

Abstract
BACKGROUND AND PURPOSE
Pancreatic cancer is one of the leading cancer-related causes of death due to high chemo-resistance and fast metastasation. Nemorosone, a polycyclic polyprenylated acylphloroglucinol, has recently been identified as a promising anticancer agent. Here, we examine its growth-inhibitory effects on pancreatic cancer cells. Based on transcription profiling, a molecular mode of action is proposed.
EXPERIMENTAL APPROACH
Nemorosone cytotoxicity was assessed by the resazurin proliferation assay on pancreatic cancer cells and fibroblasts. Apoptosis was determined by Annexin V/propidium iodide staining as well as cytochrome c and caspase activation assays. Staining with the voltage-dependent dye JC-1 and fluorescence microscopy were used to detect effects on mitochondrial membrane potential. Total RNA was isolated from treated cell lines and subjected to microarray analysis, subsequent pathway identification and modelling. Gene expression data were validated by quantitative polymerase chain reaction and siRNA-mediated gene knock-down.
KEY RESULTS
Nemorosone significantly inhibited cancer cell growth, induced cytochrome c release and subsequent caspase-dependent apoptosis, rapidly abolished mitochondrial membrane potential and elevated cytosolic calcium levels, while fibroblasts were largely unaffected. Expression profiling revealed 336 genes to be affected by nemorosone. A total of 75 genes were altered in all three cell lines, many of which were within the unfolded protein response (UPR) network. DNA damage inducible transcript 3 was identified as a key regulator in UPR-mediated cell death.
CONCLUSIONS AND IMPLICATIONS
Nemorosone could be a lead compound for the development of novel anticancer drugs amplifying the already elevated UPR level in solid tumours, thus driving them into apoptosis. This study forms the basis for further investigations identifying nemorosone's direct molecular target(s).
Keywords: nemorosone, polycyclic polyprenylated acylphloroglucinols, pancreatic cancer, endoplasmic reticulum stress, unfolded protein response, mitochondrial membrane potential
Introduction
Pancreatic cancer is the fourth leading cancer-related cause of death in the developed world with more than 36 000 deaths in the USA in 2010 and a 5 year survival rate of only 4% (Jemal et al., 2010). The poor prognosis is directly linked to the inability of diagnosing pancreatic cancer in an early resectable state and its tendency towards fast dissemination into regional lymph nodes and the liver, thereby evading chemotherapeutic approaches (Keleg et al., 2003). The current standard therapy with gemcitabine leads to only modest median survival times between 5 and 6 months (El-Rayes and Philip, 2003; Sultana et al., 2007), thus emphasizing the need for drugs with novel mechanisms of action.
In recent years, the identification of hyperforin as a mild antidepressant directed attention towards polycyclic polyprenylated acylphloroglucinols (PPAPs), a novel class of secondary metabolites with diverse biological activities derived from plants of the Guttiferae family (Ciochina and Grossman, 2006). Hyperforin is isolated from St John's wort (Hypericum perforatum) and was subsequently been shown to also possess anticancer activity (Schempp et al., 2002). Another PPAP, nemorosone was first isolated from flowers of the Clusia species and subsequently found to be the major constituent of Cuban propolis (Cuesta-Rubio et al., 2001; 2002;). It was active against a panel of cancer cell lines, exhibiting a mean IC50 of 2.6 µM without any cross-resistances to clinically important cytostatics (Diaz-Carballo et al., 2003). In more detailed studies, cytotoxic effects of nemorosone on neuroblastoma and leukaemia cell lines were demonstrated with IC50 values between 2 and 4 µM (Diaz-Carballo et al., 2008a,b;). It was shown that nemorosone caused significant dephosphorylation of ERK1/2 and activated the AKT/PKB signalling pathway, thus inducing growth arrest and apoptosis. However, more detailed insights into its molecular mechanism of action are still missing.
In this study, we demonstrate that nemorosone is active against pancreatic cancer cells and analysed its molecular mode of action. The endoplasmic reticulum (ER) stress response was shown to be amplified by nemorosone, possibly through disturbance of the ER calcium homeostasis.
Methods
Isolation of nemorosone
Nemorosone was isolated from an ethanolic flower extract of Clusia rosea in one of our laboratories using reversed-phase high pressure liquid chromatography with high purity (>97%) as reported previously (Diaz-Carballo et al., 2008a). It was suspended in dimethylsulphoxide (DMSO) to obtain a stock solution stored at −20°C. Each treatment was performed with the same amount of DMSO, and DMSO concentration did not exceed 0.25% in any experiment.
Cell culture and determination of IC50 values
Three pancreatic cancer cell lines were chosen based on their degree of differentiation and obtained from the American Type Culture Collection (ATCC; Rockville, MD, USA). Capan-1 (liver metastasis) represents a well-differentiated cell line with WHO tumour classification G1. AsPC-1 was isolated from ascites and is moderately differentiated (G2), whereas MIA-PaCa-2 represents a poorly differentiated (G3) cell line obtained from a primary tumour. All cell lines carry mutations in the p53 and p16INK4 genes, key regulators in the cell cycle. Capan-1 and AsPC-1 cell lines were maintained in RPMI 1640 medium, MIA-PaCa-2 in Dulbecco's modified Eagle's minimal essential medium supplemented with 10% heat-inactivated fetal calf serum (FCS) as well as 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin in a humid environment and 5% CO2. Non-transformed human dermal (HDF) and foreskin fibroblasts were incorporated in this study and cultured in Dulbecco's modified Eagle's minimal essential medium as control cell lines with normal growth characteristics.
IC50 values were determined using the resazurin proliferation assay (Nakayama et al., 1997). Briefly, 2 × 103 cells were seeded into the wells of a 96-well cell culture plate and allowed to settle for 24 h. Cells were then treated with increasing nemorosone concentrations and cytotoxicity was analysed after 24 and 72 h of incubation respectively. IC50 values were interpolated from the resulting dose–response plots.
Flow-cytometric analysis of cell cycle, cell death and cytochrome c release
Cell cycle analysis
Cells, 2 × 105, were seeded into the wells of a six-well cell culture plate and grown for 24 h. Cells were then treated with 0.5× and 1× IC50 of nemorosone as well as vehicle alone for 24 h. After being harvested by short trypsination and washing in Dulbecco's-PBS (Invitrogen, Karlsruhe, Germany), cells were fixed by adding cold 100% ethanol and incubated for 24 h at 4°C. Cells were then resuspended in staining buffer (50 µg·mL−1 propidium iodide (PI), 100 µg·mL−1 RNAse A, in D-PBS) and the DNA content of single nuclei was measured on a FACSCanto II flow cytometer (Beckton-Dickinson, Germany) after a 20 min incubation. Data analysis was performed using FlowJo (Treestar, Ashland, OR, USA) and the programme's Watson algorithm for cell cycle analysis.
Analysis of cell death
For detection of nemorosone-induced cell death, cells were incubated with vehicle alone, 10, 20 or 50 µM nemorosone for 48 h. Cells were harvested by short trypsination and subsequently washed in D-PBS. After incubation in Nicoletti buffer [50 µg·mL−1 PI, 0.1% (w·v−1) sodium citrate, 0.1% (v·v−1) Triton-X100, in dH2O] for 30 min, the DNA content of single nuclei was determined on a FACSCanto II flow cytometer (Riccardi and Nicoletti, 2006). Nemorosone-specific cell death was determined using the FlowJo software by elimination of background in control cells.
Analysis of caspase activation
Activity of caspases 9, 3/7 and 8 were determined using the respective carboxyfluorescein FLICA assays (Immunochemistry Technologies, Bloomington, MN, USA). Briefly, cells were treated as indicated for 24 h, trypsinized and incubated in FLICA reagent for 1 h. After two washes, cells were resuspended in assay wash buffer and analysed by flow cytometry.
Analysis of cytochrome c release
Cytochrome c release was assessed using the InnoCyte Flow Cytometric Cytochrome C Release Kit (Calbiochem, Darmstadt, Germany) according to the manufacturer's instructions. Plasma membranes were selectively permeabilized in order to wash out cytochrome c released from mitochondria into the cytoplasm in apoptotic cells. Cytochrome c was then stained using a specific antibody and a fluorescein isothiocyanate (FITC)-labelled secondary antibody. Fluorescence intensity of stained cells was measured flow-cytometrically. A shift in fluorescence intensity indicates cytochrome c release.
Annexin V and PI staining
Apoptosis was induced with the indicated concentrations of nemorosone in cells seeded in 96-well microtritre plates. After 24 h, cells were carefully washed in Annexin V binding buffer (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) supplemented with 10% FCS. Annexin V-FITC (Immunotools, Friesoythe, Germany) and PI (Sigma Aldrich, Munich, Germany) were diluted to 0.15 µg·mL−1 and 2.5 µg·mL−1, respectively, in binding buffer. Cells were stained in 100 µL staining solution for 15 min and then analysed on a Zeiss Cell Observer fluorescence microscope using appropriate filters for FITC and PI. Brightfield images served as control to determine total cell number, and the percentage of annexin V-positive and annexin V/PI double positive cells was assessed by counting a total of at least 300 cells in four random fields.
Detection of mitochondrial membrane potential
Cells were seeded in a 96-well microtitre plate at a density of 5000 cells per well and grown for 24 h. After treatment with indicated concentrations for 4 h, cells were pelleted and the supernatant was exchanged with 100 µL DePsipher (JC-1) staining solution (R&D Systems, Abingdon, UK). Incubation was for 30 min according to the manufacturer's instructions. The cells were subsequently washed twice with D-PBS, and staining of intact mitochondria (red fluorescing aggregates, emission at 590 nm) versus cytoplasmic staining (green fluorescing monomers, emission at 527 nm) was visualized using a Zeiss Cell Observer fluorescence microscope equipped with a GFP long-pass filter (Zeiss, Jena, Germany). For kinetic analysis, staining preceded treatment.
Calcium assay
The cytosolic calcium concentration was determined using the Fluo4-NW assay (Invitrogen, Germany). Briefly, 5000 cells were seeded in a 96-well plate and grown to 80% confluence for 24 h. Cells were then loaded with Fluo4 dye according to the recommended protocol for 45 min at 37°C and 5% CO2. Loading buffer was exchanged with calcium-free phosphate-buffered saline (PBS) and background fluorescence was recorded until stabilized. Cells were then treated with nemorosone and Fluo4 fluorescence was recorded every 5 s in a FluoStar Galaxy plate reader (BMG Labtech, Offenburg, Germany). Fluorescence values were normalized to the median of the base line values.
Microarray analysis
Sample preparation and analysis
Cell lines were treated for 24 h with their respective 0.5×, 1× and 2× IC50 of nemorosone in duplicates in 175 cm2 cell culture flasks. Total RNA was isolated using the RNeasy Midi Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. For microarray assay only, RNA of biological replicates was pooled to account for the minor biological variations expected under standard cell culture conditions. RNA quality and quantity were routinely checked by gel electrophoresis and photometrically on a NanoDrop ND-1000 (Peqlab, Erlangen, Germany). Labelled cDNA was synthesized out of 15 µg total RNA using SuperScript II reverse transcriptase and Oligo(dT) primers (Invitrogen, Germany) according to the manufacturer's procedures, thereby incorporating Cy3 (test samples) or Cy5 (control samples) labelled dCTPs (GE Healthcare, Buckinghamshire, UK). The resulting cDNA was purified with polymerase chain reaction (PCR) clean-up spin columns (Qiagen, Hilden, Germany), dried, resuspended in hybridization buffer [5 µL 10 mM EDTA, 20 µL SlideHyb #1 solution (Ambion, Darmstadt, Germany)] and subsequently hybridized to spotted DNA microarray slides as reported in detail earlier (Bauer et al., 2008). Microarrays covered approximately 4000 genes, mainly involved in pancreatic cancer, cell cycle control, apoptosis, response to stress and metabolization. Three technical replicates comprising cDNA labelling and hybridization steps were performed for each experimental condition because technical variation was expected to be greater than biological variation.
Data analysis
Spot intensities of Cy3/Cy5 (control/test sample) microarray images were calculated with GenePix Pro 6.0 software (Molecular Devices, Sunnyvale, CA, USA) using a constant feature diameter for all spots. Data were subsequently loaded into the MIAME-compliant Multi-Conditional Hybridization Intensity Processing System for further data analysis (Fellenberg et al., 2002). Local background-corrected data were normalized applying the locally weighted scatterplot smoothing algorithm, LOWESS (Cleveland, 1981). Hybridizations with correlation coefficients lower than 0.8 (between the red and green channel) were excluded from further analysis. Genes whose signals never exceeded twofold standard deviation of background intensities were excluded. Genes with twofold regulation in at least one condition were considered for downstream analysis. To filter for reproducible expression changes between replicates, outliers were excluded using a very restrictive ‘min-max separation’ filter (Beissbarth et al., 2000) accounting for the low number of replicates. In combination, only about 10% reproducibly differential genes survived all filter criteria. P-values for each condition versus the control condition were calculated using the empirical Bayes method in the LIMMA package (Bioconductor) (Smyth, 2004). Data were visualized in a correspondence analysis (CA) plot (Fellenberg et al., 2001) allowing for simultaneous presentation of experimental conditions and genes (Figure S1). Differences between conditions as well as genes associated with certain conditions are represented by their relative position within the CA plot. Differentially expressed genes were subjected to further analysis using Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA, USA) and DAVID (Dennis et al., 2003; Huang da et al., 2009) to find pathways or gene ontology terms exhibiting a significant number of differentially regulated genes. For pathways modelling, a knowledge-driven approach was used applying Ingenuity Pathway Analysis's gene and protein interaction database.
Microarray data were deposited in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress; Accession No. E-MEXP-1869).
Real-time quantitative RT-PCR
Total RNA (25–50 ng in a 10 µL reaction) was reverse-transcribed and subsequently amplified using the QuantiFast SYBR Green RT-PCR kit (Qiagen, Hilden, Germany) on a 384-well plate in a LightCycler 480 (Roche Applied Science, Mannheim, Germany) according to the manufacturer's protocol. QuantiTect primers (Qiagen, Hilden, Germany) were used for amplification in triplicates, and α-Tubulin served as housekeeping control gene for normalization.
DNA damage inducible transcript 3 (DDIT3) mRNA silencing
Cells were seeded in six-well cell culture plates and grown to 50% confluence for 24 h before transfection with HP GenomeWide siRNA duplexes corresponding to human DDIT3 gene (5′-GGG UCA AGC AGG AAA UCG A-3′; Qiagen, Hilden, Germany) using HiPerFect transfection reagent (Qiagen, Hilden, Germany) as recommended by the manufacturer. Control cells were transfected with scrambled AllStars control siRNA (Qiagen, Hilden, Germany). The final siRNA concentration was 5 nM. Cells were changed into fresh media containing 20 µM nemorosone or vehicle-containing medium 24 h after transfection and incubated for 24 or 48 h respectively. mRNA silencing was checked using quantitative RT-PCR and immunofluorescence. The proportion of apoptotic cells was determined flow-cytometry as described above.
Immunostaining of DDIT3
Cells were seeded in a 96-well plate and transfected with siRNA as mentioned above. After treatment with 20 µM nemorosone for 24 h, cells were fixed with 3.7% formaldehyde, 0.1% Tween-20 in PBS for 10 min. After washing in PBS and blocking with 1% FCS in PBS, cells were incubated in a 1:200 dilution of mouse anti-DDIT3 antibody (Abcam, Cambridge, UK) in 0.5% FCS in PBS for 1 h. Cells were subsequently washed and incubated in a 1:2000 dilution of Alexa-488 conjugated goat anti-mouse IgG (Invitrogen, Germany) in 0.5% FBS in PBS for 1 h. Fluorescence images were recorded with a CellObserver microscope (Zeiss, Germany) after excessive washes with PBS.
Western blotting
For detection of DDIT3 protein, cells were seeded in 100 mm cell culture dishes, transfected with control or DDIT3 siRNA and treated with vehicle or 20 µM nemorosone. Cells were then collected by treatment with accutase (Sigma Aldrich, Germany) and lysed in Laemmli buffer (63 mM Tris-HCl, 10% glycerol, 2% SDS, pH 6.8). Lysates were centrifuged at 16 000 g to remove insoluble material and quantified by a BCA assay; 30 µg of protein was resolved by a 12% SDS gel, transferred to a nitrocellulose membrane, blocked with 5% non-fat milk and probed with an anti-DDIT3 antibody (F-168; Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:200 overnight at 4°C. The blot was washed, exposed to a horse-radish peroxidize-conjugated secondary antibody for 1 h and developed with POD substrate (Roche Applied Science, Germany). Horse-radish peroxidize-conjugated anti-GAPDH antibody (Sigma Aldrich, Germany) was used to confirm equal loading.
Statistical tests
All experiments were performed at least in triplicate unless stated otherwise. For all experiments other than microarray experiments, Student's t-test (in SigmaPlot 10.0) was used for significance analysis of the difference between two groups. P-values for genes of microarray experiments and gene ontology terms were corrected for multiple testing using the Benjamini–Hochberg algorithm (Benjamini and Hochberg, 1995).
Results
Nemorosone treatment inhibits growth and induces cell cycle arrest
Cytotoxic activity of nemorosone has previously been demonstrated on several cancer cell lines (Diaz-Carballo et al., 2003; 2008a,b;). However, cytotoxicity data are still lacking for pancreatic cancer cells. Hence, three pancreatic cancer cell lines, Capan-1, AsPC-1 and MIA-PaCa-2, as well as HDF and human foreskin fibroblasts were exposed to increasing nemorosone concentrations for 24 and 72 h respectively. IC50 values were interpolated from dose–response plots. Nemorosone was shown to inhibit cellular growth in a time- and concentration-dependent manner with cancer cell lines exhibiting a maximum growth inhibition of >90% and fibroblasts of 65–72% (Table 1). After 72 h of incubation, all pancreatic cancer cell lines demonstrated an IC50 at or below 5 µM whereas the non-transformed fibroblasts showed much higher IC50 values of 30–40 µM (Figure 1A). These results are comparable to previous investigations on other cancer cell lines (Diaz-Carballo et al., 2003). At 10 µM, our results indicate a negligible impact of nemorosone on the viability of fibroblasts but a significant reduction of pancreatic cancer cell growth by at least 80%.
Table 1.
IC50 values and maximum growth inhibition for pancreatic cancer cell lines and fibroblasts after treatment with nemorosone for 24 and 72 h
| IC50 value (µM) | Maximum growth inhibition (%) | |||
|---|---|---|---|---|
| Cell line | 24 h | 72 h | 24 h at 200 µM | 72 h at 80 µM |
| MIA-PaCa-2 | 16 (±1.0) | 4.5 (±0.5) | 98 (±5.5) | 98 (±0.5) |
| AsPC-1 | 16 (±1.5) | 5 (±0.5) | 91 (±5.0) | 93 (±0.5) |
| Capan-1 | 25 (±2.0) | 5 (±0.5) | 95 (±3.0) | 98 (±0.5) |
| Human dermal fibroblasts (HDF) | 80 (±4.5) | 40 (±3.5) | 65 (±2.5) | 71 (±2.0) |
| Human foreskin fibroblasts (HFF) | 73 (±4.0) | 35 (±3.0) | 70 (±3.5) | 72 (±6.5) |
Figure 1.
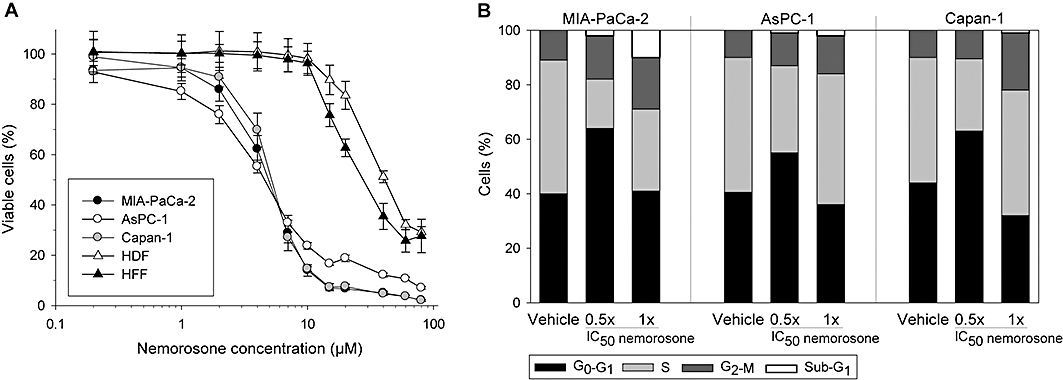
Effects of nemorosone on the viability and cell cycle of pancreatic cancer cells and fibroblasts. (A) Cell viability assay: pancreatic cancer cells and fibroblasts were treated with increasing nemorosone concentrations for 72 h. Cell viability was determined using a fluorescent resazurin assay. Values are mean ± SD of three independent experiments in sextuplets. (B) Cell cycle analysis: cells were treated with the indicated concentrations of nemorosone for 24 h, then fixed and stained with propidium idodide for flow-cytometric cell cycle analysis. The distribution of cells in different cell cycle phases is shown as mean of three independent experiments. HDF, human dermal fibroblasts; HFF, human foreskin fibroblasts.
As analysed by flow cytometry, nemorosone-induced growth reduction of pancreatic cancer cells was due to inhibition of cell cycle progression. When treated with 0.5× IC50 for 24 h, a significant increase of cells in the G1 phase of the cell cycle was observed, pointing towards early events interfering with DNA synthesis (Figure 1B). When treated with 1× IC50, however, a slight G2 arrest was observed instead of cells accumulating in G1 phase. Because cell viability is severely impaired at 1× IC50, this indicates an impact on all cell cycle phases at higher nemorosone concentrations.
Nemorosone induces cytochrome c release and caspase-dependent apoptosis in pancreatic cancer cells but not in fibroblasts
To investigate whether nemorosone-induced cytotoxicity is associated with the onset of apoptosis, cells were incubated with different nemorosone concentrations for 24 or 48 h and subsequently analysed for annexin V/PI staining or apoptotic DNA fragmentation by flow cytometry respectively (Figure 2A and B). Apoptosis induction was found to be dose- and time-dependent with onset of early apoptosis characterized by annexin V staining after 24 h and late apoptosis characterized by DNA fragmentation after 48 h. The apoptotic effect was most prominent on the poorly differentiated MIA-PaCa-2 cell line, with late-apoptotic (annexin V + /PI+) cells being detectable already after 24 h. Interestingly, dermal fibroblasts did not show any significant signs of apoptosis even when treated with 50 µM nemorosone.
Figure 2.
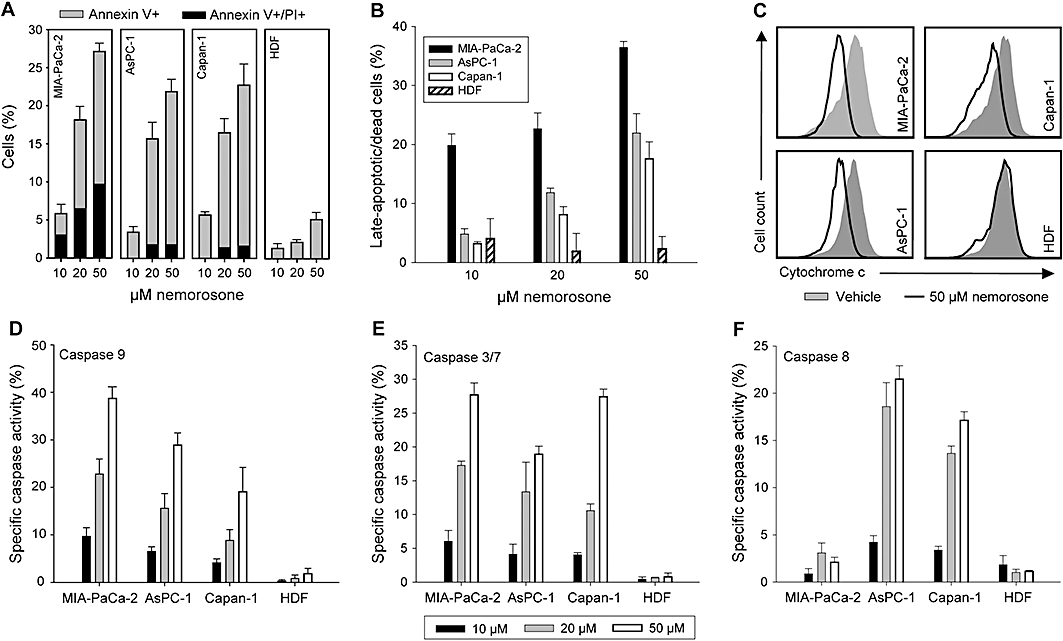
Analysis of nemorosone-induced apoptosis, cytochrome c release and caspase activation. Cells were treated with the indicated concentrations for 24 h (A) or 48 h (B) before being washed and stained with annexin V and propidium iodide (A) or propidium iodide alone according to the Nicoletti method (B). Percentage of cells demonstrating nemorosone-specific early (annexin V+) and late (PI+) apoptosis is shown. (C) Cytochrome c release was measured by flow-cytometry after treatment with 50 µM nemorosone or vehicle for 24 h. (D–E) Percentage of cells demonstrating nemorosone-specific activation of caspases 9, 3/7 and 8 after treatment with the indicated concentrations for 24 h. Values are mean ± SD of three independent experiments. HDF, human dermal fibroblasts.
It is well established that mitochondrial cytochrome c release and activation of the caspase cascade precede DNA fragmentation. Therefore, cytochrome c release was assessed by flow-cytometry and found to be induced after 24 h of nemorosone treatment in cancer cell lines but not in fibroblasts (Figure 2C). Activation of caspases 9, 3/7 and 8 was determined after 24 h of incubation with different nemorosone concentrations using FLICA caspase activation assays on a flow cytometer. Our results demonstrate that nemorosone treatment induced caspase 9 and 3/7 activation in all cancer cell lines, but not in fibroblasts, in a dose-dependent manner (Figure 2D and E). Interestingly, caspase 8 activation was only observed in better differentiated Capan-1 and AsPC-1 cells, pointing towards induction of the extrinsic apoptotic pathway in these two cell lines (Figure 2F). These results show that nemorosone-induced apoptosis is, at least in part, due to the final activation of the effector caspase 3/7 by intrinsic (involving cytochrome c release and caspase 9) and extrinsic (involving caspase 8; Capan-1 and AsPC-1) signalling cascades.
Nemorosone rapidly abolishes the mitochondrial membrane potential and elevates cytosolic calcium concentration
One of the earliest events in apoptosis is a process called mitochondrial membrane permeability transition involving reduction or abolition of the mitochondrial membrane potential ΔΨm. Staining with the voltage-dependent fluorescent dye JC-1 following treatment with nemorosone for 4 h revealed a dose-dependent reduction of ΔΨm in all pancreatic cancer cells with complete abolition of ΔΨm at 50 µM nemorosone (Figure 3A). In contrast, mitochondrial JC-1 staining was stable in dermal fibroblasts at up to 50 µM nemorosone and only fully abolished at 160 µM.
Figure 3.
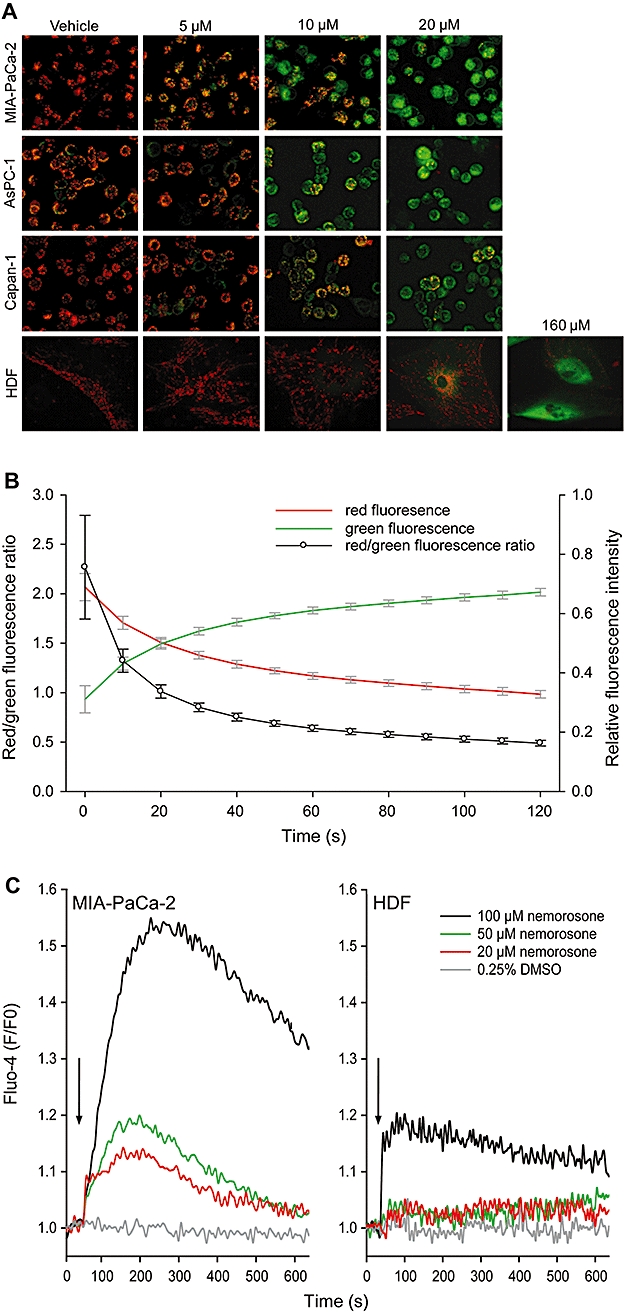
Analysis of effects of nemorosone on mitochondrial membrane potential ΔΨm and cytosolic calcium concentration [Ca2+]c in pancreatic cancer cells and fibroblasts. (A) Cells were treated with the indicated concentrations of nemorosone or vehicle for 4 h and stained with the voltage-dependent dye JC-1 prior to fluorescence microscopy. Intact mitochondria fluoresce red while cells with atteunuated mitochondrial membrane potential ΔΨm exhibit cytosolic green fluorescence. Images are representative of three independent experiments with similar results. (B) Kinetic analysis of ΔΨm upon nemorosone treatment. Cells were prestained with JC-1 and subsequently treated with 20 µM nemorosone. Fluorescent images were recorded every 10 s after onset of treatment. Development of relative red and green fluorescence as well as red/green fluorescence ratio is shown as mean ± SD of three independent experiments. (C) Analysis of nemorosone-induced changes in cytosolic calcium concentration [Ca2+]c in MIA-PaCa-2 cells and human dermal fibroblasts (HDF). Cells were loaded with the calcium-sensitive Fluo-4 dye and subsequently treated with the indicated concentrations of nemorosone or vehicle after recording a baseline for 30 s (black arrows). Development of Fluo-4 fluorescence relative to baseline fluorescence (F/F0) is shown as mean of three independent experiments. DMSO, dimethylsulphoxide.
Interestingly, a more detailed study on nemorosone-induced ΔΨm kinetics using MIA-PaCa-2 cells revealed that the mitochondrial potential was reduced within seconds when prestained cells were treated with 20 µM nemorosone and JC-1 fluorescence was recorded every 10 s (Figure 3B). The red/green fluorescence ratio was inverted within 2 min. This result points towards very rapid processes being induced in pancreatic cancer cells upon nemorosone treatment.
As a second messenger, calcium plays a vital role in cellular viability, and small changes in subcellular Ca2+ distribution can have severe effects. Because nemorosone seemed to induce rapid changes within the cell, we hypothesized that it might alter Ca2+ homeostasis. We used the Fluo-4 fluorescent calcium indicator assay to detect changes in cytosolic calcium concentration [Ca2+]c upon nemorosone treatment. A rapid dose-dependent increase in [Ca2+]c was detected in MIA-PaCa-2 cells already at 20 µM nemorosone (Figure 3C). A similar increase was observed in Capan-1 and AsPC-1 cells (data not shown). In HDF cells, however, only 100 µM, but not 50 or 20 µM nemorosone was able to elevate [Ca2+]c (Figure 3C).
Microarray analysis indentifies key players in nemorosone-induced cytotoxicity on pancreatic cancer cells
To investigate the molecular mechanism of action underlying nemorosone-induced growth inhibition of pancreatic cancer cells in more detail, MIA-PaCa-2, AsPC-1 and Capan-1 cells were treated with their respective 0.5×, 1× and 2× IC50 as well as vehicle only for 24 h. Total RNA was isolated and hybridized to DNA microarrays. After highly stringent filtering for genes that were significantly regulated (P < 0.05) in at least one condition, 336 genes were found to be associated with nemorosone treatment in all three cell lines (Table S1). MIA-PaCa-2 cells exhibited the lowest number of significantly regulated genes, and a dose-dependent effect on gene expression was observed for MIA-PaCa-2 and AsPC-1 (Figure 4A). Interestingly, MIA-PaCa-2 cells showed more up-regulated than down-regulated genes in each condition.
Figure 4.
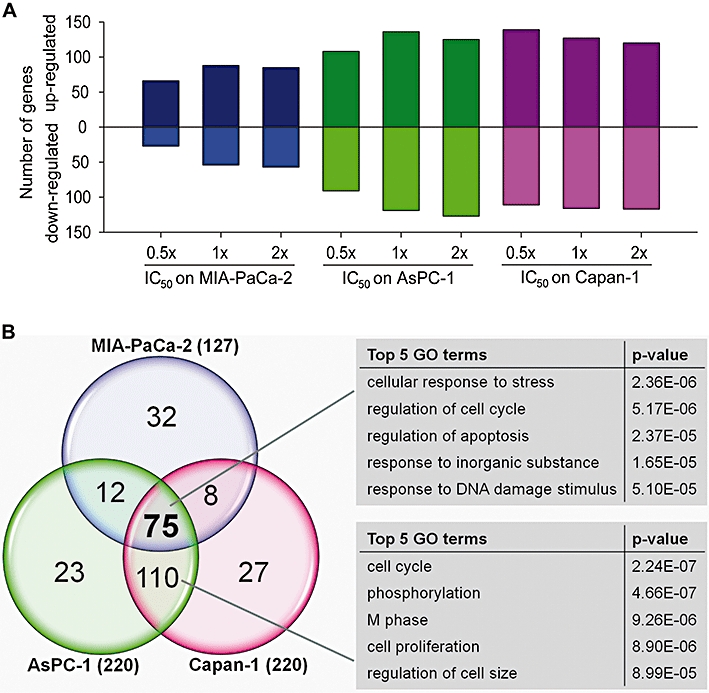
Comparison of the number and overlap of differentially expressed genes in pancreatic cancer cells after nemorosone treatment, and analysis of gene ontology (GO). (A) Shown is the number of significantly (P < 0.05) up- and down-regulated genes per cell line and condition. (B) The number of genes common in any two or all three cell lines is depicted in the intersections of the circles. GO analysis was carried out on the gene sets common to all cell lines and common to AsPC-1 and Capan-1 using the DAVID gene ontology website. The top five GO terms with lowest P-value are displayed.
For further interpretation of the results, they were subjected to CA (Fellenberg et al., 2001). In the resulting CA plot, replicate experiments always form clusters, indicating a high degree of reproducibility when using their genewise median (Figure S1). Experiments with vehicle were located next to the medium control, demonstrating no changes in gene expression upon treatment with vehicle only. Gene expression was found to be dose-dependent in Capan-1 and AsPC-1. For MIA-PaCa-2, however, all treatment conditions form one and the same cluster.
In order to bring these results in a biological context, the overlap of differentially expressed genes between the tested cell lines was calculated (Figure 4B). All three cell lines shared 75 differentially expressed genes in the 1× and 2× IC50 conditions (genes marked grey in Table S1). These genes are most likely associated with the mechanism of action of nemorosone in all three cell lines, and gene ontology analysis revealed them to be significantly associated with response to cellular stress as well as regulation of apoptosis and cell cycle (Figure 4B). One hundred and ten genes were found to be shared by Capan-1 and AsPC-1 only and are associated with cell cycle and proliferation. Thus, the differences between MIA-PaCa-2 and the other two cell lines observed in the CA plot were mainly due to differential expression of cell cycle regulatory genes as indicated by the expression values in Table 2.
Table 2.
Cell cycle genes that are differentially regulated in pancreatic cancer cells after treatment with the respective IC50 of nemorosone for 24 h
| Fold change | |||||
|---|---|---|---|---|---|
| EntrezGene ID | Gene symbol | Description | MIA-PaCa-2 | AsPC-1 | Capan-1 |
| 472 | ATM | Ataxia telangiectasia mutated | 1.75 | 2.64 | 1.77 |
| 699 | BUB1 | BUB1 budding uninhibited by benzimidazoles 1 homologue | 1.87 | −2.82 | −3.12 |
| 890 | CCNA2 | Cyclin A2 | −3.25 | −2.51 | −2.54 |
| 891 | CCNB1 | Cyclin B1 | −1.28 | −21.08 | −3.40 |
| 9133 | CCNB2 | Cyclin B2 | −1.66 | −14.24 | −6.87 |
| 595 | CCND1 | Cyclin D1 | −1.84 | −1.88 | −1.66 |
| 894 | CCND2 | Cyclin D2 | −1.23 | −1.38 | −1.01 |
| 983 | CDC2 | Cell division cycle 2, G1 to S and G2 to M | 1.23 | −7.32 | −6.06 |
| 991 | CDC20 | Cell division cycle 20 homologue (S. cerevisiae) | −1.78 | 1.20 | 1.16 |
| 994 | CDC25B | Cell division cycle 25 homologue B (S. pombe) | −1.27 | −1.03 | −1.49 |
| 1019 | CDK4 | Cyclin-dependent kinase 4 | −1.64 | −2.52 | −2.37 |
| 1022 | CDK7 | Cyclin-dependent kinase 7 | 2.67 | 1.54 | 2.03 |
| 1026 | CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 11.35 | 54.27 | 21.58 |
| 1027 | CDKN1B | Cyclin-dependent kinase inhibitor 1B (p27, Kip1) | −1.48 | −3.74 | −2.63 |
| 1032 | CDKN2D | Cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) | −1.02 | −1.29 | −1.40 |
| 8454 | CUL1 | Cullin 1 | 1.12 | 1.99 | 1.30 |
| 1869 | E2F1 | E2F transcription factor 1 | −1.96 | −2.00 | −1.95 |
| 1647 | GADD45A | Growth arrest and DNA-damage-inducible, alpha | 4.06 | 1.29 | 1.88 |
| 4085 | MAD2L1 | MAD2 mitotic arrest deficient-like 1 (yeast) | −3.93 | −8.06 | −12.69 |
| 4171 | MCM2 | Minichromosome maintenance complex component 2 | −3.85 | −5.70 | −4.53 |
| 4172 | MCM3 | Minichromosome maintenance complex component 3 | 1.63 | 1.74 | 1.94 |
| 4175 | MCM6 | Minichromosome maintenance complex component 6 | −1.98 | −4.69 | −3.30 |
| 4176 | MCM7 | Minichromosome maintenance complex component 7 | −1.84 | −3.20 | −3.77 |
| 5111 | PCNA | Proliferating cell nuclear antigen | −5.50 | −23.10 | −15.83 |
| 5347 | PLK1 | Polo-like kinase 1 (Drosophila) | −1.69 | −13.68 | −8.23 |
| 5591 | PRKDC | Protein kinase, DNA-activated, catalytic polypeptide | 1.69 | −1.18 | −1.48 |
| 9232 | PTTG1 | Pituitary tumor-transforming 1 | −1.35 | −11.31 | −7.29 |
| 5934 | RBL2 | Retinoblastoma-like 2 (p130) | 3.01 | 3.80 | 3.75 |
| 2810 | SFN | Stratifin | −1.11 | 3.30 | 3.21 |
| 6500 | SKP1 | S-phase kinase-associated protein 1 | −1.06 | −2.33 | −2.96 |
| 6502 | SKP2 | S-phase kinase-associated protein 2 (p45) | 2.33 | 2.63 | 2.30 |
| 7027 | TFDP1 | Transcription factor Dp-1 | −1.12 | −8.88 | −3.71 |
A detailed list with all treatment conditions and P-values is provided in the supporting information.
Using a pathway modelling approach, the majority of highly induced genes shared by all cell lines was found to be either directly involved in the signalling network of ER stress and the unfolded protein response (UPR) or downstream of it (Figure 5). Many of them are induced by ATF4 and resemble pro-survival genes of the UPR involved in amino acid synthesis (ASNS), protein folding (HSPA5), growth control (VEGFA, AREG, GDF15) or transcription factors like DDIT3 and ATF3 controlling cell cycle and apoptosis. Indeed, DDIT3 (also known as CHOP or GADD153) was found to be one of the most strongly induced genes upon nemorosone treatment. It acts as a signal integrator of all three ER stress sensors and controls the expression of BCL2 family member genes, initiating apoptosis if ER stress can not be resolved (Figure 5). Thus, DDIT3 seems to be an important mediator of nemorosone-induced cytotoxicity and cell death. The array-based gene expression data were validated by performing quantitative RT-PCR on 10 selected genes yielding approximately 80 data points with a correlation coefficient of 0.89 between microarray and quantitative PCR expression results (Figure S2).
Figure 5.
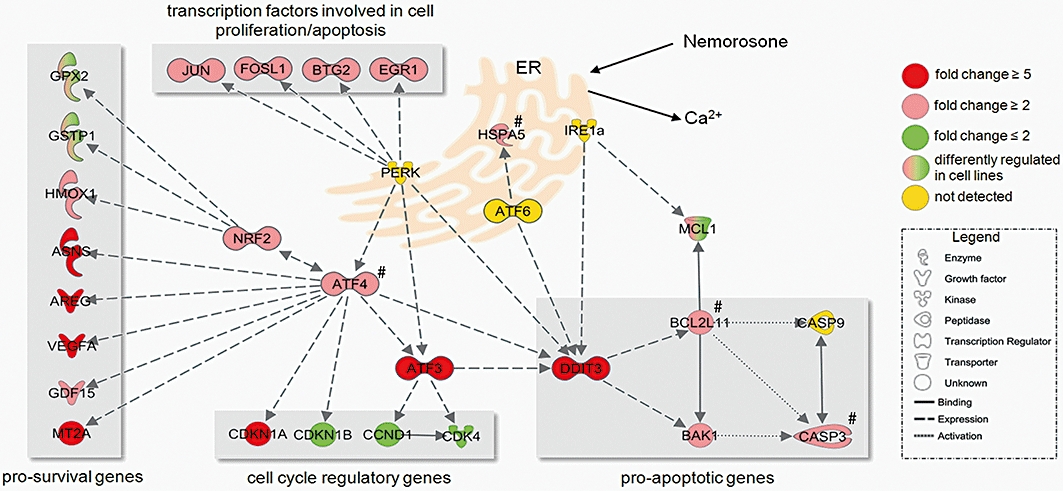
Resulting endoplasmic reticulum (ER) stress and unfolded protein response (UPR) gene network from a modelling approach using the set of differentially regulated genes common to all pancreatic cancer cell lines after nemorosone treatment. The genes showing strongest regulation (DDIT3, ATF3, CDKN1A, AREG, ASNS, VEGFA) were used as starting points for expanding the gene regulatory network in Ingenuity Pathway Analysis software using a knowledge-based algorithm applied to all 336 genes regulated by nemorosone treatment. The gene expression fold changes subsequent to the hypothetical nemorosone-induced calcium release from the ER are depicted. Red genes are up-regulated, green genes are down-regulated. Nemorosone-induced ER stress is sensed by three ER-resident proteins (PERK, ATF6, IRE1α), thus inducing downstream transcription factors ATF4, ATF3 and DDIT3 mainly involved in cell cycle regulation and apoptosis. If ER stress cannot be resolved through induction of pro-survival genes and growth factors, DDIT3 induced pro-apoptotic BCL2 family members like BAK1 and BCL2L11, thus initiating the caspase cascade. Expression levels of genes marked with # were inferred by quantitative PCR rather than microarray measurements (Figure 6C). GO, gene ontology.
Silencing of DDIT3 partially inhibits nemorosone-induced UPR and cell death
In order to test our hypothesis that nemorosone-induced ER stress causes DDIT3 up-regulation and apoptosis, we silenced DDIT3 expression in MIA-PaCa-2 and Capan-1 cells using siRNA. Quantitative real-time PCR, Western blotting and immunostaining confirmed that transfection with DDIT3 siRNA resulted in only 10–20% residual mRNA expression and a complete inhibition of protein expression (Figure 6A, B and D).
Figure 6.
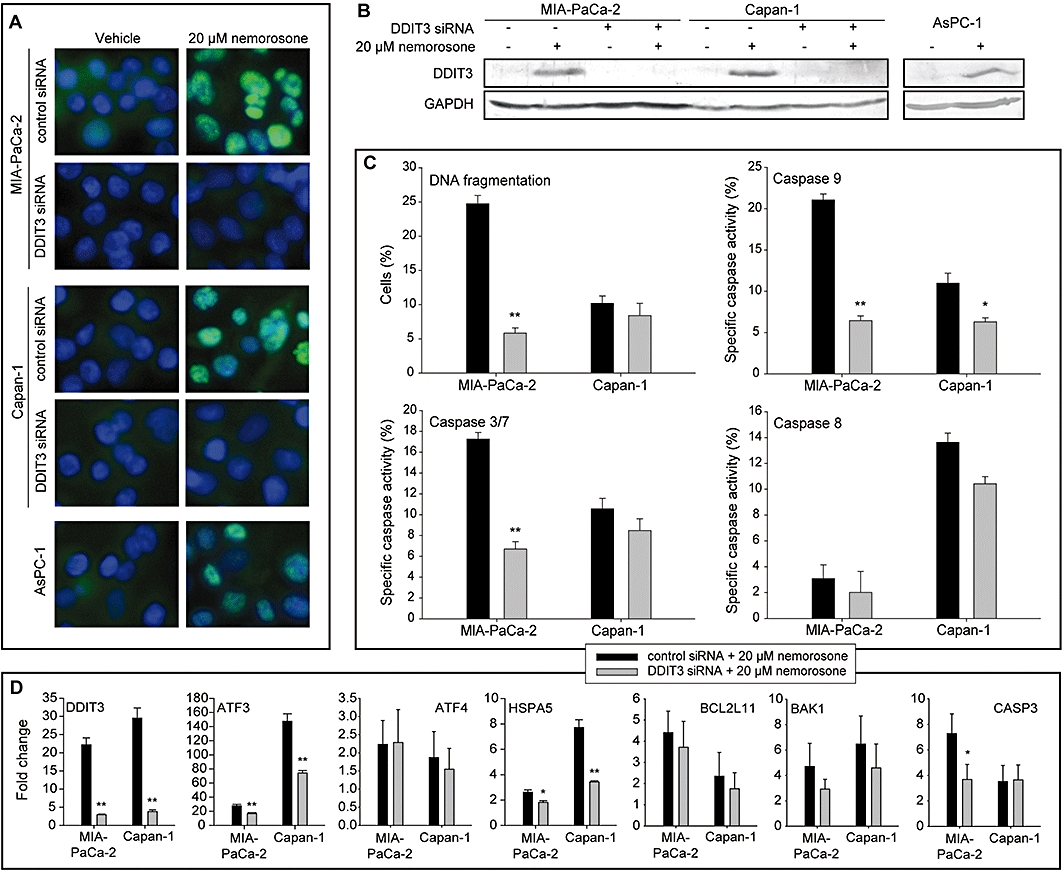
Validation of DNA damage inducible transcript 3 (DDIT3) up-regulation, knock-down and analysis of the effects of DDIT3 knock-down on apoptotic DNA fragmentation, caspase activity and expression of selected genes in pancreatic cancer cells. Cells were transfected with DDIT3 siRNA or scrambled control siRNA and treated with 20 µM nemorosone or vehicle for 24 h. (A) Immunostaining of DDIT3 in control and silenced cells. DDIT3 was labelled with an Alexa-488-conjugated secondary antibody detecting a specific primary antibody directed against DDIT3 protein (green). Nuclei were stained with Hoechst 33342 (blue). (B) Western blot confirming up-regulation of DDIT3 protein in all pancreatic cancer cell lines and DDIT3 knock-down in MIA-PaCa-2 and Capan-1 cells. (C) Analysis of apoptosis and caspase activity. Cells were assayed for subG1 DNA content (DNA fragmentation) using the Nicoletti method and activity of caspases 9, 3/7 and 8. Nemorosone-specific DNA fragmentation or caspase activity is shown as mean ± SD of three independent experiments. (D) Quantitative RT-PCR. Total RNA of treated control and silenced cells was subjected to quantitative RT-PCR using specific primers for selected genes involved in the unfolded protein response pathway. The columns represent fold changes relative to the untreated (vehicle) control. Statistically significant differences are marked: *P < 0.05, **P < 0.01.
After silencing, the percentage of MIA-PaCa-2 cells showing DNA fragmentation was markedly decreased by 75% after a 48 h incubation with 20 µM nemorosone (Figure 6C). Additionally, a significantly lower percentage of cells displayed caspase 9 and 3/7 activation after 24 h, indicating a direct impact of DDIT3 silencing on initiation of apoptosis (Figure 6C). Capan-1 cells, however, did not show any signs of reduced DNA fragmentation after DDIT3 knock-down. Although caspase 9 activation was found to be significantly decreased in Capan-1 cells, caspases 3/7 and 8 still exhibited activities comparable to those in the non-silenced control.
DNA damage inducible transcript 3 silencing also resulted in slightly reduced BCL2L11 (BIM) and BAK1 expression (Figure 6D). Consistent with our caspase 3/7 assay results, DDIT3 knock-down significantly down-regulated expression of CASP3 in MIA-PaCa-2 but not in Capan-1 cells. Interestingly, the expression of ATF3 and HSPA5 was also reduced by DDIT3 silencing although it has not been demonstrated, so far that DDIT3 controls expression of ATF3 and HSPA5. None of the tested genes was found to be significantly regulated in fibroblasts after treatment with 20 µM nemorosone for 24 h (Figure S3).
Discussion and conclusion
Polycyclic polyprenylated acylphloroglucinols are a diverse class of secondary plant metabolites with a broad range of biological activities including antimicrobial, anti-inflammatory, anti-oxidant and anticancer effects (Ciochina and Grossman, 2006). Hyperforin, garcinol and nemorosone are among the best studied PPAPs and all exhibit antitumour activities in vitro or in vivo (Pan et al., 2001; Schempp et al., 2002; Dona et al., 2004; Diaz-Carballo et al., 2008a,b; Prasad et al., 2010). However, a detailed molecular mode of action with functional evidence was still missing for these compounds although large-scale expression data are available for cells treated with hyperforin and garcinol (Balasubramanyam et al., 2004; Krusekopf and Roots, 2005). Thus, our study aimed at identifying gene expression changes induced by nemorosone, building up hypotheses about its molecular mode of action and evaluating this compound for the treatment of pancreatic cancer. Microarray experiments were performed after treatment with nemorosone for 24 h in order to evaluate gene expression changes at a time point, which was also used for the assessment of growth inhibition and apoptosis. This time point additionally ensured transcriptional changes to be measured before the onset of late apoptosis presumably biasing expression profiling.
Our results indicate that nemorosone reduced cell viability of pancreatic cancer cells in the low micromolar range and induced cell cycle arrest as well as caspase-dependent apoptosis, consistent with previous studies on neuroblastoma and leukaemia cell lines (Diaz-Carballo et al., 2008a,b;). Cell proliferation and division is controlled by a sequential activation and inactivation of cyclin-dependent kinases (CDKs). Transition from G1 to S phase of the cell cycle is driven by the active cyclin D1, 2, 3/CDK4, 6 complex which phosphorylates Rb, leading to the activation and expression of E2F and its downstream targets involved in DNA synthesis (Lodish et al., 2004). G2/M transition is controlled by cdc25-mediated activation of the cyclin B/cdc2 complex unless this complex is not dissociated by GADD45α (Stark and Taylor, 2006). The growth-inhibitory effects of nemorosone are supported by our gene expression data, indicating a general repression of cell cycle progression genes like cyclins and CDKs in all phases of the cell cycle. The up-regulation of the cell cycle inhibitor CDKN1A (p21) and the repression of E2F target genes as well as cyclin D1 and CDK4 suggest a strong impact of nemorosone on G1/S phase check points. Nemorosone-induced expression of GADD45α and repression of cdc2, cdc25 and cyclin B may block progression through G2 phase. These results are congruent with previous observations of increased p21 protein expression in neuroblastoma cells and decreased cyclin and cdc2 levels in leukaemia cell lines (Diaz-Carballo et al., 2008a,b;). Effects of nemorosone on the regulation of cell cycle genes were much stronger in Capan-1 and AsPC-1 cells than in MIA-PaCa-2 cells. Thus, the differences observed between these two cohorts in the CA are most likely due to the differential regulation of cell cycle genes. Because nemorosone-induced alterations in gene expression were found to be dose-dependent, cells might simply arrest cell cycle in either phase when treated with higher nemorosone concentrations.
More detailed studies on the mechanism of action revealed that nemorosone rapidly altered intracellular processes. The mitochondrial membrane potential ΔΨm of cancer cells was almost completely inhibited already after 2 min when treated with 20 µM nemorosone. Interestingly, we also observed a concomitant increase of [Ca2+]c in cancer cells at the same concentration. Thus, it can be hypothesized that loss of ΔΨm is a direct cause of nemorosone-induced calcium release from intracellular stores. It is well established that upon increase of [Ca2+]c mitochondria take up calcium which may lead to calcium overload and subsequent loss of ΔΨm, interfering with proper mitochondrial function (Duchen, 2000; Ly et al., 2003). For example, thapsigargin, an inhibitor of the sarcoplasmic and ER calcium ATPase, SERCA, has been shown to deplete ER calcium stores and abrogate ΔΨm (Beaver and Waring, 1996). PPAPs like hyperforin and garcinol have also been demonstrated to disturb the mitochondrial membrane potential in cancer cells within 30 and 60 min after treatment respectively (Pan et al., 2001; Schempp et al., 2002). This suggests a similar mechanism of action for structurally related PPAPs. Although hyperforin exerts its antidepressant effects by regulating calcium flux through activation of non-selective cation channels in neuronal cells (Treiber et al., 2005), it is not able to release calcium from intracellular stores in pancreatic cancer cells or fibroblasts (unpublished results). Thus, our study is the first to draw a possible link between nemorosone-induced calcium release and loss of ΔΨm, and to demonstrate functional differences between nemorosone and hyperforin. Because nemorosone acted differently on mitochondria from cancer cells and fibroblasts, a general ionophoric effect on membranes can be excluded. However, due to its lipophilic character, it might easily penetrate cell and organelle membranes by diffusion, thus affecting membrane-resident proteins like ion channels.
The nature of the store from which calcium is released upon nemorosone treatment remains unclear. However, our gene expression data point towards the ER as the affected organelle. The ER plays an essential role in calcium homeostasis as well as protein folding and secretion. It harbours calcium-dependent molecular chaperones like glucose-regulated protein, 78 kDa (GRP78; also known as HSPA5), and is also involved in molecular signalling processes because it is the largest intracellular calcium store and can release sequestered calcium into the cytoplasm or directly to mitochondria (Kim et al., 2008; Pinton et al., 2008). If ER homeostasis is disturbed through alterations in calcium levels, redox state or glycosylation, unfolded proteins will accumulate and trigger an ER stress-responsive pathway known as the UPR. UPR is initiated by three ER-localized stress sensors, IRE1, PERK (with its downstream transcription factor ATF4) and ATF6, which activate pro-survival genes like chaperones (HSPA5), growth factors (VEGF, AREG) and ER-assisted degradation genes (Zhao and Ackerman, 2006). All three pathways converge on the DDIT3 transcription factor, which is one of the most highly expressed genes during ER stress (Harding et al., 2000). If stress can not be resolved, DDIT3 accumulates in the cell and initiates the pro-apoptotic branch of the UPR by disturbing the balance between anti- and pro-apoptotic BCL2 family members like BCL2L11 (BIM) and BAK1, eventually activating caspases (Kim et al., 2008).
Nemorosone treatment differentially regulated many genes involved in UPR signalling, as revealed by our gene expression data and subsequent pathway modelling. Indeed, three of the most highly expressed genes on our array were ATF4 targets DDIT3, ATF3 and CDKN1A (p21). These genes are associated with cell cycle arrest and induction of apoptosis. Among them, DDIT3 is an important mediator of UPR-triggered cell death (Oyadomari and Mori, 2004) and has been demonstrated to be up-regulated upon treatment with ER stress-inducing agents like thapsigargin and bortezomib (Lin et al., 1997; Nawrocki et al., 2005), but not upon treatment with agents not targeting the ER like staurosporine and nucleoside analogues (Wang et al., 1996; Cusinato et al., 2006). Thus, up-regulation of DDIT3 is a specific indicator for ER stress-mediated onset of apoptosis. The precise mechanism by which effector caspases are activated in the DDIT3-induced pro-apoptotic pathway is still unclear but probably involves the mitochondrial route and caspase 9 (Di Sano et al., 2006). This is consistent with the fact that siRNA-mediated knock-down of DDIT3 was able to partially inhibit nemorosone-induced caspase 9 and 3/7 activation and subsequent DNA fragmentation in MIA-PaCa-2 cells, probably through reduction of pro-apoptotic BCL2-family gene products BAK1 and BCL2L11. In Capan-1 cells, however, no signs of reduced DNA fragmentation were visible although caspase 9 activity was reduced after DDIT3 knock-down. This is probably due to the activation of caspase 8 in this cell line, but not in MIA-PaCa-2, because ER stress has also been demonstrated to trigger the extrinsic pathway of apoptosis via death receptors (Hu et al., 2006). Indeed, garcinol was shown to potentiate TRAIL-induced extrinsic apoptosis in cancer cells via up-regulation of death receptors DR4 and DR5, but did not induce DDIT3 expression (Prasad et al., 2010). Interstingly, hyperforin also did not induce members of the ER stress response like HSPA5 in HepG2 cells (Krusekopf and Roots, 2005), again emphasizing the fact that structurally very related PPAPs do not share a common mechanism of action.
Due to the hypoxic and nutrient-deprived environment, ER stress levels have been demonstrated to be elevated in solid tumours to drive pro-survival signals and subsequent angiogenesis through expression of vascular endothelial growth factors (Duffy et al., 2003; Feldman et al., 2005). Thus, it has recently been proposed that either inhibition or amplification of UPR signalling might represent a powerful anticancer strategy to prevent pro-survival signals or induce UPR-mediated apoptosis (Kim et al., 2008; Healy et al., 2009). Nemorosone seems to amplify all branches of the UPR, including pro-survival and angiogenic mediators like ATF4-induced AREG, HMOX1 and VEGF as well as pro-apoptotic signals via the extrinsic and intrinsic pathways of programmed cell death, with an eventually dominating pro-apoptotic branch. It can be hypothesized that due to their lower demand for energy, non-transformed fibroblasts are more resistant to nemorosone treatment than pancreatic cancer cells, as also demonstrated by our experiments.
In conclusion, our results demonstrate that nemorosone affects the cell cycle and UPR regulatory pathways in pancreatic cancer cells but not in fibroblasts. Nemorosone-induced calcium release seems to disturb ER homeostasis with subsequent depolarization of the mitochondrial membrane and apoptosis through activation of the UPR. Thus, it is plausible that nemorosone acts ionophorically on the ER membrane through a yet to be identified mechanism – possibly by acting on ER-resident calcium channels. Due to its lipophilicity, however, nemorosone might well act on several other cellular targets, thus interfering with membrane proteins or affecting the lipophilic core of cytosolic or nuclear proteins, for example, transcription factors (Wink, 2008). The almost global change in gene expression demonstrated in this study supports this hypothesis.
Because normal fibroblasts are largely unaffected by nemorosone, this PPAP might resemble a lead compound for the development of drugs with a novel mechanism of action directed against pancreatic cancer and other solid tumours. Until then, however, in vivo experiments demonstrating bioavailability and efficacy as well as more detailed insights on nemorosone's molecular target(s) are needed, to determine why it acts preferentially on cancer cells.
Acknowledgments
This work was financially supported by the PaCaNet project within the NGFNplus programme and a 3 year research fellowship from the Helmholtz International Graduate School for Cancer Research to F.H. The authors gratefully acknowledge technical assistance by Sven Rüffer and the genomics & proteomics, flow cytometry and light microscopy core facilities at DKFZ.
Glossary
Abbreviations
- CA
correspondence analysis
- DDIT3
DNA damage inducible transcript 3
- ER
endoplasmic reticulum
- HDF
human dermal fibroblasts
- PI
propidium iodide
- PPAP
polycyclic polyprenylated acylphloroglucinol
- UPR
unfolded protein response
Conflicts of interest
None to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Correspondence analysis of 336 differentially regulated genes and corresponding conditions of treated/untreated pancreatic cancer cells. After filtering for significantly regulated genes, a correspondence analysis plot was calculated in M-CHiPS. It simultaneously displays hybridizations (conditions, coloured symbols) and genes (grey dots). The relative position of conditions indicates their similarities or differences. Conditions clustering closely together exhibit similar gene expression patterns while conditions located opposite of the centroid (0/0) exhibit substantial differences in gene expression. Genes strongly associated with a condition are located in the same direction from the centroid. The vehicle only control conditions cluster together and are located next to the medium only control, indicating that treatment with vehicle only did not change gene expression. AsPC-1 and Capan-1 treatment conditions demonstrate a similar localization, thus exhibiting comparable gene expression patterns. Localization of treatment clusters is concentration-dependent with the largest differences observed between treated and untreated samples at 2× IC50 nemorosone. MIA-PaCa-2 conditions cluster closely together and opposite of AsPC-1 and Capan-2 conditions, indicating that NEM-induced gene expression in some respect is different from that in AsPC-1 and Capan-1 cells.
Figure S2 Correlation between microarray and quantitative RT-PCR expression results. Expression of 10 selected genes in all treatment conditions was validated using quantitative RT-PCR. Fold changes relative to medium control were plotted for microarray and qRT-PCR results.
Figure S3 Expression of selected genes from the UPR network in human dermal fibroblasts after treatment with 20 µM nemorosone for 24 h. Expression values are shown relative to the untreated (vehicle) control and represent the mean ± SD of three independent measurements.
Table S1 Expression values of 336 genes found to be differentially regulated by nemorosone at least in one condition
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004;279:33716–33726. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- Bauer A, Kleeff J, Bier M, Wirtz M, Kayed H, Esposito I, et al. Identification of malignancy factors by analyzing cystic tumors of the pancreas. Pancreatology. 2008;9:34–44. doi: 10.1159/000178873. [DOI] [PubMed] [Google Scholar]
- Beaver JP, Waring P. Thapsigargin induces mitochondrial dysfunction and apoptosis in the mastocytoma P815 cell line and in mouse thymocytes. Cell Death Differ. 1996;3:415–424. [PubMed] [Google Scholar]
- Beissbarth T, Fellenberg K, Brors B, Arribas-Prat R, Boer J, Hauser NC, et al. Processing and quality control of DNA array hybridization data. Bioinformatics. 2000;16:1014–1022. doi: 10.1093/bioinformatics/16.11.1014. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Ciochina R, Grossman RB. Polycyclic polyprenylated acylphloroglucinols. Chem Rev. 2006;106:3963–3986. doi: 10.1021/cr0500582. [DOI] [PubMed] [Google Scholar]
- Cleveland WS. LOWESS: a program for smoothing scatterplots by robust locally weighted regression. Am Stat. 1981;35:54. [Google Scholar]
- Cuesta-Rubio O, Velez-Castro H, Frontana-Uribe BA, Cardenas J. Nemorosone, the major constituent of floral resins of Clusia rosea. Phytochemistry. 2001;57:279–283. doi: 10.1016/s0031-9422(00)00510-0. [DOI] [PubMed] [Google Scholar]
- Cuesta-Rubio O, Frontana-Uribe BA, Ramirez-Apan T, Cardenas J. Polyisoprenylated benzophenones in Cuban propolis; biological activity of nemorosone. Z Naturforsch C. 2002;57:372–378. doi: 10.1515/znc-2002-3-429. [DOI] [PubMed] [Google Scholar]
- Cusinato F, Pighin I, Luciani S, Trevisi L. Synergism between staurosporine and drugs inducing endoplasmic reticulum stress. Biochem Pharmacol. 2006;71:1562–1569. doi: 10.1016/j.bcp.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M, Cecconi F. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J Biol Chem. 2006;281:2693–2700. doi: 10.1074/jbc.M509110200. [DOI] [PubMed] [Google Scholar]
- Diaz-Carballo D, Seeber S, Strumberg D, Hilger RA. Novel antitumoral compound isolated from Clusia rosea. Int J Clin Pharmacol Ther. 2003;41:622–623. doi: 10.5414/cpp41622. [DOI] [PubMed] [Google Scholar]
- Diaz-Carballo D, Malak S, Bardenheuer W, Freistuehler M, Reusch HP. Cytotoxic activity of nemorosone in neuroblastoma cells. J Cell Mol Med. 2008a;12:2598–2608. doi: 10.1111/j.1582-4934.2008.00232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Carballo D, Malak S, Freistuhler M, Elmaagacli A, Bardenheuer W, Reusch HP. Nemorosone blocks proliferation and induces apoptosis in leukemia cells. Int J Clin Pharmacol Ther. 2008b;46:428–439. doi: 10.5414/cpp46428. [DOI] [PubMed] [Google Scholar]
- Dona M, Dell'Aica I, Pezzato E, Sartor L, Calabrese F, Della Barbera M, et al. Hyperforin inhibits cancer invasion and metastasis. Cancer Res. 2004;64:6225–6232. doi: 10.1158/0008-5472.CAN-04-0280. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy JP, Eibl G, Reber HA, Hines OJ. Influence of hypoxia and neoangiogenesis on the growth of pancreatic cancer. Mol Cancer. 2003;2:12. doi: 10.1186/1476-4598-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Rayes BF, Philip PA. A review of systemic therapy for advanced pancreatic cancer. Clin Adv Hematol Oncol. 2003;1:430–434. [PubMed] [Google Scholar]
- Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005;3:597–605. doi: 10.1158/1541-7786.MCR-05-0221. [DOI] [PubMed] [Google Scholar]
- Fellenberg K, Hauser NC, Brors B, Neutzner A, Hoheisel JD, Vingron M. Correspondence analysis applied to microarray data. Proc Natl Acad Sci U S A. 2001;98:10781–10786. doi: 10.1073/pnas.181597298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellenberg K, Hauser NC, Brors B, Hoheisel JD, Vingron M. Microarray data warehouse allowing for inclusion of experiment annotations in statistical analysis. Bioinformatics. 2002;18:423–433. doi: 10.1093/bioinformatics/18.3.423. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol. 2009;625:234–246. doi: 10.1016/j.ejphar.2009.06.064. [DOI] [PubMed] [Google Scholar]
- Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Keleg S, Buchler P, Ludwig R, Buchler MW, Friess H. Invasion and metastasis in pancreatic cancer. Mol Cancer. 2003;2:14. doi: 10.1186/1476-4598-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Krusekopf S, Roots I. St. John's wort and its constituent hyperforin concordantly regulate expression of genes encoding enzymes involved in basic cellular pathways. Pharmacogenet Genomics. 2005;15:817–829. doi: 10.1097/01.fpc.0000175597.60066.3d. [DOI] [PubMed] [Google Scholar]
- Lin XS, Denmeade SR, Cisek L, Isaacs JT. Mechanism and role of growth arrest in programmed (apoptotic) death of prostatic cancer cells induced by thapsigargin. Prostate. 1997;33:201–207. doi: 10.1002/(sici)1097-0045(19971101)33:3<201::aid-pros9>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP. Regulating the eukaryotic cell cycle: cell-cycle control in mammalian cells. In: Byrd ML, editor. Molecular Cell Biology. 5th edn. New York: W. H. Freeman; 2004. pp. 881–886. [Google Scholar]
- Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8:115–128. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- Nakayama GR, Caton MC, Nova MP, Parandoosh Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J Immunol Methods. 1997;204:205–208. doi: 10.1016/s0022-1759(97)00043-4. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K, Jr, Huang P, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65:11658–11666. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Pan MH, Chang WL, Lin-Shiau SY, Ho CT, Lin JK. Induction of apoptosis by garcinol and curcumin through cytochrome c release and activation of caspases in human leukemia HL-60 cells. J Agric Food Chem. 2001;49:1464–1474. doi: 10.1021/jf001129v. [DOI] [PubMed] [Google Scholar]
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S, Ravindran J, Sung B, Pandey MK, Aggarwal BB. Garcinol potentiates TRAIL-induced apoptosis through modulation of death receptors and antiapoptotic proteins. Mol Cancer Ther. 2010;9:856–868. doi: 10.1158/1535-7163.MCT-09-1113. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1:1458–1461. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- Schempp CM, Kirkin V, Simon-Haarhaus B, Kersten A, Kiss J, Termeer CC, et al. Inhibition of tumour cell growth by hyperforin, a novel anticancer drug from St. John's wort that acts by induction of apoptosis. Oncogene. 2002;21:1242–1250. doi: 10.1038/sj.onc.1205190. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Stark GR, Taylor WR. Control of the G2/M transition. Mol Biotechnol. 2006;32:227–248. doi: 10.1385/MB:32:3:227. [DOI] [PubMed] [Google Scholar]
- Sultana A, Smith CT, Cunningham D, Starling N, Neoptolemos JP, Ghaneh P. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer. J Clin Oncol. 2007;25:2607–2615. doi: 10.1200/JCO.2006.09.2551. [DOI] [PubMed] [Google Scholar]
- Treiber K, Singer A, Henke B, Muller WE. Hyperforin activates nonselective cation channels (NSCCs) Br J Pharmacol. 2005;145:75–83. doi: 10.1038/sj.bjp.0706155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wink M. Evolutionary advantage and molecular modes of action of multi-component mixtures used in phytomedicine. Curr Drug Metab. 2008;9:996–1009. doi: 10.2174/138920008786927794. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.