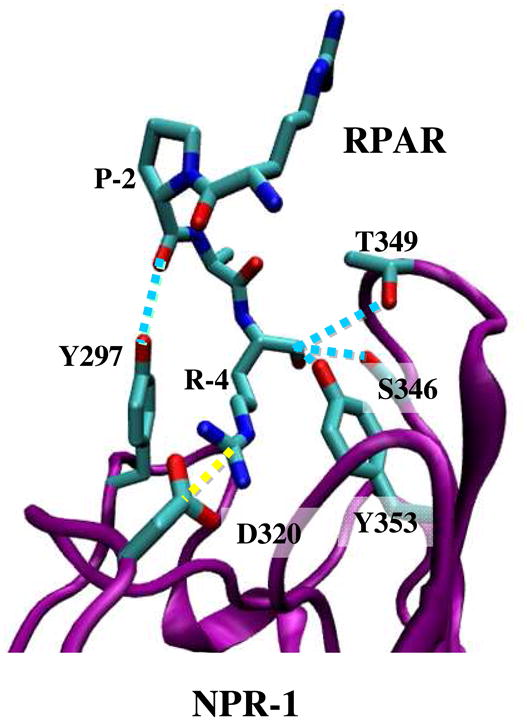
Ribbon representation of the NRP-1 – RPAR complex showing the most notable interactions found between the peptide and the binding pocket of NRP-1. The ligand and the interacting side chains of the receptor are depicted as solid lines. NRP-1 backbone is shown in purple and RPAR backbone in green Hydrogen atoms are omitted for clarity. Specific interactions are drawn: hydrogen bonds are shown as blue discontinuous lines while salt bridges are marked by yellow discontinuous lines.
Table 2.
Averaged interaction energies for the specific interactions observed in the NRP1 – RPAR complex. The energies, which were averaged over the whole trajectories, were calculated considering the non-bonding contributions: van de Waals and electrostatics. The first amino acid refers to NRP-1 and the second refers to RPAR. C-Arg corresponds to the C-terminal Arg in RPAR. Interaction energies were evaluated considering all the atoms in the participating amino acids and not just the interacting centers.
| Interaction in NRP1 – RPAR | Average energy (kcal·mol−1) |
|---|---|
| Ser 346 – C-Arg | −25.2±0.6 |
| Thr349 – C-Arg | −23.4±0.7 |
| Asp320 – C-Arg | −78.7±0.9 |
| Tyr297 – Pro2 | −6.0±0.2 |
Almost all the interactions discussed above were also observed in the crystal structure of the NRP1 – tuftsin complex (PDB code 2ORZ) (11), which was used to model NRP-1 – RPAR in this study. Thus, these findings suggest that the interactions identified in both NRP-1 – tuftsin and NRP-1 – RPAR are crucial for the binding of NRP-1 with its ligands. These interactions anchor the C-terminal Arg in the binding pocket, and emphasize the importance of the C-terminal Arg in binding to NRP-1 as denoted by the C-end rule (9). The additional highly preserved hydrogen bond involving Thr-349 of NRP-1 and the Pro backbone in RPAR (Lys backbone in tuftsin, also in position 2 of the peptide) helps in maintaining the geometry of the ligand and further stabilize the complex binding. The structural differences between Lys and Pro backbones hinder the adoption of an ideal geometry for such hydrogen bond when RPAR is docked instead of tuftsin. In spite of this, the distance between the two interacting centers remains very low, as displayed in Figure 2d.
On the other hand, an additional hydrogen bond was detected in NRP-1 – tuftsin complex (11) but not in NRP-1 – RPAR. This corresponds to the hydrogen bond between the backbone C-terminal Arg of the peptide and the side chain of Tyr-353. This interaction did not exist in our simulation, the average distance between the donor and acceptor atoms being approximately 3.8 Å. The absence of this additional hydrogen bond has been attributed to the different geometry of the RPAR peptide with respect to tuftsin, which is a consequence of the rigidity of the Pro residue in RPAR. Since the binding of RPAR and NRP-1 remains strong, as indicated by the RMSD and the other well-preserved interactions, we conclude that one missing interaction is not critical for the binding, especially if the two other interactions exist between the backbone of the C-terminal Arg of RPAR and the side chains of NRP-1. Moreover, even though the distance between Tyr-353 and the C-terminal of Arg is too large to be considered as a strong interaction, both polar groups form a weak charge – dipole interaction that evidently contributes to the global stability of the complex.
Most recently, we began expanding the search for other molecular organizations that could act as NRP-1 ligand. Our preliminary results are quite unexpected. Simplified models that mimic protein loops and that were based on an oligopeptide rich in Glycine residues suggested potential binding between the NRP-1 receptor and species that do not follow the C-end rule. Thus, three different heptapeptides (G7, G3RG3 and G4RG2), all of them featuring a β-turn type I, were probed to see if they could dock in the receptor binding site. Not only was G7 able to dock, causing a minimal deformation to the receptor topology, but also peptides that presented Arginine residues (such as G3RG3 or G4RG2) could be inserted into the binding site with small deformation to the receptor. The average RMSD difference between the topology of the binding site when bound to RPAR, comparing to the heptapeptides, was under 0.30 Å for the G7 and 0.45 Å for the other Glycine based peptides. These results may suggest alternative therapeutic approaches beyond the mutant analogs that are presented below.
New RPAR analogs resistant to proteolysis
Rational design of synthetic residues
In order to explore possible chemical modifications that would enhance the RPAR affinity toward NRP-1 while hindering its proteolytic degradation, we focused on two main approaches. In the first, we attempted to optimize the docking of the homing peptide with the receptor by introducing chemical modifications in the side chain of the most active residue, the C-terminal Arg. Specifically, we modified the aliphatic segment of the Arg side group, which should not dramatically affect the interaction between the charged guanidinium group and the carboxylate side chain of the Asp-320. The investigated analogs are schematically represented in Figure 4a, which have been labeled as II–VI. The modifications inserted in the Arg template were either based on the incorporation of an unsaturated bond in the aliphatic segment (Figure 4a, analogs II to IV) or on the variation of the number of methylene units (Figure 4a, analogs V and VI). Two isomers, which differ in the relative position of the non-hydrogen atoms attached to the doubly bounded carbon atoms, i.e. cis and trans isomers, are obtained by incorporation an unsaturated aliphatic segment. The two isomers have been investigated in all cases.
In the second approach, we tried to make the peptide more resistant to proteases by following two strategies. In the first one we considered chirality: if the chirality of one or more residues of the RPAR peptide is to be inverted, cleavage could be prevented due to the loss of the protease recognition pattern. However, in a previous study it was shown that a D-peptide, in which the chirality of all the residues was simultaneously inverted, did not interact with NRP-1 (9). This made us reduce the modification range to single mutations (i.e. one-by-one residue). Another potential tool to obstruct the proteolytic action is the modification of the amide bond. Accordingly, we examined the possibility of changing the directionality of the amide bond by introducing a retro-modification, which consists of the substitution of two consecutive amino acids by the gem-diamine and the acid, respectively, as indicated in Figure 4b. The net effect over the peptide scaffold is a partial inversion of the local directionality of the peptide main chain, the resulting compound being known as retropeptide (15).
All these analogs were systematically tested by running MD simulations, using our simplified receptor model. A complete list of the simulations is provided in Table 3. For each analog family, its potential usefulness was assessed based on the requirements noted above: a good candidate should not distort the receptor organization around the binding site and should preserve the interactions that characterized the association of the RPAR peptide with the receptor.
Table 3.
Most relevant structural and chemical properties featured by each RPAR analog interacting with NRP-1 averaged over 5 ns of MD simulation. Averaged values for wild type RPAR (system I) were obtained using the last 5 ns of the 40 ns simulation.
| RPAR Analog | Average RMSD (Å) | % Preserved interactionsa | Total interaction energy (kcal·mol−1)b | |
|---|---|---|---|---|
| (D)RPAR | 1.25±0.9 | 97% | −125.3±3.4 | |
| R(D)PAR | 2.01±1.0 | 85% | −94.0±7.7 | |
| RP(D)AR | 2.21±1.1 | 79% | −88.4±5.6 | |
| RPA(D)R | 1.31±0.5 | 96% | −129.9±3.2 | |
| I | 1.24±0.2c | 99%d | −133.6±2.5e | |
| II | cis | 1.44±0.7 | 78% | −98.5±3.3 |
| trans | 1.29±0.6 | 98% | −129.3±3.9 | |
| III | cis | 1.85±0.4 | 96% | −111.1±5.1 |
| trans | 1.92±0.5 | 93% | −109.4±4.7 | |
| IV | cis | 1.99±0.6 | 91% | −101.0±2.3 |
| trans | 1.34±0.3 | 97% | −122.6±3.5 | |
| V | 1.81±0.91 | 72% | −77.2±5.7 | |
| VI | 2.18 ±0.94 | 73% | −89.2±6.1 | |
| VII | 1.25±0.3 | 99% | −131.3±2.9 |
Computed as the percentage of preserved specific interactions analyzed in Table 2 over 5 ns of simulation.
The total interaction energy was determined by summing the individual interaction energies, which were obtained for each specific interaction as was indicated in Table 2.
Values extracted for those residues that constitute the receptor model.
Values extracted combining information from Figures 1c and 1d.
The values of the individual interactions energies were extracted from Table 2
Simulations of RPAR analogs
Table 3 lists the averaged RMSD, the percentage of preserved interactions and the total interaction energy values, which were calculated by summing the interaction energies determined for all the specific interactions (i.e. adding the pair-wise interaction energies between the amino acids participating in all salt bridges and hydrogen bonds), for all the investigated systems As expected, results for D-amino acid replacements depended on the D-residue position: the potential affinity for the receptor decreased when the mutated residues were in the central segment of the peptide because they affected the orientation of the RPAR analog in the binding pocket. Even though in all cases the interactions pattern was acceptably preserved, with percentages above 80% of the total amount for 5 ns, only (D)RPAR and RPA(D)R satisfied all the screening criteria. Replacements in the central site distorted the receptor organization, as reflected by the higher values of RMSD, reducing the interaction strength as the computed higher energy suggests (system I in Table 3). On the other hand, replacement of any terminal Arg residue does not perturb the formation of the complex receptor – peptide, giving very similar results in both cases. A detailed view of the organization obtained for system RPA(D)R is shown in Figure 5a.
Figure 5.

Ribbon representation of the NRP-1 binding site with RPAR analogs docking in NRP-1 – RPAX complexes: (a) X= (D)R; (b) X= trans-II; (c) X= trans-IV; and (d) X= retro-Arg. The ligand and the interacting side chains of the receptor are depicted as solid lines. NRP-1 backbone is shown in purple and RPAX backbone in green. Hydrogen atoms are omitted for clarity. Specific interactions are drawn: hydrogen bonds are shown as blue discontinuous lines while salt bridges are marked by yellow discontinuous lines.
The modeling of all the remaining analogs was based on the introduction of several chemical modifications at the C-terminal Arg. Thus, all present the RPAX motif, where X corresponds to a new non-coded amino acid that replaces a natural Arg in RPAR peptide. Within this set of molecules, when the length of the methylene segment in the C-terminal Arg was modified most of the necessary conditions for successful interaction were not satisfied (V and VI in Figure 4a and Table 3). Under such conditions, the relocation of the guanidinium group in the binding pocket, which is guided by the interaction between such group and the Asp 320 side chain, induces the loss of some interactions between RPAX and the side chains of Thr-349 and Ser-346. Table 2 shows that the percentage of preserved interactions drops below 75% in both studied analogs for the 5 ns of simulation. This loss results in important changes in the receptor topology indicating that both analogs, with an average RMSD that almost doubles the structural fluctuations observed in the wild type segment for the same amount of time, are unlikely.
On the other hand, insertion of unsaturated bonds along the aliphatic segment of Arg leads to binding modes that depend strongly on both the particular topology of the double bond (i.e. cis or trans) and its position in the alkyl segment. The only analogs that were able to fulfill the selection criteria were those in which the geometrical restrictions of the new constitution drove the side chain towards an organization that resembles the conformation adopted by the side chain of the coded Arg in RPAR. Among six possible analogs (II, III and IV in Figure 4a, each one with cis and trans isomers), only three formed the proper interactions with the binding site residues: trans-II (Figure 5b), cis-III and trans-IV (Figure 5c). However, cis-III should be discarded since its docking produced rearrangements in the receptor organization (with RMSD fluctuations 45% higher than in the wild type peptide) and diminish the interaction strength between the ligand and the receptor. However, both trans-II and trans-IV satisfy the requirements, and are good candidates for an RPAR analog, reaching interaction energies close to the values computed for the wild type peptide.
Thus, peptide VII, which consists of a RPAX analog with X= retro-Arg (Figure 4b), becomes the most promising candidate. This is because the majority of the interactions that stabilize the NRP-1 – RPAR complex were supplied either by the Arg side chain or by the terminal carboxylate and, therefore, alteration of the main chain at the N terminus does not preclude binding NRP-1. Indeed, the results obtained with retro-Arg proved that this analog perfectly suits all the previously imposed selection conditions (Figure 5d), showing structural fluctuations and interaction energies almost identical to those observed with the wild type peptide (Table 3).
Conclusions
In this study we used computer simulation methods to model the binding of several peptides to the NRP-1 receptor, which plays an important role in angiogenesis and vascular permeability. Previous work showed that peptides containing the consensus sequences K/RXXK/R promote binding and internalization into tumor cells, while blocking the C-terminal Arg prevented its internalization. This property was coined the CendR rule. The molecular details that allow binding of RPAR to NRP-1 have been characterized in this work. The structural features of the complex have been modeled using as molecular template the crystal structure of NRP-1 bound to tuftsin, a tetrapeptide that mimics the C-terminal sequence of the tumor-secreted vascular permeability factor VEGF (11). Furthermore, the specific interactions found in the RPAR – NRP-1 complex have been used to suggest RPAR analogs with improved functional features.
Our simulations have demonstrated that the RPAR – NRP-1 complex is supported by a network of stabilizing hydrogen bonds and salt bridges, similar to that previously observed in the cocrystal NRP-1 – tuftsin. The most relevant interaction is a salt bridge between the C-terminal Arg of RPAR and Asp230 of NRP-1. The stabilization of the RPAR C-terminus is further enhanced with a bifurcated hydrogen bond between the carboxylate group of the terminal Arg and the side chains of Ser-346 and Thr-349. Finally, RPAR features an extra anchoring point in the NRP-1 pocket through a hydrogen bond between the backbone of the Pro residue of RPAR and the side chain of Tyr-297 of NRP-1. However, this interaction, which is also present in the crystal template NRP-1 – tuftsin, is weaker than the other three since it does not involve any charged center, which suggests a secondary role.
The potential therapeutic use of the RPAR peptide as internalization trigger in anticancer nanodevices requires increasing its resistance to protease cleavage. The use of a reduced model for the NRP-1 receptor combined with short molecular simulations has allowed a quick screening of potential RPAR analogs. The analogs that show a rapid destabilization can be immediately discarded. Those that formed a stable molecular complex should be further studied as potential therapeutic approaches. In this context, we have explored potential RPAR analogs that are the outcome of chemical modifications aimed to make it less sensitive to proteolytic action. The suitability of each modification has been evaluated using molecular simulations by comparing the molecular features of each studied analog when binding NRP-1 with those characterized for the original RPAR segment. Thus, modifications that disrupted the formation of the previously described interactions with NRP-1 have been discarded. Among all potential chemical modifications that have been introduced in the RPAR peptide, only those modifications in the C- terminal Arg that either did not alter the shape of the C-terminus (D-Arg and retro-Arg) or did not restrict the orientation of the guanidinium group upon adding a double bond in the side chain of the C-terminal Arg (trans-II and trans-IV) have been categorized as suitable candidates. When any of those features were absent in a tested analog, the resulting interaction pattern between the NRP-1 receptor and the modified peptide did not present all the necessary interacting groups and the internalization activity may diminish. Therefore, chemical changes in RPAR for increasing the proteolytic resistance need to maintain the carboxylate moiety and should not excessively restrict the conformational freedom of the C-terminal Arg side chain.
Acknowledgments
The research was supported in part by the National Science Foundation through TeraGrid resources provided by the Texas Advanced Computing Center (TACC) under grant number TG-MCB100025 (N.H.). Computer resources were generously provided by the Centre de Supercomputació de Catalunya (CESCA), the National Cancer Institute for partial allocation of computing time and staff support at the Advanced Biomedical Computing Center of the Frederick Cancer Research and Development Center and the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). Financial support from Generalitat de Catalunya (research group 2009 SGR 925; XRQTC; ICREA Academia prize for excellence in research to C.A.) is gratefully acknowledged. This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the view of the policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- NRP-1
Neuropilin-1
- RPAR
peptide Arg-Pro-Ala-Arg
- VEGF
Vascular permeability factor
- CendR
C end rule
- NVT constant Volume
Temperature and Number of moles
- NPT constant Pressure
Temperature and Number of moles
- MD
Molecular Dynamics
- RMSD
Root Mean Square Deviation
References
- 1.Rabbani ML, Rogers PA. Role of vascular endothelial growth factor in endometrial vascular events before implantation in rats. Reproduction. 2001;122:85–90. doi: 10.1530/rep.0.1220085. [DOI] [PubMed] [Google Scholar]
- 2.Maeda H, Fang J, Inutsuka T, Kitamoto Y. Vascular permeability enhancement in solid tumor: Various factors, mechanisms involved and its implications. Int Immunopharmacol. 2003;3:319–328. doi: 10.1016/S1567-5769(02)00271-0. [DOI] [PubMed] [Google Scholar]
- 3.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 4.Baban DF, Seymour LW. Control of tumour vascular permeability. Adv Drug Deliv Rev. 1998;34:109–119. doi: 10.1016/s0169-409x(98)00003-9. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161:851–858. doi: 10.1016/0006-291x(89)92678-8. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 7.Takashima S, Kitakaze M, Asakura M, Asanuma H, Sanada S, Tashiro F, Niwa H, Miyazaki Ji J, Hirota S, Kitamura Y, Kitsukawa T, Fujisawa H, Klagsbrun M, Hori M. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc Natl Acad Sci USA. 2002;99:3657–3662. doi: 10.1073/pnas.022017899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staton CA, Kumar I, Reed MWR, Brown NJ. Neuropilins in physiological and pathological angiogenesis. J Pathol. 2007;212:237–248. doi: 10.1002/path.2182. [DOI] [PubMed] [Google Scholar]
- 9.Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1 dependent cell, vascular and tissue penetration. Proc Natl Acad Sci USA. 2009;106:16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science. 2010;328:1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, Leahy DJ. Structural basis for ligand and heparin binding to neuropilin B domains. Proc Natl Acad Sci USA. 2007;104:6152–6157. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Wronski MA, Raju N, Pillai R, Bogdan NJ, Marinelli ER, Nanjappan P, Ramalingam K, Arunachalam T, Eaton S, Linder KE, Yan F, Pochon S, Tweedle MF, Nunn AD. Tuftsin binds neuropilin-1 through a sequence similar to that encoded by exon 8 of vascular endothelial growth factor. J Biol Chem. 2006;281:5702–5710. doi: 10.1074/jbc.M511941200. [DOI] [PubMed] [Google Scholar]
- 13.Agemy L, Sugaharai KN, Kotamraju VR, Gujraty K, Girard OM, Kono Y, Mattrey RF, Park JH, Sailor MJ, Jiménez AI, Cativiela C, Zanuy D, Sayago FJ, Alemán C, Nussinov R, Ruoslahti E. Nanoparticles-induced vascular blockade in human prostate cancer. Blood. 2010;116:2847 – 2856. doi: 10.1182/blood-2010-03-274258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang L, Derfus AM, Yang M, Hoffman RM, Bathia S, Sailor MJ, Ruoslahti E. Biomimetic amplification of nanoparticle homing to tumors. Proc Natl Acad Sci USA. 2007;104:932–936. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alemán C, Puiggali J. Retromodified Residues: Small Peptides and Polymers. Interactions, Force-Field Parametrization and Conformational Analyses. J Org Chem. 1995;60:910–924. [Google Scholar]
- 16.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J of Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second Generation Force-Field For The Simulation of Proteins, Nucleic-Acids, and Organic-Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 18.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong GM, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman PA. Point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 19.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 20.Darden T, Perer L, Li L, Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7:R55–R60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- 21.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 22.Zanuy D, Flores-Ortega A, Casanovas J, Curco D, Nussinov R, Aleman C. The Energy Landscape of a Selective Tumor-Homing Pentapeptide. J Phys Chem B. 2008;112:8692–8700. doi: 10.1021/jp711477k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Matthews DH, Schafmeister C, Ross WS, Kollman PA. AMBER. Vol. 9. University of California; San Francisco: 2006. [Google Scholar]