

Abstract
A large amount of experimental evidence is available for the effects of magnesium ions on the structure and the stability of the DNA double helix. Less is known, however, on how these ions affect the dynamics of the molecule and the stability of each individual base pair. The present work addresses these questions by a study of the DNA duplex [dCGCAGATCTGCG]2, and its interactions with magnesium ions, using nuclear magnetic resonance (NMR) spectroscopy and proton exchange. Two-dimensional NMR experiments indicate that binding of magnesium to this DNA duplex does not affect its structure. However, even in the absence of structural changes, magnesium ions specifically affect the exchange properties of imino protons in the two GC/CG base pairs that are located in the interior of the double helix. These specific changes do not result from alterations in the rates of spontaneous opening of these base pairs. Instead, the changes most likely reflect an enhancement in the energetic propensity for spontaneous opening of the GC/CG base pairs that is induced by the binding of magnesium ions.
Keywords: proton exchange, base-pair opening, divalent metal ions, NMR spectroscopy, structural energetics
INTRODUCTION
Magnesium ions play key roles in the structure and the enzymology of DNA. As counterions, they interact with the phosphate groups, thus neutralizing the negative charges on the DNA backbone.1 The ions can also bind to specific functional groups on DNA nucleobases, and this mode of binding is finely modulated by the DNA base sequence.2-4 The sequence also determines, in large part, the changes in DNA structure resulting from binding of the ions. For example, it has been shown that Mg2+ induces the B-to-Z transition of poly(GC)5 and the bending of the helix axis for DNA sequences containing tracts of AT base pairs.6 Regardless of the base sequence, the binding of Mg2+ ions profoundly affects the physicochemical properties of the DNA. The largest effects are on the stability of the double helix. Early investigations have established that the melting temperature of the DNA increases with increasing Mg2+ concentration.7 This stabilization has been attributed to the decrease in electrostatic repulsion between phosphate groups upon binding of the positively charged Mg2+ counterions.8 Molecular dynamics simulations have augmented this picture by showing that the presence of Mg2+ also attenuates the fluctuations of the phosphate backbone and of the water molecules associated with it.9 Furthermore, high-precision ultrasonic velocity measurements have demonstrated that binding of Mg2+ to DNA also results in a dehydration effect, which depends on the structure of the Mg2+-DNA complex.10
The present work investigates the effects of Mg2+ ions upon another fundamental property of the DNA, namely, the spontaneous opening of individual base pairs. The opening of base pairs in DNA, at temperatures below the melting temperature, is a prerequisite for many key biological processes. Among these, replication of DNA into two daughter molecules and transcription of the DNA base sequence into messenger RNA both require the local opening of the double helix such that the base sequence of one strand can be used as a template. The repair of damage in DNA also relies upon opening of single base pairs: to be acted on, the damaged base must be displaced from the helical stack into a position where it is accessible to the repair machinery of the cell. The enzymes involved in these and many other processes use Mg2+ as a cofactor.11 The catalytic role of the ion in many of these reactions is, in large part, understood. However, it is not yet known if the ion itself also affects the opening of base pairs that is required for these reactions. The present work addresses this question using nuclear magnetic resonance (NMR) spectroscopy and proton exchange. The DNA investigated (Figure 1) is a dodecamer, and its base sequence is palindromic. The DNA was chosen for investigation because the resolution of its imino proton NMR resonances, on which the investigation relies, is excellent. The processes of base-pair opening in this DNA, in the absence of Mg2+ ions, have been previously characterized by our laboratory.12-14
Figure 1.

A: The base sequence and numbering of base pairs in the DNA dodecamer investigated. B: The structures of AT base pair (right) and GC base pair (left) with the imino protons highlighted.
EXPERIMENTAL METHODS
DNA samples
The DNA oligonucleotide was synthesized on a DNA synthesizer (Applied Biosystems. Model 381A) using solid-support phosphoramidite chemistry. The crude DNA was purified by reverse-phase HPLC in formate buffer at pH 7 (with a gradient of 5 to 32% acetonitrile in 39 minutes). The counterions were replaced with Na+ ions by repeated centrifugation (5 times) through Centricon YM-3 tubes (Amicon) using 0.5 M NaCl. The sample was then desalted by repeated centrifugation (8 times) with water. The DNA concentration in the NMR samples was 1.3 mM (duplex). The samples also contained 2 mM triethanolamine, which was used to determine the pH of the sample directly inside the NMR tube as we described.15 In all experiments reported in the paper the pH of the DNA samples was 8.6±0.1.
NMR experiments
The NMR experiments were performed on a Varian INOVA 500 spectrometer operating at 11.75 T. One-dimensional NMR spectra for samples in H2O were obtained using the Jump-and-Return pulse sequence.16 The proton exchange rates were measured by transfer of magnetization from water. The water proton resonance was selectively inverted using a Gaussian 180° pulse (5.8 ms). This was followed by a variable delay in which the magnetization is transferred from water protons to DNA imino protons. In order to prevent the effects of radiation damping upon the recovery of water magnetization to equilibrium a weak gradient (0.21 gauss/cm) was applied during this exchange delay. At the end of the exchange delay, a second Gaussian pulse (2.9 ms) was applied to bring the water magnetization back to the z-axis. The observation was with the Jump-and-Return pulse sequence. Twenty-four values of the exchange delay in the range from 2 to 600 ms were used in each experiment. The exchange rates were calculated from the dependence of the intensity of the imino proton resonance of interest on the exchange delay as we have described.17,18 Two-dimensional nuclear Overhauser effect spectroscopy (NOESY) spectra were obtained for a DNA sample in D2O (1.3 mM DNA duplex) at a mixing time of 150 ms.
Determination of Base-Pair Opening Parameters
The opening of individual base pairs in DNA is generally characterized from the exchange of imino protons (Figure 1) with solvent protons.18,19 In the native DNA double helix, the imino protons are not accessible to solvent due to their location in the center of the structure and to their participation in hydrogen bonds. For the exchange to occur the base pairs must open. In this opening reaction, the hydrogen bond holding the imino proton is transiently broken, and the proton is moved into a solvent accessible state where it can be transferred to acceptors present in solution.20,21 The exchange rate observed experimentally depends upon the kinetic parameters of the opening reaction as:20
| (1) |
where kop and kcl are the rates of opening and closing of the base pair, and kex,open is the rate of exchange from the open state. To determine the opening and closing rates one varies the rate of exchange from the open state, kex,open, by adding to the DNA solution increasing concentrations of a proton acceptor. In the present work the proton acceptor used was Tris-base. Increasing concentrations of Tris-base were obtained by adding to the sample small aliquots of a stock Tris solution (1 M) at pH 8.6. The concentration of Tris-base was calculated from the total Tris concentration and the pK value of Tris. The latter was determined by NMR at 15°C, by monitoring the chemical shifts of selected Tris proton resonances as a function of pH. The following values were found: pK= 8.55±0.01 in the absence of Mg2+ and Na+ ions, pK=8.40±0.02 in the presence of 200 mM Mg2+ ions, and pK=8.46±0.01 in the presence of 500 mM Na+ ions.
The rate of exchange of the imino proton from the open state of the base pair depends on the concentration of Tris-base B as:
| (2) |
where k0 is the rate of exchange from the open state in the absence of Tris, kB is the rate constant for transfer of the imino proton to Tris-base in isolated nucleotides, and α is a factor that accounts for any differences in the rate of proton transfer between isolated nucleotides and open DNA base pairs, e. g., restricted accessibility of the proton acceptor to the imino proton in the open base. Previous investigations have shown that, for Tris-base, α is close to 1.21 The rate constant kB depends on the pK values of the imino group (pKNH) and of Tris (pKB) as:22
| (3) |
where kcoll is the diffusion-controlled rate of collision between the imino group (in the open state of the base pair) and the proton acceptor (e.g., 4.6·108 M−1 s−1 for Tris-base at 15°C). The rate constants calculated based on Eq. 3 for Tris in the absence of Mg2+ ions are: 9.82·106 M−1 s−1 for the thymine imino proton and 3.07·107 M−1 s−1 for the guanine imino proton, at 15°C.
The final equation for the exchange rate is obtained by inserting Eq. 2 into Eq. 1 (with α=1):
| (4) |
Two kinetic regimes can be distinguished depending on how the rate of exchange from the open state compares with the rate of closing of the base pair. The EX2 regime occurs when the concentration of proton acceptor is low such that kex,open << kcl. In this case, the observed rate of exchange is proportional to the rate of exchange from the open state (Eq. 1), and thus to the concentration of proton acceptor B:
| (5) |
where is the equilibrium constant for opening of the base that contains the imino proton. The other regime, called EX1 regime, occurs at high concentrations of proton acceptor, when kex,open >> kcl. In this regime, the exchange occurs in each opening event and
| (6) |
RESULTS
The effects of Mg2+ ions upon the structure of the DNA dodecamer were investigated in two-dimensional NOESY experiments. Figure 2 illustrates the NOESY spectra in the absence and in the presence of Mg2+ ions. It is readily seen that the spectral positions of the cross-peaks in the two spectra are similar. The largest changes are observed for the terminal C1 base, namely: upon adding 200 mM Mg2+, C1-H8 resonance shifts from 7.61 to 7.48 ppm, C1-H2′ resonance shifts from 1.96 to 1.84 ppm, and C1-H2″ resonance shifts from 2.37 to 2.29 ppm. A full comparison of the spectral positions of NOESY cross-peaks in the absence and in the presence of Mg2+ is shown in Figure 3. The figure contains 63 resolved cross-peaks corresponding to NOESY intra- and inter-nucleotide connectivities between H8, H6, H5, and CH3 protons on the nucleobases, between nucleobase protons and deoxyribose H1′, H2′, H2″, H3′ and H4′ protons, and between deoxyribose H1′ and H2′/H2″ protons. The chemical shifts in the presence of 200 mM Mg2+ are within 0.15 ppm from those in the absence of Mg2+. These results clearly indicate that Mg2+ ions do not induce significant changes in the structure of the DNA dodecamer.
Figure 2.

Selected region of the NOESY spectra of the DNA dodecamer in 50 mM Tris buffer in D2O at pH 8.5 and at 15°C. The left spectrum is in the absence of Mg2+ and the right spectrum is in the presence of 200 mM Mg2+ ions. The cross-peaks are NOESY connectivities between H8/H6 protons on nucleobases (F1 dimension) and deoxyribose H2′/H2″ protons (F2 dimension).
Figure 3.
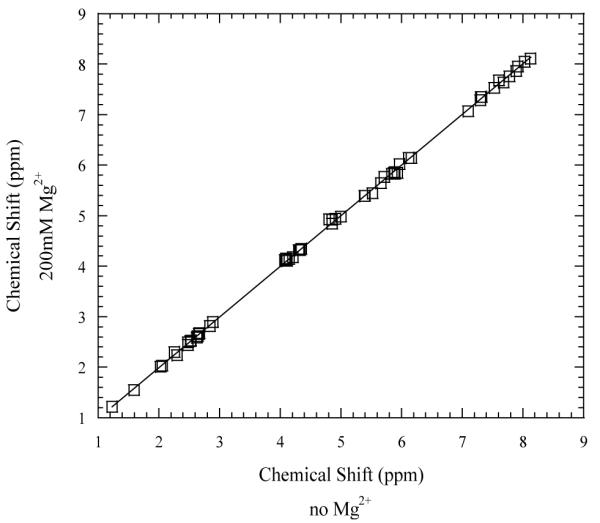
Comparison of the chemical shifts of 63 NOESY cross peaks in the absence and in the presence of 200 mM Mg2+ ions. The data were fitted with a straight line with the slope 1.002±0.002.
To investigate the effects of Mg2+ ions on base-pair opening we used the NMR resonances of the imino protons (i.e., H1 in guanine and H3 in thymine, Figure 1). These resonances are shown in Figure 4. Since the base sequence of the DNA is palindromic six imino proton resonances are expected. However, the resonance from the terminal CG1 base pairs is not observed because, at 15°C, it is broadened by fraying at the ends of the helix.
Figure 4.
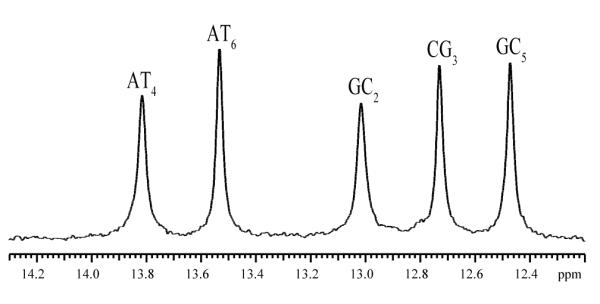
The NMR resonances of the imino protons of the DNA dodecamer in 50 mM Tris with 2 mM triethanolamine, in 90%H2O/10%D2O at pH 8.6 and at 15°C. The assignments of the resonances to individual protons were previously obtained by this laboratory.12
The exchange of imino protons in the dodecamer has been previously characterized by our laboratory, in the absence of Mg2+ ions.12,14 A summary of our previous results at 15°C is given in Figure 5. For the imino protons in AT6, CG3 and GC5 the dependence of the exchange rates on Tris-base concentration is that predicted by Eq. 4 and, at high concentrations of Tris-base, the exchange approaches the limiting EX1 regime. The opening rates and equilibrium constants, obtained from fits to Eq. 4, are shown in the figure for each of these three base pairs. For the AT4 base pair, the exchange rate of the imino proton at 0.2 M Tris-base (~ 80 s−1) is the highest rate that can be measured accurately by NMR using transfer of magnetization from water.18 Due to this technical limitation we were unable to measure the exchange rates of this proton at Tris-base concentrations exceeding 0.2 M. The exchange rates of the GC2 imino proton at Tris-base concentrations higher than 0.1 M (not shown) are also increased beyond the range suitable for NMR measurement due to fraying at the ends of the helix.
Figure 5.

Dependence of imino proton exchange rates on the concentration of Tris-base in the DNA dodecamer in the absence of Mg2+ ions, in 90%H2O/10%D2O at pH 8.6 and at 15°C.14 The curves are non-linear least-squares fits to Eq. 4. The ranges of Tris-base concentrations investigated in the present work (EX2 and EX1 regimes, respectively) are shaded.
To characterize the effects of Mg2+ ions upon the opening of individual base pairs in the DNA dodecamer we have selected for investigation the two regimes of imino proton exchange discussed in the Materials and Methods section. The rationale for this choice is two-fold. In the EX2 regime, the equilibrium constant for the opening reaction, Kop, can be directly obtained from the linear dependence of the exchange rate on proton acceptor concentration (Eq. 5). In the DNA dodecamer, the EX2 regime is observed at Tris-base concentrations less than 0.15 M (shown shaded in Figure 5 and expanded in Figure 6). The EX1 regime is of interest because the exchange rate in these conditions provides the rate of opening of the base pair (Eq. 6). This regime is reached asymptotically at high Tris-base concentrations. Since these high concentrations cannot be readily reached experimentally, we have chosen as an approximation of the EX1 regime the exchange rates at the highest Tris-base concentration investigated, namely, 1 M (shown shaded in Figure 5).
Figure 6.

Dependence of imino proton exchange rates on the concentration of Tris-base in the EX2 regime, in the absence of Mg2+ ions (×), in the presence of 200 mM Mg2+ ions (■ ) and in the presence of 500 mM Na+ ions (□ ). For CG3 and GC5 in the presence of 200 mM Mg2+ the curves are non-linear least-squares fits to Eq. 4. For all other exchange rates the lines are least-squares fits to Eq. 5.
The effects of Mg2+ ions upon the exchange of imino protons in the EX2 regime are shown in Figure 6. For both, AT4 and AT6 base pairs the exchange rates in the presence of 200 mM Mg2+ ions decrease such that the slope of the linear dependence of the exchange rate on Tris-base concentration becomes smaller. In contrast, for the CG3 and GC5 base pairs, Mg2+ ions increase the exchange rates of imino protons. Furthermore, the shape of the exchange curve changes such that the dependence of the exchange rate on Tris-base concentration is no longer linear over the range of Tris-base concentrations up to 0.15 M. Instead, due to the increases in the exchange rates, the EX2 regime is now restricted to Tris-base concentrations less than ~ 0.01M.
The effects of Mg2+ ions upon the exchange of imino protons of CG3, GC5 and AT6 at 1M Tris-base concentration (i.e., at or near the EX1 regime) are shown in Figure 7. The results clearly show that, for the CG3 and GC5 imino protons, the exchange rates do not change significantly upon increasing the concentration of Mg2+ ions up to 200 mM. A decrease in the exchange rate (~ 10 s−1) is observed for the AT6 imino proton.
Figure 7.

Dependence of imino proton exchange rates on the concentration of Mg2+ ions (lower x-axis and ■ ) and Na+ ions (upper x-axis and □ ) in the EX1 regime (i.e., 1M Tris-base concentration).
In order to determine if the effects shown in Figures 6 and 7 are specific for Mg2+ ions we have also carried out a parallel investigation of the effects of Na+ ions upon the imino proton exchange in the dodecamer. The concentration of Na+ ions chosen for this investigation (i.e., 500 mM) provides an ionic strength that is comparable to that from 200 mM Mg2+. The results show that Na+ ions have no effect upon the exchange of CG3 and GC5 imino protons in the EX2 regime (Figure 6) or in the EX1 regime (Figure 7). For the AT4 and AT6 base pairs, the effects of Na+ upon imino proton exchange in the EX2 regime are in the same direction as those of Mg2+ ions, but they are smaller. Similarly, in the EX1 regime, the effect of Na+ upon the exchange of AT6 imino proton parallels that of Mg2+ ions (Figure 7).
DISCUSSION
The changes in the exchange properties of imino protons that we observed in the presence of Mg2+ ions (Figure 6) can originate from an effect of Mg2+ ions on the stability of DNA base pairs. A change in base pair stability alters the equilibrium constant for opening of the base pair, Kop and thus, the exchange rate of the imino proton (Eqs. 4 and 5). To define the magnitude of this effect the exchange rates were fitted to Eq. 5. The equilibrium constants were obtained from the slopes of these linear fits (with kB= 9.82·106 M−1 s−1 for thymine and kB=3.07·107 M−1 s−1 for guanine). For the CG3 and GC5 base pairs in the presence of Mg2+ ions, the exchange rates were fitted to Eq. 4. The opening rates were assumed to be the same as those in the absence of Mg2+ ions. This assumption is justified by our observation that, in (or close to) the EX1 regime (Figure 7), the exchange rates of guanine imino protons are constant at increasing Mg2+ concentrations. Hence, it is likely that Mg2+ does not affect the opening rates of the two base pairs. The equilibrium constants were calculated as Kop = kop /kcl, using the closing rates obtained from the fits.
The results for the calculated opening equilibrium constants are summarized in Table 1. For the AT base pairs, the equilibrium constants in the presence of 200 mM Mg2+ ions () are smaller than those in the absence of the ions (), namely, . Decreases in the equilibrium constants are also observed in the presence of 500 mM Na+ ions, namely, . Hence, the Mg2+-induced stabilization of the AT base pairs is a non-specific effect due, in large part, to the higher ionic strength of the solution in the presence of the ions. For the GC/CG base pairs, the effects of Mg2+ are in opposite direction and are much larger than those for AT base pairs. For CG3, the opening equilibrium constant in the presence of Mg2+ is ~3-fold larger than that in the absence of the ions. Similarly, for GC5, the equilibrium constant in the presence of Mg2+ is ~8-fold larger than that in the absence of the ions. By comparison, Na+ ions decrease the opening equilibrium constants of these two base pairs and the effect is much smaller. Therefore, for the CG3 and GC5 base pairs, the effects of Mg2+ are specific to these ions. An insight into the origin of these Mg2+-induced changes in the opening equilibrium constants for CG3 and GC5 is provided by our results in the EX1 regime. As discussed above, the results show that Mg2+ does not affect the opening rates of the two base pairs. Hence, the increases in the equilibrium constants cannot originate from increases in the opening rates. Instead, it is likely that the opening equilibrium constants in the presence of Mg2+ are larger because the rates of closing of the two base pairs are smaller.
Table 1.
Equilibrium constants for opening of individual base pairs in the DNA dodecamer in the absence of Na+ and Mg2+ ions (), in the presence of 200 mM Mg2+ ions (), and in the presence of 500 mM Na+ ions ().
| Base Pair | |||
|---|---|---|---|
| CG3 | (7.5±0.8)×10−7 | (2.5±0.5)×10−6 | (5.9±0.1)×10−7 |
| AT4 | (6.34±0.03)×10−5 | (3.88±0.06)×10−5 | (5.00±0.04)×10−5 |
| GC5 | (2.3±0.2)×10−7 | (1.8±0.3)×10−6 | (1.49±0.07)×10−7 |
| AT6 | (1.54±0.04)×10−5 | (9.7±0.2)×10−6 | (1.050±0.006)×10−5 |
The rate of closing of a DNA base pair is related to the equilibrium constant for opening by the equation:
| (7) |
In the absence of Mg2+ ions, the rates of closing for CG3 and GC5 are 2.2·107 and 1.7·107 s−1, respectively (Figure 5). Our present results suggest that Mg2+ ions decrease the closing rate for CG3 ~3-fold, to 6.6·106 s−1. Similarly, the closing rate for GC5 is decreased ~8-fold, to 2.2·10 s−1. The mechanisms responsible for these Mg2+-induced effects are not yet understood. Nevertheless, since the observed effects are specific for GC/CG base pairs, the mechanisms most likely involve the interaction of Mg2+ ions with guanine. In B-DNA, Mg2+ ions bind preferentially to guanines.8,23 High-resolution crystallographic structures of DNA have shown that the hexahydrated Mg2+ ion is coordinated to guanines in the outer sphere mode, via water molecules in its primary hydration shell.2,4,24 The bound ion is stabilized by an extensive network of hydrogen bonds from water molecules to the N7 and O6 atoms of guanine as well as to neighboring bases and phosphate groups.2,4,24 The base-pair opening reaction clearly perturbs this network of hydrogen bonds. In the closing reaction, the correct network of hydrogen bonds at the bound ion must be restored. Formation of these hydrogen bonds may require a longer time, thus explaining why, in the presence of the ions, the closing reaction is slower than that in the absence of the ions.
The changes in the exchange properties of imino protons that we observed may also result from a change in the rate constant kB for the transfer of the imino proton to Tris-base, in the presence of Mg2+ ions (Eqs. 4 and 5). Quantum mechanical calculations suggest that direct, inner sphere coordination of Mg2+ ion to the guanine N7 should increase the acidity of the imino group.25 In turn, such an increase in acidity would facilitate the exchange of the imino protons. To analyze this effect we have fitted the exchange rates for CG3 and GC5 in the presence of Mg2+ to Eq. 4, while keeping the opening and closing rates the same as those in the absence of Mg2+. The kB values obtained from the fits are: (3.8±0.7)·108 M−1s−1 for CG3 and (6±1)·108 M−1s−1 for GC5. These values are comparable to the rate of collision between the imino group and Tris-base, e. g., 4.6·108 M−1s−1 in the absence of Mg2+ ions at 15°C.22 Hence, to account for our results, the rate constants kB should increase into the diffusion-controlled limit. Such large increases would require substantial decreases in the pK value of the guanine imino group (Eq. 3). This fact is illustrated in Figure 8. One can readily see that the rate constant kB reaches, for example, 90% of the collision rate when the pK value of guanine decreases by more than 2 pH units, namely from 9.46 to 7.2. At present, no experimental evidence is available to support such changes in the guanine pK value upon Mg2+ binding. Furthermore, it is unlikely that the outer sphere coordination of Mg2+, which is generally observed in B-DNA structures, could result in such a large change in the guanine pK. One should also note that this mechanism requires that the Mg2+ ions induce a change in the pK in the open state of the base pair, which is the state from which the exchange occurs. This is unlikely since, as discussed above, the base opening reaction is expected to weaken the binding of Mg2+ to the guanine. Taken together, these arguments thus suggest that the effects of Mg2+ upon the rate constant for proton transfer, kB, are probably small, and can not account for the Mg2+-induced changes in the exchange of guanine imino protons that we observed.
Figure 8.
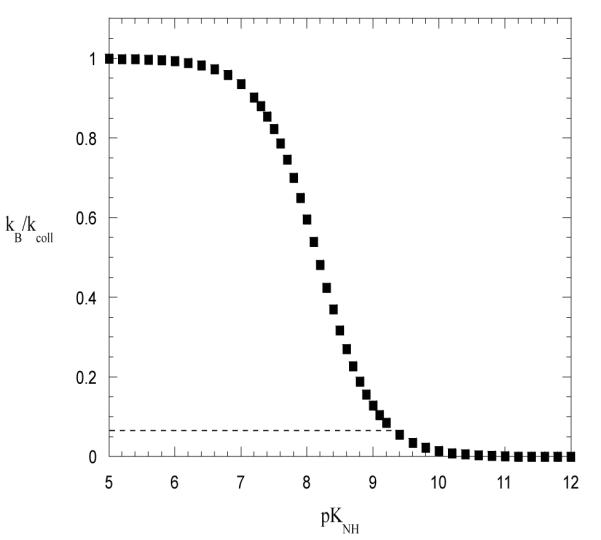
Dependence of the ratio kB /kcoll on the pK value of the imino group. The curve was obtained based on Eq. 3 using the pK value of Tris that we measured by NMR (pK = 8.40) and the pK value of the imino group in guanine (pK=9.46).26 The horizontal line indicates the value of the kB /kcoll ratio for the guanine imino proton in the absence of Mg2+ ions.
CONCLUSIONS
In the present work we have shown that Mg2+ ions specifically affect the exchange properties of the guanine imino protons in DNA. The effects are most likely due to a thermodynamic enhancement of the spontaneous opening of GC/CG base pairs upon binding of Mg2+. This enhancement is not accompanied by an increase in the rates of base-pair opening. Instead, our results suggest that Mg2+ ions slow down the closing reactions, and thus increase the lifetimes of the GC/CG base pairs in the open, extra-helical state. These changes point to a possible role of Mg2+ ions in modulating the accessibility of individual guanine bases to enzymes that act upon the open states of the DNA double helix.
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Institutes of Health (GM65159). A.E.E. was supported by a training grant from the National Institutes of Health (GM08271).
REFERENCES
- (1).Manning GS. Q. Rev. Biophys. 1978;11:179. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- (2).Chiu TK, Dickerson RE. J. Mol. Biol. 2000;301:915. doi: 10.1006/jmbi.2000.4012. [DOI] [PubMed] [Google Scholar]
- (3).Hud NV, Polak M. Curr. Opin. Struct. Biol. 2001;11:293. doi: 10.1016/s0959-440x(00)00205-0. [DOI] [PubMed] [Google Scholar]
- (4).Egli M. Chem. Biol. 2002;9:277. doi: 10.1016/s1074-5521(02)00116-3. [DOI] [PubMed] [Google Scholar]
- (5).Behe M, Felsenfeld G. Proc. Natl. Acad. Sci. U.S.A. 1981;78:1619. doi: 10.1073/pnas.78.3.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Diekmann S, Wang JC. J. Mol. Biol. 1985;186:1. doi: 10.1016/0022-2836(85)90251-7. [DOI] [PubMed] [Google Scholar]
- (7).Eichhorn GL, Shin YA. J. Am. Chem. Soc. 1968;90:7323. doi: 10.1021/ja01028a024. [DOI] [PubMed] [Google Scholar]
- (8).Cowan JA, editor. The Biological Chemistry of Magnesium. VCH Publishers, Inc.; New York, NY: 1995. [Google Scholar]
- (9).MacKerell AD., Jr. J. Phys. Chem. B. 1997;101:646. [Google Scholar]
- (10).Buckin AA, Kankiya BI, Rentzeperis D, Marky LA. J. Am. Chem. Soc. 1994;116:9423. [Google Scholar]
- (11).Sreedhara A, Cowan JA. BioMetals. 2002;15:211. doi: 10.1023/a:1016070614042. [DOI] [PubMed] [Google Scholar]
- (12).Folta-Stogniew EJ, Russu IM. Biochemistry. 1994;33:11016. doi: 10.1021/bi00202a022. [DOI] [PubMed] [Google Scholar]
- (13).Folta-Stogniew E, Russu IM. Biochemistry. 1996;35:8439. doi: 10.1021/bi952932z. [DOI] [PubMed] [Google Scholar]
- (14).Every AE, Russu IM. Biopolymers. 2007;87:165. doi: 10.1002/bip.20811. [DOI] [PubMed] [Google Scholar]
- (15).Chen C, Russu IM. Biophys. J. 2004;87:1. doi: 10.1529/biophysj.104.045179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Plateau P, Gueron M. J. Am. Chem. Soc. 1982;104:7310. [Google Scholar]
- (17).Mihailescu MR, Russu IM. Proc. Natl. Acad. Sci. USA. 2001;98:3773. doi: 10.1073/pnas.071493598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Russu IM. Methods Enzymol. 2004;379:152. doi: 10.1016/S0076-6879(04)79009-6. [DOI] [PubMed] [Google Scholar]
- (19).Gueron M, Leroy J-L. Methods Enzymol. 1995;261:383. doi: 10.1016/s0076-6879(95)61018-9. [DOI] [PubMed] [Google Scholar]
- (20).Englander SW, Kallenbach NR. Q. Rev. Biophys. 1984;16:521. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- (21).Gueron M, Charretier E, Hagerhorst J, Kochoyan M, Leroy JL, Moraillon A. In: Structure & Methods. Sarma R, Sarma MH, editors. Vol. 3. Adenine Press; NY: 1990. p. 113. [Google Scholar]
- (22).Benight AS, Schurr JM, Flynn PF, Reid BR, Wemmer DE. J. Mol. Biol. 1988;200:377. doi: 10.1016/0022-2836(88)90248-3. [DOI] [PubMed] [Google Scholar]
- (23).Cowan JA, Huang H-W, Hsu L-Y. J. Inorg. Biochem. 1993;52:121. doi: 10.1016/0162-0134(93)85028-7. [DOI] [PubMed] [Google Scholar]
- (24).Subirana JA, Soler-Lopez M. Annu. Rev. Biophys. Biomol. Struct. 2003;32:27. doi: 10.1146/annurev.biophys.32.110601.141726. [DOI] [PubMed] [Google Scholar]
- (25).Sponer J, Sabat M, Gorb L, Leszczynski J, Lippert B, Hobza P. J. Phys. Chem. B. 2000;104:7535. [Google Scholar]
- (26).Ts’o POP. Basic Principles in Nucleic Acid Chemistry. Vol. I. Academic Press; New York: 1974. [Google Scholar]