

Abstract
Cells respond to a variety of secreted molecules by modifying their physiology, growth patterns, and behavior. Motile bacteria and eukaryotic cells can sense extracellular chemoattractants and chemorepellents and alter their movement. In this way fibroblasts and leukocytes can find their ways to sites of injury and cancer cells can home in on sites that are releasing growth factors. Social amoebae such as Dictyostelium are chemotactic to cAMP which they secrete several hours after they have initiated development. These eukaryotic cells are known to be able to sense extremely shallow gradients but the processes underlying their exquisite sensitivity are still largely unknown. In this study we determine the responses of developed cells of Dictyostelium discoideum to stable linear gradients of cAMP of varying steepness generated in 2 μm deep gradient chambers of microfluidic devices. The gradients are generated by molecular diffusion between two 50 μm deep flow-through channels, one of which is perfused with a solution of cAMP and the other with buffer, serving as continuously replenished source and sink. These low ceiling gradient chambers constrained the cells in the vertical dimension, facilitating confocal imaging, such that subcellular localization of fluorescently tagged proteins could be followed for up to 30 minutes without noticeable phototoxicity. Chemotactic cells enter these low ceiling chambers by flattening and elongating and then move almost as rapidly as unconstrained cells. By following the localization of activated Ras (RasGTP) using a Ras Binding Domain fused to Green Fluorescent Protein (RBD-GFP), we observed the rapid appearance of membrane associated patches at the tips of pseudopods. These patches remained associated with pseudopods while they continued to extend but were rapidly disassembled when pseudopods stalled and the cell moved past them. Likewise, fluorescence associated with localized RasGTP rapidly disappeared when the gradient was turned off. Correlation of the size and persistence of RasGTP patches with extension of pseudopods may set the rules for understanding how the signal transduction mechanisms convert a weak external signal to a strong directional bias.
Introduction
Eukaryotic cells signal each other and respond to a wide variety of metabolites and peptides. For instance, yeast cells respond to peptide mating factors by polarizing in the direction of the source to facilitate fusion with cells of the opposite mating type. 1,2 Likewise, embryogenesis of many metazoans is orchestrated by the localized release of morphogenetic factors from a subset of cells which determine the transcriptional profile and shape of adjacent cells.3,4 Motile cells such as fibroblasts, leukocytes and cancer cells can sense shallow gradients of peptides and respond by moving toward sites of injury or infection.5,6,7 One of the most dramatic responses can be seen in social amoebae as they move chemotactically to form aggregates of thousands of cells. In the well-studied social amoeba Dictyostelium discodieum it is known that the chemoattractant cAMP is recognized by the G-protein linked receptor, CAR1, and the signal is transduced within the cells to regulate the cytoskeleton such that the cells move towards regions of higher level of camp.8,9,10,11 Under optimal conditions the cells will move their own length every minute. They also respond by synthesizing and secreting more cAMP thereby relaying the signal as a non-dissipating wave resulting in complex motile behaviors that can extend for several centimeters on a moist surface.11,12,13,14,15 To quantitatively study directional sensing, polarity and motility in this model chemotactic organism, we have built several microfluidic devices that generate stable gradients with known fields of cAMP concentration.16,17 In all these devices, chemoattractant gradients were imbedded in a stream of a buffer continuously flowing over cells adhered to a substrate. Therefore, chemotactic cells in these gradients did not significantly affect the local concentration, either by release of cAMP or degradation of the gradient by the action of secreted cAMP phosphodiesterase. Such devices may also be useful for the study of polarized responses of embryonic cells, leukocytes or fibroblasts.
The gradient channel in our initial microfluidic devices was 525 μm wide, 50 μm high and 5 mm long.17 These devices established a stable linear gradient of cAMP across the stream. Chemotaxis was observed when the gradient, ΔC/Δx, was steeper than 10−3 nM/μm and was optimal at ΔC/Δx between 0.01 nM/μm and 0.1nM/μm, in excellent agreement with previous measurements under stationary conditions.18,19,20 These results established that developed Dictyostelium cells can respond to a stable spatial gradient by persistent chemotaxis. We calculated that in the middle of a 0–100 nM gradient only about 375 more receptors were occupied in the front half of the cell than in the back half.17 However, this number changes as the cells traverse the channel, since the concentration of cAMP at the low concentration end of the gradient is negligible while it is up to 100 nM at the high concentration end. As a result, the relative concentration difference across a cell, ΔC/Clocal, depends on where it is in the gradient. To overcome this problem we shifted to using microfluidic devices that generate exponential gradients such that cells are exposed to the same relative slope over nearly the entire width of the gradient field.16
Exponential gradients were established in devices with chambers that were 800 μm wide, 30 μm high and 2 mm long by mixing input from three reservoirs containing cAMP at concentrations forming geometric sequences such as 1 nM, 2nM and 4 nM or 1 nM, 4 nM and 16 nM.16 The relative concentration difference [ΔC/Clocal] under these conditions was 2.5% and 5%, respectively, across the width of a 10 μM cell. We could also control the concentration range while maintaining the values of ΔC/Clocal by multipling the concentrations in the reservoirs equally eg. 10 nM, 20 nM, and 40 nM. Using these devices we determined that cells that had developed in suspension for 5 hours with pulses of 50 nM cAMP every 6 minutes could respond to gradients that generate ΔC/Clocal as small as 1.25%, as long as the median concentration was above 5 nM.16 At these low concentrations and shallow gradients the accuracy of chemotaxis as judged from the ratio of distance moved up the gradient divided by the total distance moved (chemotactic index) was limited by stochastic variations in the number of external molecules of cAMP that bound to the finite number of receptors. At higher concentrations and steeper gradients, these variations were less important to accurate translocation of the cells than the internal limits.16 Maximal chemotactic indices observed in the exponential gradient devices reached 0.7 ± 0.1, rivaling those observed in the absence of flow.
Since amoeboid movement is driven by polymerization of actin at the anterior, we attempted to follow the internal localization of F-actin by transforming the cells with a construct that generates Green Fluorescent Protein (GFP) linked to a protein domain that binds specifically to F-actin (ΔlimE - GFP).21 Confocal fluorescent images (488 nm excitation) were captured every 5 seconds. Since pseudopods often extended in the vertical plane, we collected z-sections at 1 μm intervals and reconstructed the cell in 3D. We could clearly see increased fluorescence intensity at the anterior of extending pseudopods, both before and after switching on the gradient. There was also accumulation of ΔlimE - GFP on the membrane towards the back. However, we were only able to collect 4D information for a few minutes before the cells showed clear signs of phototoxicity induced by the fluorescence imaging.
Since several genetic lines of evidence indicate that activation of Ras by ligand bound CAR1 is an early step in gradient sensing 11,22,23,24, we also explored the localization of RasGTP using a portion of Raf1 which includes the Ras Binding Domain linked to GFP (RBD- GFP). In the absence of exogenous cAMP, only transient patches of fluorescence were seen at the cell periphery, but within 4 seconds of introducing a 5% exponential gradient, RBD-GFP accumulated at the tips of extending pseudopods. When we collected images in a single confocal plane, these patches often disappeared within a few seconds. But when we took multiple z-sections and reconstructed the cell, it became apparent that the patch had just followed the tip, which moved out of the original confocal plane. However, the necessity of taking multiple z-sections led to high phototoxicity that once again limited the time span of tracking of individual cells to only a few minutes. Therefore, to collect images over longer periods with high temporal resolution we needed to constrain cells in the vertical direction so that most of the membrane associated RBD-GFP could be imaged in a single confocal plane.
The task of flattening cells for high resolution microscopy was previously approached using “under agar” chemotactic migration.25,26 In this technique a thin layer of agar containing 50 nM cAMP with a well in the center is prepared on a cover slip. Developed cells in buffer are positioned in the well where they are exposed to the cAMP diffusing in from the sides. The cells also release cAMP phosphodiesterase which reduces the concentration in the well and sharpens the gradient. Within 10 minutes the cells migrate to the sides of the well and squeeze underneath. They will sometimes migrate outward for 10 minutes or more, but stop as the gradient degrades. Besides the problem of not having a stable and well-defined gradient, the thickness of the cell can vary day to day depending on how the agar layer is prepared. Often the cells can be seen to lift the agar, protruding upwards almost as high as unconstrained cells. Therefore, we turned to silicone rubber (PDMS) microfluidic devices with gradient chambers only 2 μm high. By chemotactically enticing cells into these shallow chambers, we found we could image “2D” cells with the high time resolution necessary to follow moving cells for 30 minutes with no signs of phototoxicity as judged by the rate of translocation.
Materials and methods
Growth and Development
Transformed AX4 cells carrying constructs in which the regulatory region of actin 15 drives genes encoding a fusion of GFP to LimE (ΔLimE-GFP) 21 or a fusion of GFP to the Ras Binding Domain 23 were grown in suspension in HL5 medium.27 Plasmid pDM115, a non-integrating vector containing a truncated version of the Ras binding domain of Raf1 tagged with GFP and driven by the actin15 promoter, was the kind gift of Peter Van Haastert. Only cultures of these strains that had mass doubling times less than 10 hrs were used since we found that slower growing cells were less chemotactically responsive. When exponentially growing cells reached 1–2 × 106 cells/ml, they were harvested by centrifugation, washed in KN2/Ca buffer (14.6 mM KH2PO4, 5.4 mM Na2HPO4, 100 μM CaCl2, pH 6.4), and resuspended in KN2/Ca at 107 cells/ml. Shaken cells were developed for 5hrs with pulses of 50nM cAMP added every 6 minutes. Most cells at this time had a length to width ratio >3 and appeared to be polarized.
Low ceiling microfluidic devices
Microfluidic devices were made of silicon elastomer PDMS (Sylgard 184) chips sealed to microscope coverslips. The channel structure was formed by soft lithography. The design was similar to one used to study gradient sensing in yeast.2 The functional region of the device consisted of two 80 μm deep, 500 μm wide flow-through channels (long vertical channels with kinks in Figure 1) and five clusters of 2 μm deep gradient chambers between the flow-through channels. The gradient chamber lengths (horizontal dimensions in Figure 1) were identical within a given cluster and were 585, 390, 240, 150, 100 μm for different clusters from top to bottom (from the most upstream to the most downstream). The depth of the gradient chambers was reduced from 5 μm used in the previous devices of Paliwal et al. to 2 μm to study amoeboid cell migration in a “2D” environment. Stable linear gradients of cAMP in the gradient chambers are generated by molecular diffusion between the two flow-through channels, which are continuously perfused with buffer with and without cAMP at a mean velocity of ~200 μm/s.
Figure 1.

The microfluidic device. (A) Layout of microchannels in the device. Buffer is introduced through “buffer inlet”, while buffer with chemoattractant is introduced through “attractant inlet”. Linear concentration profiles in 2μm deep gradient chambers (shown in blue) are established by molecular diffusion between two 500μm wide and 50μm deep flow-through channels (long vertical channels with steps). The flow-through channels are connected by clusters of gradient chambers that are 585, 390, 240, 150, and 100μm in length (horizontal dimension). Cells are loaded into the microchannels through the “cells in” port with only the “cells out” port open. Those ports are then closed and the other three ports opened to generate the gradients. (B) Concentration profile of cAMP in a gradient chamber of length L = 160μm and width 50μm (vertical dimension) with a circular cell of diameter d = 10μm in the middle. The profile was obtained from a steady-state two-dimensional numerical simulation in COMSOL. The boundary conditions were C0 = 0 nM at the left boundary (corresponding to a flow-through channel with plain buffer), C0 = 100 nM at the right boundary (flow-through channel with 100 nM solution of cAMP), and no flux through the other boundaries (solid walls of the gradient chamber). The concentration of cAMP is color-coded with the legend (in nM) shown at the bottom. (C) Concentration of cAMP along the segment marked by the white dashed line in B for the gradient chamber with an occluding cell (black curves) and for the same chamber with no cell (gray line). The difference in cAMP concentration across the cell is 12.6 nM, which is twice the difference across the same 10 μm segment in a chamber with no cell (given by ΔC = 2C0R/L = 6.3 nM).
A short distance downstream of the last gradient chamber, the two flow-through channels merge in a T-junction, where pressures in the channels are equilibrated. Equal pressures applied to the cAMP and buffer inlets (ports 1 and 2) and equal flow resistances of the microchannels connecting the inlets with the respective flow-through channels result in nearly equal flow velocities in the flow-through channels and nearly equal pressures at opposite ends of gradient chambers. Some pressure differences across the chambers may still result from imperfections of fabrication of the device. Nevertheless, because of the large ratio between the depths of the flow-through channels and perfusion chambers (80 μm: 2 μm), these unwanted differences have negligible effect on the gradients in the chambers. Indeed, at the flow velocity of ~200 μm/s, the pressure at the most upstream chamber is ~2 Pa higher than at the T-junction. Even if the cross-chamber pressure difference were 5% of this number, corresponding to very poor fabrication, the flow velocity across the chamber would be 0.05 μm/s. This number is much smaller than the characteristic velocity associated with molecular diffusion, D/L ≈ 0.67 μm/s, where D = 400 μm2/s is the diffusion coefficient of cAMP and L = 600 μm is the chamber length.
With no cells inside, the slope of the linear gradient in a chamber is determined by the concentration of cAMP in the flow-through channel, C0, divided by the length of the chamber, ΔC/x = ΔC0/L. The presence of a cell in a gradient chamber affects the diffusive flux and the concentration profile around the cell because it occludes the chamber and blocks diffusion across the cell footprint (Fig. 1B). This can be easily seen by solving the steady-state diffusion equation for a two-dimensional circular cell in a linear gradient with an infinite spatial extent and slope C0/L. The solution, in polar coordinates, can be found by using the zero-flux boundary condition at the cell perimeter and is given by . Here, R is the cell radius, r=0 is the cell’s center and the gradient is in the θ = 0 direction. Thus, the difference in concentration across a cell is equal to , which is twice that across a length equal to the cell diameter in the absence of the cell. Two-dimensional numerical simulations for circular footprints indicated that this two-fold difference in concentration across a cell is essentially independent of the diameter of the cell, its movement or its location in the gradient chamber, as long as R is substantially smaller than the width of the chamber (50 or 80 μm) (Fig. 1C).
Both flow-through channels were initially filled with buffer with no cAMP before the cells were introduced and allowed to settle onto the coverslip for 5 minutes in the absence of flow. Flow down the side channels with buffer on one side and a cAMP solution in the other then established the gradient by diffusion across the connecting chambers. Cells were imaged as they migrated across the test chambers. DIC images were taken on a spinning-disk confocal Zeiss Axio Observer inverted microscope using a 10X objective and a Roper Cascade QuantEM 512SC camera. Fluorescent images (488 nm and 561 nm excitation) were captured every 2 seconds with a 63X oil objective on the same microscope equipped with a pair of Roper Quantum 512SC cameras using filters to simultaneously collect light at 500 to 550 nm (green) as well as at 575 to 650 nm (red). Images were collected in Slidebook 5 (Intelligent Imaging Innovations, Inc.).
Cell tracking
To characterize cell movement in the 2D devices in a high-throughput manner, all gradient chambers were imaged in DIC every 15 s at 10× magnification using a motorized stage for 2 hours. To ensure that cells did not affect the gradient by their own production of cAMP, 3 mM caffeine was added to all solutions. Cells were tracked across the gradient chambers using Matlab (The MathWorks, Natick, MA 2008) and the chemotactic index and average velocity were computed for each cell. The chemotactic index (CI) was calculated by dividing the distance traveled up the gradient by the total distance traveled. Cells that came into contact with another cell were manually identified and discarded.
To study the correlation between membrane fluorescence intensity and membrane extension in chemotaxing cells at high resolution, an automated cell-tracking and analysis program was developed in Matlab and made freely available on our Web site. The fluorescence images were first segmented by an active contour method to identify the cell.28 To reduce the effects of pixelation, the pixels comprising the perimeter of the cell were next subsampled at a ratio 5:1 and the resulting pixels interpolated by a closed cubic spline to give a smooth representation of the cell membrane. The membrane was then parameterized by 100–200 nodes, which were evolved in time as a coupled spring system.29 At each frame, nodes were first initialized along the membrane to best match the spacing and number of nodes in the previous frame. Each node was connected to its two nearest neighbors, as well as its previous position, by linear springs with spring constants kn and kprev, respectively, and constrained to move on the 1-dimensional spline contour representing the membrane. The constraint was enforced by projecting the net force on a given node onto the local tangent to the spline contour. The final positions of the nodes were determined by force balance using a nonlinear least squares (Marquard-Levenberg) algorithm.
The ratio of the spring constants, kn and kprev, governed how the nodes evolved in time. Empirically we found that to keep the nodes from crossing required that the ratio of spring constants (kn/kprev) be greater than 5. When kn/kprev < 10, the nodes remained roughly stationary with respect to the substrate as the cell moved. In this case, nodes became wider spaced at protruding fronts and more densely spaced at the rear of the cell. To keep the distance between nodes within a given range, nodes were inserted when the distance between two nodes reached an upper threshold and were deleted when this distance reached a lower threshold. When kn/kprev ≫ 10, the nodes flowed with the cell and nodes were rarely inserted or deleted. We used kn/kprev = 10 in the current study.
Various quantities were measured for each node, such as intensity, curvature, and local membrane extension. Intensity was sampled by giving the membrane a specified depth through repeated erosions and assigning pixels in this region to the nearest node. The intensity at each node was the average of the pixels assigned to it. We found all our results to be insensitive to the details of the erosion and assignment. Membrane curvature was measured locally for each node by taking appropriate derivatives of the spline contour. Local membrane extension was defined as the displacement of a node from frame t to frame t+1 in the direction of the local normal to the membrane at frame t.
Patch and extension identification and correlation
RasGTP patches were identified in a given frame by first selecting all pixels with intensity greater than Icyto+3 δIcyto, where Icyto and δIcyto are the mean and standard deviations, respectively, of the intensity of the cytoplasm. These pixels were then clustered using a hierarchical clustering scheme based on the Euclidean distance between pixel positions. Clusters containing four or more pixels were identified as patches of membrane associated RasGTP. The nearest nodes to each pixel in a given patch were associated with the patch and used to correlate patches with extensions.
Extensions were identified by first selecting all nodes with local membrane extension greater than 0.1 μm/s and positive local curvature, then using hierarchical clustering on the selected nodes based on the Euclidean distance of node positions, and finally retaining clusters containing two or more nodes with at least one node having extension greater than 0.2 μm/s. The correlation between extensions and patches was calculated by associating a patch to an extension if they shared at least one common node and finding the frequency of extensions with associated patches.
Membrane associated RasGTP
To quantify membrane localization of RasGTP, cells expressing both RBD-GFP and cytosolic free RFP were simultaneously imaged for GFP and RFP in chemotaxing cells. The GFP and RFP were first aligned in Slidebook 5 via an affine transformation. The parameters of the transformation were inferred from imaging beads which were fluorescent in both channels. The RFP signal was then used to correct the GFP signal for contributions from cytosolic RBD-GFP to give only contributions from membrane RBD-GFP, as described in Bosgraaf et al., 2008. In brief, the RFP signal was multiplied by the ratio of average GFP to RFP in an inner region of the cell containing only cytosol and then the normalized RFP signal for each pixel was subtracted from the GFP signal for that pixel. Cells were also corrected for photobleaching in each channel by normalizing each image such that the total intensity of the cell stayed constant over time.
Results
Gradients in the microfluidic device
To achieve a mean flow velocity of 200 μm/s in the two flow-through channels, both inlets were pressurized at 5 KPa above the outlet by holding the level of liquids in the reservoirs connected to the inlets at 50 cm above the level of liquid in the reservoir connected to the outlet. To monitor gradient formation, we added the red fluorescent dye Alexa 594 to the cAMP solution. With continuous flow of buffer in one flow-through channel and buffer with 100 nM cAMP in the other, the cAMP gradient in the 400 μm long chamber is ΔC/Δx = 0.25 nM/μm (100 nM/400 μm), the midpoint concentration is 50 nM, and the ΔC/Clocal over a 10 μm cell at the midpoint is 5%. Shorter chambers have steeper gradients (Table 1). Gradients with lower mean concentrations were generated by using 10 nM cAMP in one flow channel and buffer in the other.
Table 1.
Motility in the different chambers. A. Gradients established between 0 nM and 100 nM. B. Gradients established between 0 nM and 10 nM. The relative gradient, ΔC/Clocal, is given in percentage at the midpoint of the chambers. ΔC is the concentration difference between the front and back of a 10 μm long cell and Clocal is the concentration at the midpoint of the chambers. Relative gradient in a given chamber is the same whether the chemoattractant solution contains 100 nM or 10 nM cAMP although the midpoint concentration differs by a factor of 10 [50 nM and 5 nM, respectively].
| A.) 0 nM to 100 nM | |||
|---|---|---|---|
| Width of chamber (μm) | Relative gradient | chemotactic index | rate of translocation (μm/min) |
| 150 | 13.2 | 0.96 ± .03 | 8.9 ± 1.9 |
| 240 | 8.0 | 0.95 ± .04 | 9.8 ± 2.1 |
| 390 | 5.0 | 0.96 ± .02 | 11 ± 2.7 |
| 585 | 3.2 | 0.93 ± .08 | 10.7 ± 2.4 |
| B.) 0 nM to 10 nM | |||
|---|---|---|---|
| Width of chamber (μm) | Relative gradient | chemotactic index | rate of translocation (μm/min) |
| 150 | 13.2 | 0.99 ± .02 | 10.3 ± 1.8 |
| 240 | 8.0 | 0.96 ± .03 | 8.9 ± 2.3 |
| 390 | 5.0 | 0.93 ± .06 | 8.8 ± 2.2 |
| 585 | 3.2 | 0.93 ± .05 | 11.0 ± 3.3 |
In order to test the effect of unintended differences in pressure between the ends of the gradient chambers, which may arise because of imperfections in the device fabrication and incomplete balance of the inlet pressures, we measured concentration profiles of Alexa 594 in a 400 μm long chamber at different inlet pressure settings. The pressure at the buffer inlet was constant at 5 kPa, and the pressure at the attractant inlet (where the Alexa 594 solution was fed) was decreased. A pressure difference of 2 kPa (40%) was required for the concentration at the center of the channel to change by 10 percent. The results of this test indicated that the concentration profiles in the gradient chambers are robust, remaining practically unchanged under substantial changes in the driving pressures, and are also likely to have high tolerance to imprecision of fabrication.
Chemotaxis of vertically constrained cells
In the absence of a cAMP gradient, cells seldom entered the cross-chambers. However, soon after a gradient was established, cells were enticed to enter the gradient chambers. When high concentrations of cAMP were used as the source (e.g. in 0–100 nM gradients), almost all cells that got within 30 microns of the entrance were quickly lured inside. However, the frequency of cells entering the gradient chambers was reduced at lower concentrations of cAMP (e.g. in 0–10 nM gradients). Using DIC microscopy with a 10X objective we could follow cells all the way across a chamber which took 5 to 60 minutes depending on the length of the chamber. Cells flattened as they entered the chambers and sometimes had difficulty in pulling their posterior in with them. Once they had fully entered, they moved rapidly (10 ± 2 μm/min) up the gradient with exceptionally high chemotactic indices irrespective of the steepness of the gradient or the local concentration over the measured range (Table 1). We previously found that unconstrained cells in a gradient move at 15 ± 2 μm/minute.16 The high accuracy of chemotactic translocation relative to a population of cells in high ceiling microfluidic devices may be partly the consequence of our tracking cells which were properly oriented to start with and occasionally guided by the walls of the chamber. We adjusted the density of cells loaded into the device such that most of the time only a single cell was present in a gradient chamber at any given time. This precluded chemotactic signaling between cells which might occur in the gradient chambers where there is no flow. We also found that we could eliminate signaling between cells at higher density by adding 3 mM caffeine, which blocks the stimulation of cAMP synthesis in Dictyostelium. Caffeine had no detectable effect on the chemotactic index or the rate of translocation in the gradient chambers.
Actin foci and cell movement
To get some idea how the cells were affected by the confined environment, we collected fluorescent images of cells expressing ΔlimE - GFP in the gradient chambers. By focusing first on the floor and then on the ceiling of the chamber it was clear that actin foci were present on both surfaces, indicating that the cells were pressed to both the ceiling and the floor over most of their area (movie S1). The top and bottom foci were seldom colocalized as would be expected if F-actin filaments stretched vertically across the cell. It has been suggested that these foci are points where internal F-actin pushes on the surroundings to generate motile force.30,31,32 The foci that we observed persisted for up to 20 seconds before disappearing. New foci would appear in this time such that the surface had 10 to 30 such localizations of F-actin at any given time (Figure 2).
Figure 2.
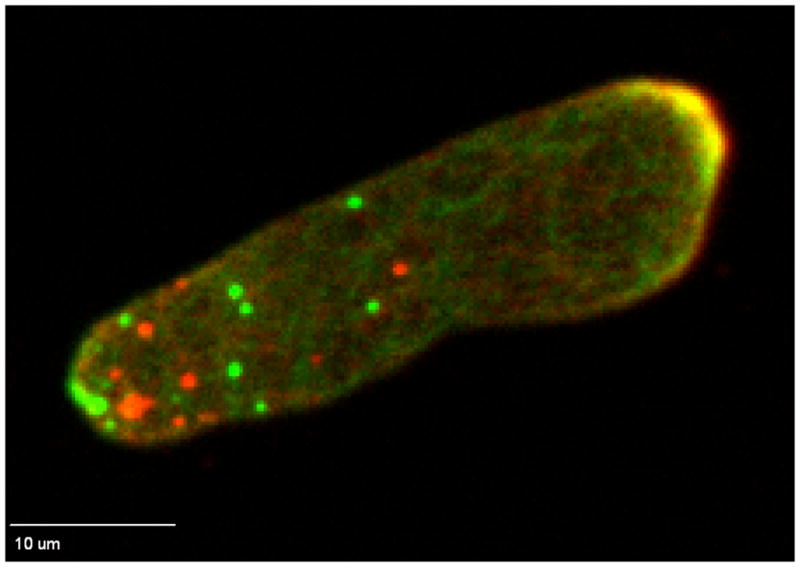
Actin foci. A cell expressing ΔlimE-GFP migrated into a low ceiling gradient chamber and moved up the gradient to the right. Fluorescence from the ΔlimE-GFP bound to F-actin was visualized either on the upper surface of the chamber (red) or on the bottom surface of the chamber (green). F-actin was seen to accumulate at the anterior as well as in foci at both the upper and lower surfaces.
Movement was driven by extending pseudopodia at regions of positive membrane curvature, though occasionally regions of negative curvature showed sudden broad expansions. Once pseudopodia stopped extending, they remained stationary with respect to the substrate as the cell moved past them and were only retracted once the cell rear approached them, as shown for a representative cell in Figure 3.
Figure 3.

Movement of pseudpods with respect to the substrate. Overlay of contours of a representative cell chemotaxing in a gradient chamber. The cell outline was plotted every 2 seconds for 10 minutes and color coded by time (time increases from green to red). Two smaller time intervals are enlarged to show in further detail that upon stalling, pseudopods remained stationary with respect to the substrate while the cell moved past them. They were retracted only when the rear came abreast.
RasGTP patches
To follow the localization of activated Ras, fluorescent images of cells carrying the RBD-GFP reporter construct were collected every 2 seconds as they crossed the gradient chambers. We found that patches of RasGTP were present at the membrane of the pseudopods and that these patches were almost always found at the apex of a pseudopod (Figure 4 and movie S2). Often, chemotactically moving cells had two or more leading pseudopods and each pseudopod had a RasGTP patch. Some of these patches persisted for several minutes while others were more short-lived.
Figure 4.
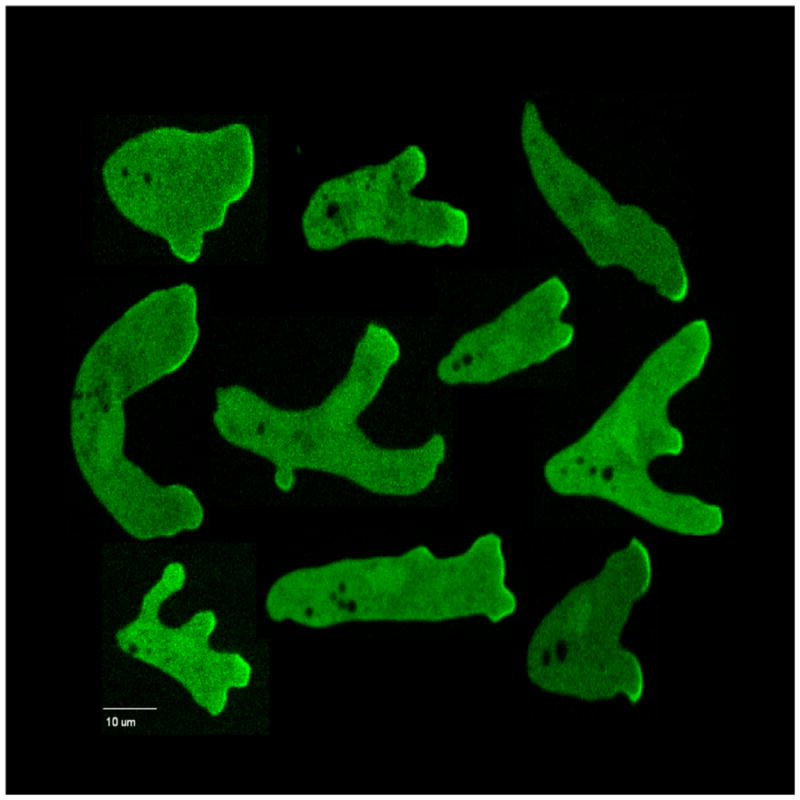
Activated Ras. A collection of images of cells expressing RBD-GFP as they migrated in the gradient chambers to showcase diversity of cell shape. Fluorescence of RBD-GFP bound to RasGTP was seen to be more intense at the leading edge of pseudopods.
The design of the microfluidic devices allowed us to rapidly replace the stream of buffer containing cAMP with a stream of buffer without cAMP by opening the “cells out” port and closing the “outlet” (Figure 1). Under these conditions the buffer flowed down the left channel and then up the right channel before exiting at the “cells out” port. When we turned the gradient off, cells that were traversing the gradient chambers rounded up or set out on random walks while the RasGTP patches disappeared within a few seconds. Cells that had their posteriors stuck at the chamber entrances, rapidly retracted their anteriors as they rounded up. This is one of the few cases where anterior retraction can be observed.
When we turned off the gradient for several minutes, cells expressing RBD-GFP had patches at random orientation. When the gradient was re-established, RasGTP accumulated within 2 seconds at the side facing the high end of the gradient. It appears that the system that controls localization of activated Ras has fast kinetics and can turn on and off rapidly.
Quantification of membrane signals
Inspection of cells moving in the low ceiling gradient chambers indicated that patches occur preferentially on anterior pseudopods. Patches were seen to rapidly appear and disappear along the membrane at the front of the cell but to persist longer when associated with an extension that became the leading pseudopod. To quantify the intensity of the membrane RasGTP signal, we used the technique of Bosgraaf, Keizer-Gunnink and Van Haastert 33 to delineate the membrane (Figure 5A,B). The intensity of the membrane fluorescence was measured throughout the period the cells were crossing the middle half of the gradient chambers and the values at each node on the membrane were averaged. In Figure 5C we plot the patch frequency, defined as the proportion of nodes that are part of patches, as a function of the distance of a node from the cell’s centroid in the direction of the gradient, Δx (open symbols). For the three different cells we analyzed we found that the patch frequency at the back is not statistically different from zero. The time-averaged GFP intensity for our three cells shows a qualitatively similar dependence on Δx with a zero signal in the back (data not shown). We note that the GFP values without RFP signal correction show an apparent non-zero signal at the rear of the cell (data not shown), demonstrating the necessity of subtracting the cytosolic RBD-GFP contribution from membrane RBD-GFP.
Figure 5.

Membrane associated Ras GTP in chemotaxing cells. Cells expressing both RBD-GFP and cytosolic free RFP were imaged as they chemotaxed across a chamber of width 150 um. A) Fluorescence of GFP. B) Merged fluorescence of RFP and corrected GFP. C) Corrected membrane fluorescence at each node was used to compute the patch frequency (the proportion of nodes that are part of patches). This patch frequency is plotted as a function of the distance of a node from the cell’s centroid in the direction of the gradient for three cells (open symbols). The black curve corresponds to the cell shown in panels A and B. The patch frequency directly away from the gradient is not statistically different from zero. The red line (solid symbols) shows the time-averaged distribution of bound receptors as the cell travels across the middle-half of the channel (assuming a dissociation value Kd=30 nM).
To compare the cell’s response to the input it receives from the gradient, we have also computed the distribution of bound receptors by averaging over the distributions as the cell travels across the middle half of the channel (red line; solid symbols). This time-averaged distribution is essentially linear and mimics the linear external gradient. This is in sharp contrast to the patch distribution which is highly non-linear with the vast majority of patches occurring in the front half of the cell and almost no patches at the back. This switch-like response must be the consequence of intracellular signal processing and we plan to further explore and quantify this response.
RasGTP and pseudopods
If localization of RasGTP at the membrane is used as a signal for the formation of a pseudopod, then we would expect a high correlation of extensions with RasGTP patches. We analyzed the membrane nodes of 4 cells every two seconds for those included in extensions and correlated them with nodes included in patches (see Methods and movie S3). We found that 93 ± 3% of the extensions were associated with a patch and this number increased to 99 ± 1% when the same criteria were applied to cytosol-subtracted images. Using slightly different criteria for extensions and patches for cells in these devices, we found that 92 ± 3% of the extensions were associated with patches.34
Discussion
Studies on chemotaxis carried out over the last 60 years have relied almost exclusively on diffusion without active flow to establish chemotactic gradients. One of the few exceptions was the experiment of John Bonner that first demonstrated that cells can respond chemotactically to a signal released by other cells.35 He deposited a few hundred Dictyostelium cells in buffer on a glass slide and added a coverslip. The buffer was allowed to flow slowly under the coverslip from a reservoir at one end to a slightly lower sink at the other end. After a few hours, cells in the middle were observed to move upstream towards other cells but not downstream where the signal would be weaker. This was essentially a microfluidic device but the gradient was ill-defined since it was generated by the upstream cells. Subsequent work often used diffusion of chemoattractant from a small drop deposited on the surface of agar nearby a small population of developing Dictyostelium cells.14,36 This assay continues to be used in a semi-quantitative manner by scoring the response to droplets with various concentration of cAMP put at various distances from the test cells.18,33 Gradients of different steepness and concentration will be generated but will not be stable in time. Moreover, such gradients are difficult to directly measure.
A major improvement in generating reproducible gradients was the adoption of the Zigmond chamber.37 In this device a source of chemoattractant is connected to a sink by a thin layer of fluid under a coverslip where the cells are deposited. Since there is no active flow in these devices, the gradient is set up by diffusion. Linear gradients are established within a few minutes that are stable for another few minutes but then become progressively more shallow. In addition to the relatively short time window when gradients have a well-defined linear shape, a limitation of analyses in Zigmond chambers is that the cells can affect the local concentration of chemoattractant by secreting or degrading it.
Another popular technique for studying chemotactic cells has been the use of a micropipette filled with chemoattractant that diffuses into a small dish filled with buffer. When the micropipette is positioned near to dispersed cells, they are exposed to a steep gradient. By adding fluorescent dye to the solution in the micropipette, the concentration field could be visualized and was found to decrease exponentially starting from a distance of 15 μm from the tip.33 Closer to the tip the gradient was steeper than exponential. By observing the behavior of cells at various distances from the micropipette, semi-quantitative measurements of responses to steep or shallow gradients could be made. However, as the cells move toward the micropipette, both the concentration and the steepness that they experience change, complicating the analyses.
Microfluidic devices can generate robust chemotactic fields by controlling the mixing of solutions with different concentrations of chemoattractant before passing into small chambers which ensure laminar flow. Both linear and exponential gradients are easily established in chambers where the cells can be imaged at high magnification.16,17 By carefully controlling the developmental stage of cells and collecting data from a large number of cells, statistically significant measurements of cell movement and chemotactic accuracy can be recovered.
Using these devices we were able to demonstrate that Dictyostelium cells that had developed for 5 hours were able to sense a gradient when the ambient concentration of cAMP was only 1 nM and the difference in concentration across a 10 μm cells was only 2.5%. The difference in number of ligand bound receptors in the front and the back halves of the cell will be less than a hundred under these conditions, yet the cells almost always move towards the high end of the gradient (C.I. = 0.42 ± 0.03). The external signal must be highly amplified within the cells to generate such exquisite sensitivity.
A similar amplification of the external signal could be seen in the positioning of patches of RasGTP relative to the gradient direction (Fig. 5C). We had to use microfluidic devices that limited the vertical extension of cells to be able to capture localized fluorescence of RBD-GFP without imposing phototoxicity on the cells. However, cells moved almost as rapidly in these devices as in full height chambers and showed excellent chemotactic accuracy even in shallow gradients. This may be the result of selection in their natural habitat, the soil, where they must often squeeze between tightly packed grains and other obstacles.
Activated Ras not only acts as a good indicator of the chemotactic response but also correlates strongly with pseudopodal extension. In fact, accumulation of RasGTP may be sufficient under certain conditions to determine whether a pseudopodal extension will form or not. However, it is still not clear if it is the size or persistence of a RasGTP patch which is important for pseudopod formation since we often observe transient small patches of fluorescence that do not result in pseudopods. Further study of the interaction of adjacent patches and correlation of their size and persistence with extension of pseudopods may set the rules for understanding how the signal transduction mechanisms convert a weak external signal to a strong directional bias.
Conclusions
We show that chemotactic Dictyostelium cells can be easily enticed into low ceiling microfluidic chambers by shallow gradients of cAMP. Although the cells are closely apposed to both the top and bottom surface, they migrate at almost the same rate as unconstrained cells and show excellent chemotactic directionality. Activated Ras was localized at the tips of pseudopodia as long as the gradient was maintained but rapidly dispersed when the gradient was removed. Our quantitative measurements show that small differences in the spatial distribution of ligand bound receptors trigger much larger differences in effectors that control directional movement.
Concentration profiles in the 2μm tall gradient chambers of the microfluidic devices are robust, have little sensitivity to imprecision of device fabrication and retain their well-defined linear shape under relatively large variations of the flow conditions, making the devices simple to manufacture and easy to operate. The low ceiling devices are well suited for high resolution fluorescent microscopy of cells in well defined stable gradients. We are presently using them to define the subcellular localization of various components of the circuits that direct chemotactic motility in both wild type and mutant strains. They should also be useful for studies of other motile eukaryotic cells and might even help in the study of bacterial chemotaxis.
Acknowledgments
We thank Danny Fuller for technical assistance and construction of the strains. Peter Van Haastert kindly provided the RBD-GFP construct. This work was supported by the National Institutes of Health (PO1 GM078586). After submitting our manuscript, Westendorf et al. reported a different technique that is able to constrain cells in the vertical dimension.38
References
- 1.O’Shea EK, Herskowitz I. The ins and outs of cell-polarity decisions. Nat Cell Biol. 2000;2:39–41. doi: 10.1038/35004065. [DOI] [PubMed] [Google Scholar]
- 2.Paliwal S, et al. MAPK-mediated bimodal gene expression and adaptive gradient sensing in yeast. Nature. 2007;446:46–51. doi: 10.1038/nature05561. [DOI] [PubMed] [Google Scholar]
- 3.Pearse RV, Esshaki D, Tabin CJ, Murray MM. Genome-wide expression analysis of intra- and extracellular connective tissue. J Orthop Res. 2009;27:427–434. doi: 10.1002/jor.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shubin N, Tabin C, Carroll S. Deep homology and the origins of evolutionary novelty. Nature. 2009;12:818–823. doi: 10.1038/nature07891. [DOI] [PubMed] [Google Scholar]
- 5.Weiner O, et al. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nature Cell Biol. 2002;4 doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inouye T, Meyer T. Synthetic activation of endogenous PI3K and Rac identifies an AND-gate switch for cell polarization and migration. PLoS ONE. 2008;3:e3068. doi: 10.1371/journal.pone.0003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kedrin D, van Rheenen J, Hernandez L, Condeelis J, Segall J. Cell motility and cytoskeletal regulation in invasion and metastasis. J Mammary Gland Biol Neoplasia. 2007;12:143–152. doi: 10.1007/s10911-007-9046-4. [DOI] [PubMed] [Google Scholar]
- 8.Charest P, Firtel RA. Big roles for small GTPases in the control of directed cell movement. Biochem J. 2007;401:377–390. doi: 10.1042/BJ20061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janetopoulos C, Firtel RA. Directional sensing during chemotaxis. FEBS Lett. 2008;582:2075–2085. doi: 10.1016/j.febslet.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parent CA, Devreotes PN. A cell’s sense of direction. Science. 1999;284:765–770. doi: 10.1126/science.284.5415.765. [DOI] [PubMed] [Google Scholar]
- 11.Swaney K, Huang C, Devreotes P. Eukaryotic Chemotaxis: A Network of Signaling Pathways Controls Motility, Directional Sensing, and Polarity. Annu Rev Biophys. 2010;39 doi: 10.1146/annurev.biophys.093008.131228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konijn TM, van de Meene JGC, Bonner JT, Barkley DS. The acrasin activity of adenosine-3′,5′-cyclic phosphate. Proc Natl Acad Sci USA. 1967;58:1152–1154. doi: 10.1073/pnas.58.3.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rappel WJ, Loomis WF. Eukaryotic chemotaxis. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2009;1:141–153. doi: 10.1002/wsbm.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaffer BM. Aggregation in cellular slime moulds: in vitro isolation of acrasin. Nature. 1953;171:975–979. doi: 10.1038/171975a0. [DOI] [PubMed] [Google Scholar]
- 15.Tomchik KJ, Devreotes PN. Adenosine 3′,5′-monophosphate waves in Dictyostelium discoideum: A demonstration by isotope dilution-fluorography technique. Science. 1981;212:443–446. doi: 10.1126/science.6259734. [DOI] [PubMed] [Google Scholar]
- 16.Fuller D, Chen W, Adler M, Groisman A, Levine H, Rappel W-J, Loomis WF. External and internal noise limits to eukaryotic chemotaxis. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.0911178107. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song L, et al. Dictyostelium discoideum chemotaxis: threshold for directed motion. Eur J Cell Biol. 2006;85:981–989. doi: 10.1016/j.ejcb.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Mato JM, Losada A, Nanjundiah V, Konijn TM. Signal input for a chemotactic response in the cellular slime mold Dictyostelium discoideum. Proc Natl Acad Sci USA. 1975;72:4991–4993. doi: 10.1073/pnas.72.12.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varnum B, Soll DR. Effects of cAMP on single cell motility in Dictyostelium. J Cell Biol. 1984;99:1151–1155. doi: 10.1083/jcb.99.3.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vicker MG, Schill W, Drescher K. Chemoattraction and chemotaxis in Dictyostelium discoideum: myxamoeba cannot read spatial gradients of cyclic adenosine monophosphate. J Cell Biol. 1984;98:2204–2214. doi: 10.1083/jcb.98.6.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider N, et al. A LIM protein involved in the progression of cytokinesis and regulation of the mitotic spindle. Cell Motil Cytoskel. 2003;56:130–139. doi: 10.1002/cm.10139. [DOI] [PubMed] [Google Scholar]
- 22.Bolourani P, Spiegelman GB, Weeks G. Delineation of the roles played by RasG and RasC in cAMP-dependent signal transduction during the early development of Dictyostelium discoideum. Mol Biol Cell. 2006;17:4543–4550. doi: 10.1091/mbc.E05-11-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kae H, et al. Cyclic AMP signalling in Dictyostelium: G-proteins activate separate ras pathways using specific RasGefs. EMBO Rep. 2007;8:477–482. doi: 10.1038/sj.embor.7400936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki AT, Chun C, Takeda K, Firtel RA. Localized Ras signaling at the leading edge regulates P13K, cell polarity, and directional cell movement. J Cell Biol. 2004;167:505–518. doi: 10.1083/jcb.200406177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andrew N, Insall RH. Chemotaxis in shallow gradients is mediated independently of PtdIns 3-kinase by biased choices between random protrusions. Nature Cell Biol. 2007;9:193–200. doi: 10.1038/ncb1536. [DOI] [PubMed] [Google Scholar]
- 26.Laevsky G, Knecht DA. Under-agarose folate chemotaxis of Dictyostelium discoideum amoebae in permissive and mechanically inhibited conditions. BioTechniques. 2001;31:1140–1149. doi: 10.2144/01315rr03. [DOI] [PubMed] [Google Scholar]
- 27.Sussman M. Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Meth Cell Biol. 1987;28:9–29. doi: 10.1016/s0091-679x(08)61635-0. [DOI] [PubMed] [Google Scholar]
- 28.Chan T, Vese L. Active contours without edges. IEEE Transactions on Image Processing. 2001;10:266–277. doi: 10.1109/83.902291. [DOI] [PubMed] [Google Scholar]
- 29.Machacek M, Danuser G. Morphodynamic profiling of protrusion phenotypes. Biophys J. 2006;90:1439–1452. doi: 10.1529/biophysj.105.070383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerisch G. Imaging actin cytoskeleton dynamics in Dictyostelium chemotaxis. Meth Mol Biol. 2009;571:385–400. doi: 10.1007/978-1-60761-198-1_26. [DOI] [PubMed] [Google Scholar]
- 31.Iwadate Y, Yumura S. Actin-based propulsive forces and myosin-II-based contractile forces in migrating Dictyostelium cells. J Cell Sci. 2008;121:1314–1324. doi: 10.1242/jcs.021576. [DOI] [PubMed] [Google Scholar]
- 32.Uchida KSK, Yumura S. Dynamics of novel feet of Dictyostelium cells during migration. J Cell Sci. 2004;117:1443–1455. doi: 10.1242/jcs.01015. [DOI] [PubMed] [Google Scholar]
- 33.Bosgraaf L, Keizer-Gunnink I, Van Haastert PJ. PI3-kinase signaling contributes to orientation in shallow gradients and enhances speed in steep chemoattractant gradients. J Cell Sci. 2008;121:3589–3597. doi: 10.1242/jcs.031781. [DOI] [PubMed] [Google Scholar]
- 34.Hecht I, et al. Activated membrane patches guid chemotactic cell motility. 2010. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonner JT. Evidence for the formation of cell aggregates by chemotaxis in the development of the slime mold Dictyostelium discoideum. J Exp Zool. 1947;106:1–26. doi: 10.1002/jez.1401060102. [DOI] [PubMed] [Google Scholar]
- 36.Sussman M, Lee F, Kerr NS. Fractionation of acrasin, a specific chemotactic agent for slime mold aggregation. Science. 1956;123:1171–1172. doi: 10.1126/science.123.3209.1171. [DOI] [PubMed] [Google Scholar]
- 37.Devreotes PN, Zigmond SH. Chemotaxis in eukaryotic cells: A focus on leukocytes and Dictyostelium. Annu Rev Cell Biol. 1988;4:649–686. doi: 10.1146/annurev.cb.04.110188.003245. [DOI] [PubMed] [Google Scholar]
- 38.Westendorf C, et al. Live cell flattening-traditional and novel approaches. PMC Biophys. 2010;19:3–9. doi: 10.1186/1757-5036-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]