

Abstract
Background
Systemic exposure to lipopolysaccharide (LPS) has been linked to clinical disease activity in adults with IBD. We hypothesized that markers of LPS exposure and the acute phase response (APR) would be increased in pediatric IBD patients with growth failure, and that LPS signaling would be required for induction of the APR in murine colitis.
Methods
Serum markers of LPS exposure, endotoxin core IgA antibody (EndoCAb), and the APR, LPS binding protein (LBP), were quantified in pediatric IBD patients and controls. LBP and cytokine production were determined after administration of trinitrobenzene sulfonic acid (TNBS) enemas to mice with genetic deletion of Toll-Like receptor 4 (TLR4), and wild type (WT) controls.
Results
Serum EndoCAb and LBP were significantly elevated in patients with Crohn’s disease (CD), compared to disease controls with Ulcerative Colitis (UC) and healthy controls (p<0.001). This was independent of disease activity or location. CD patients with elevated serum EndoCAb and LBP exhibited linear growth failure which persisted during therapy. Serum LBP increased in WT mice following TNBS administration, in conjunction with increased serum TNF-α, IL-6, and IL-10, and expansion of regulatory T cell numbers. Both the APR and expansion of foxp3+ T cells were abrogated in TLR4 deficient mice, in conjunction with a reduction in acute weight loss.
Conclusions
LPS exposure and a persistent APR are associated with growth failure in pediatric CD. LPS signaling is required for the APR in murine colitis. Therapies targeting this pathway may benefit the subset of patients with refractory growth failure.
Keywords: acute phase response, endotoxin, inflammatory bowel disease, linear growth
INTRODUCTION
The Inflammatory Bowel Diseases (IBD), ulcerative colitis (UC) and Crohn’s disease (CD), are chronic relapsing and remitting disorders of the gastrointestinal tract. The pathogenesis of IBD is complex and multi-factorial. Current evidence suggests that IBD develops in individuals with a genetic predisposition to respond aberrantly to the enteric flora, leading to an expansion of pro-inflammatory T-cells in the gut (1). In support of this, genetically susceptible mice raised in germ free conditions do not develop colitis until colonization with commensal bacteria (2). Recent studies in human IBD have focused on the luminal flora as a key environmental trigger. Specifically, these studies have implicated genotypic variation in microbial pattern recognition (NOD2 and TLR4) and autophagy (ATG16L1 and IRGM) in susceptibility (3, 4). Moreover, CD patients with increased sero-reactivity to microbial products have been shown to experience a more aggressive course complicated by increased rates of stricturing/internal penetrating behavior (5). While linear growth failure is an important complication of pediatric CD, an association with increased exposure to microbial products has not been explored.
Considering the preponderance of gram-negative bacteria in the gastrointestinal tract, the role of endotoxin (lipopolysaccharide; LPS) in activation of immunity and complications of disease in patients with IBD is important to elucidate. Increased translocation across a defective mucosal barrier is a prevailing hypothesis in the etiology of IBD (6). Previous studies have identified circulating endotoxin in adults with IBD and these endotoxin levels have correlated with disease activity, while therapeutic reductions in endotoxin resulted in symptomatic improvement (7–9). Due to its short half-life and frequent contamination of environmental surfaces, endotoxin is difficult to measure in serum; therefore, several studies have utilized surrogate markers for endotoxin exposure. Lipopoysaccharide binding protein (LBP) is an acute phase protein produced by hepatocytes and paneth cells in response to both LPS and cytokines (10). LBP catalyzes delivery of LPS monomers to membrane-bound CD14, or to soluble CD14 (sCD14), which, in turn, delivers LPS to myeloid differentiation protein-2 (MD-2). LPS/MD-2 heterodimers represent the activating ligand for the signaling LPS receptor, Toll-Like Receptor 4 (TLR4). Antibodies reactive with the core of LPS (EndoCAb) are produced by B cells in response to LPS stimulation, both under normal conditions and during enteric infections (11). These neutralize LPS activity and may be protective in conditions of acute endotoxemia, such as sepsis and cardiac surgery, where low basal levels of EndoCAb are associated with increased short and long-term mortality (12, 13). EndoCAb levels also increase under conditions of chronic infection and inflammation, such as cystic fibrosis, where they serve as a marker for chronic LPS exposure (7, 14). Each of these markers have increased half-lives compared to LPS itself (LPS = 1–3 hours, IgG EndoCAb = 10–18 hours, LBP and sCD14 = 24–48 hours)(10, 11, 15). Both LBP and EndoCAb are increased in adult-onset IBD patients, and have been been shown to correlate with circulating LPS (7, 8). Whether this was also the case in pediatric-onset IBD was not known.
Toll-like receptors (TLR) link the innate and adaptive immune systems by recognizing conserved structures on microorganisms and through a complex cascade (involving an adaptor molecule Myd88) trigger pro-inflammatory cytokine production in efforts to maintain homeostasis; however, recent studies have shown that TLR signaling may also be required for counter-regulatory immune responses, and so in some settings could limit inflammation (16, 17). Aberrancy of the TLR signaling system may therefore regulate chronic inflammation in IBD. Moreover, our recent murine studies have shown that LPS can directly suppress growth hormone (GH) signaling, by inducing proteolytic degradation of the hepatic GH receptor (GHR) (18). LPS may therefore also contribute directly to linear growth failure in children with CD.
The role of TLR signaling has recently been investigated in both murine and human IBD. Adult and pediatric patients with UC and CD have increased TLR4 expression in colonic mucosa compared to healthy controls (19, 20). Mechanistically, the role of TLR4 in this setting has not been fully characterized, although recent murine studies have suggested homeostatic effects upon epithelial regeneration and repair, and bacterial clearance (17, 21). Most recently, an adoptive transfer model of colitis utilizing Myd88 deficient mice (adaptor molecule for numerous TLRs) illustrated the importance of the TLR system directly upon pro-inflammatory T cell function (22). We chose to examine effects of TLR4 signaling upon mucosal T cell responses and injury in the Trinitrobenzene-sulfonic acid (TNBS) murine model of colitis. This model utilizes a haptenizing agent that allows for both study of acute mucosal injury and activation of the acute phase response (APR), and an effector T cell response which targets the enteric flora.
In this study, we found that pediatric patients with IBD have evidence for increased systemic endotoxin response/exposure (CD greater than UC), and that in CD this is associated with linear growth failure at diagnosis and following therapy. In the murine model, we found that TLR4 was required for activation of the systemic APR following TNBS administration, as well as a relative expansion of regulatory T cells. Collectively, these studies suggest that TLR4 signaling may contribute to both systemic complications of IBD including linear growth failure, and local counter-regulatory responses.
MATERIALS AND METHODS
Human Subjects
Our studies included 104 patients with pediatric-onset CD (72 males; mean age, 11.3 yr [range 1–18 yr], 43 patients with pediatric-onset UC (24 males; mean age, 11.7 yr [range 2–18 yr]) and 24 pediatric controls (18 males; mean age, 11.6 yr [range 3–18 yr]). Diagnosis was based on established clinical, radiological, and endoscopic criteria, supported in all cases by histopathology. Controls were enrolled from children undergoing diagnostic evaluation for IBD and found to have normal tissue. The ultimate diagnoses in the controls were either functional abdominal pain or irritable bowel syndrome.
Clinical disease activity was determined by a retrospective chart review performed by a reviewer blinded to the serum EndoCAb and LBP results. We utilized the criteria employed by Griffiths et al in a previous report of growth and clinical course in pediatric CD (23). This defined four categories of clinical disease activity: quiescent, mild, moderate, or severe. For the cross-sectional evaluation of the relationship between clinical disease activity and serum EndoCAb and LBP, patients with mild, moderate, or severe activity at the time of the blood draw were classified as active. For the longitudinal assessment of disease activity and growth, patients were classified as moderate-to-severe if they experienced this level of activity at any point during six month blocks from diagnosis through month 48, or most recent follow-up if less than 48 months. Disease location (L) and behavior (B) for CD were determined according to the Montreal classification scheme (24). Records were reviewed and surgery related to IBD was recorded. Height (cm) and weight (kg) were converted to Z scores using CDC normative charts for age and gender. Growth retardation was defined as Z score < −1 for age and gender.
Animals
TLR4-deficient mice (backcrossed 10 generations) and wild-type (WT) controls both on a C57/BL6J background were maintained under specific pathogen-free conditions at the animal facility of Cincinnati Children’s Hospital Research Foundation. Mice were allowed free access to autoclaved feed and to drinking water. All mice used were sex-matched and between 6 and 8 weeks of age.
Induction of TNBS Colitis
Colitis was induced by rectal installation of 2,4,6-trinitrobenzene sulfonic acid in 100 mcl 50% ethanol (ETOH) (TNBS; Sigma Chemical Co., St. Louis, MO) (25). Mice received either 125 mg/kg (high dose) or 50 mg/kg (low dose) on day zero and were sacrificed on day 3 or day 6. Controls received the same volume of 50% ETOH. Mortality and weight loss were recorded.
Histological scoring of distal colon
Colons were excised and the distal colonic tissue was fixed with 10% neutral formalin, paraffin embedded, sectioned at 3–6 μm, and stained with hematoxylin and eosin in the UC Pathology Core. Sections were analyzed in a blinded fashion. Scoring was performed according to Neurath, et al (25).
ELISA assays
Blood samples were collected under sterile conditions into endotoxin-free tubes, and serum was obtained and stored at −80°C until analysis. Serum lipopolysaccharide binding protein (LBP;Hycult Biotechnology, Uden, Netherlands), endotoxin core IgA antibody (EndoCAb IgA; Hycult Biotechnology, Uden, Netherlands) and soluble CD14 (sCD14; Quantikine, R&D Systems, Minneapolis, MN) were determined by solid-phase, enzyme-linked immunosorbant sandwich assay as per manufacturer’s instructions. Murine blood samples were collected by cardiac puncture into endotoxin-free tubes, and serum was obtained and stored at −80°C until analysis.
Cincinnati Cytokine Capture Assay
Biotynylated antibodies to IFNγ, IL-6, TNFα and IL-10 were injected intraperitoneally 24 hours prior to harvest as per our prior published method (26). Serum was collected at harvest and enzyme-linked immunosorbant assay was performed according to our prior published method (26).
Flow cytometry (FACS) analysis for characterization of mesenteric lymph node (MLN) cell populations
Fresh single cell suspensions were prepared from mesenteric lymph nodes (MLN) and spleen. Cells were stained for surface markers anti-mouse CD3/CD4/CD25/CD44/CD62L (BD Biosciences, San Jose, CA and eBiosciences, San Diego, CA) and intracellular staining for anti-mouse Foxp3 (eBiosciences, San Diego, CA) following manufacturer’s recommendations. Cells were acquired on a FacsCaliber BD flow cytometer and analyzed with Cell Quest BD software.
Data Analysis
Statistical analysis was performed using GraphPad PRISM Version 4.01. Continuous variables were analyzed using two-sample t test or ANOVA with Bonferroni test for multiple comparisons, or for non-parametric alternatives, the Mann-Whitney test or Kruskal-Wallis with Dunns test for multiple comparisons. Differences in frequencies between groups were tested using the Fisher Exact Test. A Kaplan-Meier curve with log rank analysis was performed to test for differences in mortality in the animal model. Differences in disease activity between groups over time in the patient-based study were tested by two-way ANOVA.
ETHICAL CONSIDERATIONS
Informed consent was obtained from all families and the Cincinnati Children’s Hospital Institutional Review Board approved the study protocol. The Cincinnati Children’s Institutional Animal Care and Use Committee approved all animal studies.
RESULTS
Clinical and demographic characteristics of the IBD patients
The clinical and demographic characteristics of the IBD patients and controls are summarized in Table I. The majority of the IBD patients and controls were male. The relative frequency of males within the CD group (69%) exceeded that within the UC group (56%, p=0.05), and did not differ from the controls (75%). The mean age at diagnosis (11.3 vs. 11.7 years) and duration of disease (39.5 months vs. 35.7 months) was comparable between the CD and UC groups. The majority of the IBD patients had clinically inactive disease at the time of blood sampling, and inflammatory disease behavior with ileo-colonic involvement. Twenty-one percent had required at least one surgery.
Table I.
Clinical and Demographic Characteristics.
| Crohn’s Disease | Ulcerative Colitis | Control | |
|---|---|---|---|
| # of patients | 104 | 43 | 24 |
| Mean age (range) [years] | 11.3 (1–18) | 11.7 (2–18) | 11.6 (3–18) |
| Gender (male) | 69% | 56%* | 75% |
| Mean duration (range) [months] | 39.5 (1–176) | 35.7 (1–160) | - |
| Disease activity | - | ||
| Active (%) | 26% | 28% | - |
| Disease location | - | ||
| L1: 11% | - | - | |
| L2: 17% | - | - | |
| L3: 72% | - | - | |
| Disease behavior | - | ||
| B1 (%) | 69% | - | - |
| B2 or B3 (%) | 31% | - | - |
| Surgery (%) | 21% | - | - |
p=0.05 vs. CD
L1:Ileal location
L2:Colon-only location
L3:Ileo-colonic location
B1:Non-stricturing, non-penetrating behavior
B2:Stricturing behavior
B3:Internal penetrating behavior
Serum EndoCAb IgA, LBP, and sCD14
We first asked whether circulating markers of endotoxin exposure and the APR would be elevated in IBD patients compared to controls. The mean (SEM) serum EndoCAb IgA concentration was increased from 27.19 ± 5.5 AU/mL in controls to 53.98 ± 8.1 AU/mL in UC and 146.6 ± 20 AU/mL in CD (see Fig. 1A, p < 0.001 for CD vs. both UC and controls). CD patients had significantly higher serum EndoCAb IgA concentration than UC patients (p < 0.001). The mean (SEM) serum LBP concentration was increased from 14.07 ± 1.08 μg/mL in controls to 29.66 ± 3.1 μg/mL in UC and 38.16 ± 2.6μg/mL in CD (see Fig. 1B, p < 0.001 for UC and CD vs. controls). The mean (SEM) serum sCD14 concentration did not differ between UC, CD and controls (2328 ± 195.7, 2124 ± 161.8, and 2619 ± 229.1 ng/mL, respectively).
Figure 1. Serum EndoCAb IgA and LBP Concentration in Pediatric IBD.
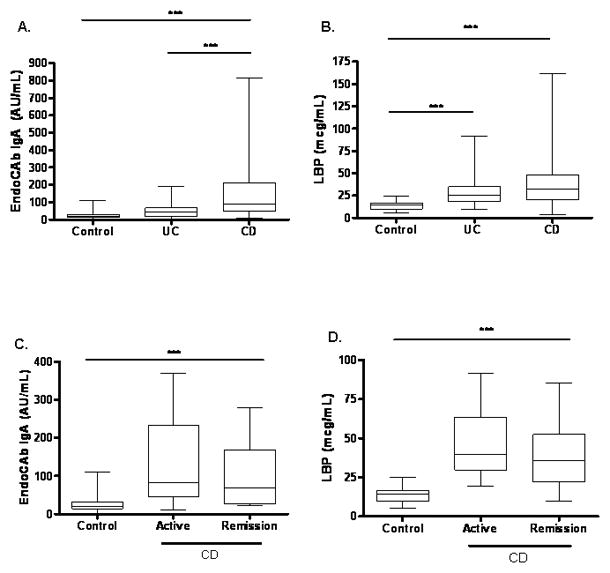
(A) Serum EndoCAb IgA concentration was determined in CD (n=65) and UC (n=34) patients and healthy controls (n=20) by ELISA. ***p < 0.001 by Kruskal-Wallis with Dunn’s multiple comparison test. (B) Serum LBP concentration was determined in CD (n=95) and UC (n=32) patients and healthy controls (n=21) by ELISA. ***p < 0.001 by Kruskal-Wallis with Dunn’s multiple comparison test. (C) Serum EndoCAb IgA concentration was determined in CD patients stratified by disease activity at the time of blood sampling (n=23) and healthy controls (n=20) by ELISA. ***p < 0.001 by unpaired t test. (D) Serum LBP concentration was determined in CD patients stratified by disease activity at the time of blood sampling (n=51) and healthy controls (n=21) by ELISA. ***p < 0.001 by unpaired t test. Data are shown as median (interquartile range) and overall range.
Circulating EndoCAb and LBP Relative to CD Activity, Location and Behavior
We then asked whether these markers for endotoxin exposure and the APR would be differ within the CD group based upon disease activity, location, or behavior. Both CD patients in remission (99.23 ± 19.34 log AU/mL, p <0.001) and those with active disease (128 ± 62.09 log AU/mL, p<0.001) at the time of the blood draw had significantly elevated serum EndoCAb IgA concentration compared to controls (see Fig. 1C). The mean (SEM) serum LBP concentration was also significantly increased in CD patients with active disease (45.9 ± 5.71 μg/mL) and those in clinical remission (39.26 ± 3.16 μg/mL) compared to controls, and did not differ between the two CD groups (see Fig. 1D, p< 0.001 for CD in remission vs. control). The serum EndoCAb IgA and LBP concentrations did not vary within the CD group with either disease location or behavior (data not shown). Consistent with this, CD patients who required surgery versus those who did not also did not vary for either mean (SEM) serum LBP concentration (34.99 ± 3.9μg/mL and 39.05 ± 3.15 μg/mL, respectively) or mean (SEM) serum EndoCAb IgA concentration (150.4 ± 36.95 AU/mL and 145.5 ± 23.59 AU/mL, respectively). Morever, these parameters did not vary with gender within the control, UC, or CD groups (data not shown). Collectively, these data confirmed that circulating markers of endotoxin exposure and the APR were elevated in pediatric-onset CD patients, and that these persisted even while in clinical remission.
Markers of Endotoxin Exposure and the APR and Linear Growth
We then asked whether linear growth or weight gain varied as a function of circulating EndoCAb IgA or LBP. The follow-up period was from diagnosis to study entry, and was equal to a mean (SD) of 4.1 (3.6) years. We did not observe a difference in HTz at follow-up as a function of age at diagnosis, gender, frequency of moderate-to-severe disease activity during follow-up, or exposure to infliximab therapy during follow-up (see Supplemental Figure 1.) We did not observe a linear correlation between serum EndoCAb IgA or LBP concentration and either HTz or WTz, (see Figs. 2A and 3A). However, the nonparametric correlation (Spearman) between serum LBP and HTz approached significance (r=−0.21, p=0.06). Therefore, a categorical analysis was performed to determine whether linear growth would vary between CD groups with high versus low values of serum EndoCAb or LBP. We stratified by the median serum EndoCAb IgA (88 AU/mL) or LBP (33 mcg/mL) concentration within the CD group (Lo vs Hi) and compared linear growth and weight gain. As shown in Table II, CD patients with elevated serum EndoCAb IgA were more likely to be female than those with lower serum EndoCAb IgA. Otherwise, age at diagnosis, race, disease location or behavior, surgery rates, or medication exposures did not vary by serum EndoCAb IgA or LBP concentration within the CD group. At diagnosis, 44% of the CD patients with elevated serum EndoCAb IgA exhibited linear growth retardation (defined as HTz < −1), compared to 23% of the group with lower serum EndoCAb IgA (see Fig. 2B, p=0.05). In contrast to the difference in linear growth, weight at diagnosis did not differ between the two groups (see Fig. 2B). This suggested that increased endotoxin exposure was associated with a specific effect upon linear growth, independent of overall malnutrition at diagnosis.
Figure 2. Serum EndoCAb IgA Concentration and Linear Growth at Diagnosis.
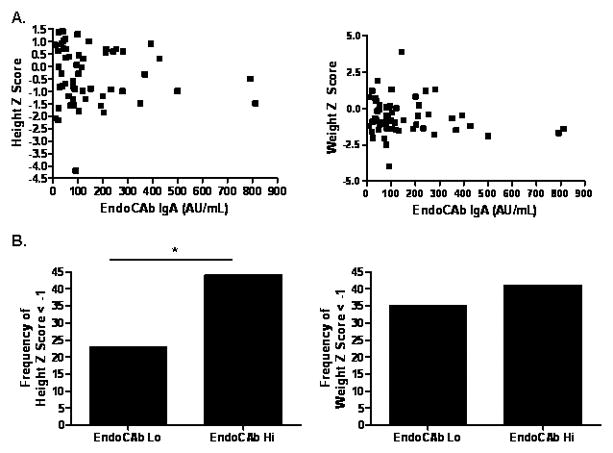
A) HTz and WTz at diagnosis within the CD group are shown relative to serum EndoCAb IgA. B) CD patients were stratified by the median serum EndoCAb IgA concentration (88 AU/mL) (n=54, Lo vs Hi), and the frequency of growth retardation at diagnosis defined as z score for age and gender < −1 was determined. Data are shown as the frequency of growth retardation for height or weight. *p≡ 0.05 by Fisher exact test.
Figure 3. Serum LBP Concentration and Linear Growth.
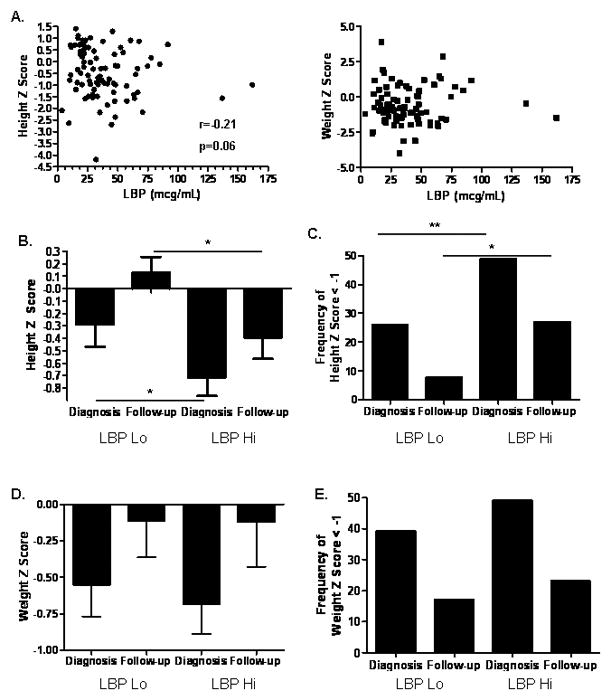
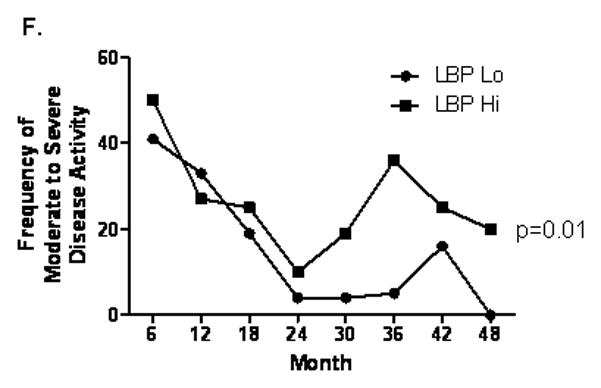
A) HTz and WTz within the CD group are shown relative to serum LBP. CD patients were stratified by the median serum LBP concentration (33 mcg/mL, Lo vs Hi), and height and weight z scores and the frequency of growth retardation defined as z score for age and gender < −1 were determined at diagnosis and most recent follow-up (n=83 and 74, respectively). Data are shown as the mean (SEM) for (B) height z score, *p < 0.05 by Mann-Whitney test, or (D) weight z score, or the frequency of growth retardation for (C) height, **p= 0.01 by Fisher exact test, *p=0.05 by Fisher exact test, or (E) weight. (F) The frequency of moderate-to-severe clinical disease activity was determined over six month intervals from diagnosis through month 48 and is shown, p=0.01 by two-way ANOVA.
Table II.
Clinical and Demographic Characteristics Stratified by Serum EndoCAb and LBP Concentration.
| EndoCAb Lo | EndoCAb Hi | LBP Lo | LBP Hi | |
|---|---|---|---|---|
| Mean age (range) | 11(3–18) | 12(5–17) | 12(3–17) | 12(5–18) |
| Gender (male) | 81% | 53%* | 63% | 71% |
| Race (white) | 97% | 84% | 91% | 96% |
| Disease location | ||||
| L1: 19% | L1: 9% | L1: 15% | L1: 11% | |
| L2: 10% | L2: 19% | L2: 20% | L2: 15% | |
| L3: 71% | L3: 72% | L3: 65% | L3: 74% | |
| Disease behavior | ||||
| B1 (%) | 74% | 62% | 78% | 70% |
| B2 or B3 (%) | 26% | 38% | 22% | 30% |
| Surgery (%) | 26% | 28% | 22% | 22% |
| Medications | ||||
| Prednisone | 83% | 84% | 76% | 89% |
| 6-Mercaptopurine/ | 83% | 90% | 85% | 77% |
| Azathioprine | ||||
| Infliximab | 48% | 44% | 35% | 43% |
p=0.01 vs. EndoCAb Lo
L1:Ileal location
L2:Colon-only location
L3:Ileo-colonic location
B1:Non-stricturing, non-penetrating behavior
B2:Stricturing behavior
B3:Internal penetrating behavior
We then asked whether differences in the APR, as measured by serum LBP, would be associated with differences in linear growth. Height z score at diagnosis and during follow-up was reduced in CD patients with elevated serum LBP (LBP Hi), compared to those with lower serum LBP (LBP Lo) (see Fig. 3B). At diagnosis, 49% of the CD patients with elevated serum LBP exhibited linear growth retardation (defined as HTz < −1), compared to 26% of the group with lower serum LBP (see Fig. 3C, p=0.01). At follow-up, this relative difference in linear growth retardation persisted (p=0.05), although both groups had experienced an improvement relative to diagnosis. In contrast to the difference in linear growth, there were no differences in weight either at diagnosis or during follow-up between the two groups (see Figs. 3D and 3E). Importantly, reduced height z score persisted in the LBP Hi group despite normalization of weight z score.
Clinical disease activity was assessed over six month periods from diagnosis through month 48, or most recent follow-up. The frequency of moderate-to-severe clinical disease activity did not differ between the groups over the first 24 months following diagnosis (see Fig. 3F). However, there was an increased frequency of moderate-to-severe activity in the group with elevated LBP from month 30 through 48 (p=0.01 by two-way ANOVA). Collectively, these studies demonstrated that a marker of the systemic APR, LBP, is associated with linear growth retardation in children with CD, in the absence of differences in weight gain.
Circulating LBP and Cytokine Levels following TNBS Administration to WT and TLR4 Deficient Mice
We then asked whether endotoxin signaling via TLR4 was required for systemic up-regulation of the pro-inflammatory cytokines TNFα and IL-6, and LBP, in the animal model of colitis due to TNBS administration. High dose TNBS administration led to severe colitis with a high level of mortality and weight loss which did not differ by genotype (data not shown). Therefore, low dose TNBS was utilized for the remainder of the studies to test the effect of TLR4 signaling upon the systemic APR to minor epithelial injury and antigenic exposure in the colon. The mean (SEM) circulating LBP concentration was increased to 84 ± 4 mcg/mL in WT mice at day three following TNBS administration, compared to 43 + 10 mcg/mL in ETOH-treated controls (p <0.01, see Fig. 4A). By day six, the serum LBP concentration had returned to basal levels in WT mice. While circulating LBP trended towards an increase following TNBS administration in TLR4 deficient mice at day 3, this did not reach significance, compared to TLR4 deficient controls, and was significantly lower than WT mice at day 3 (p<0.05). Both serum TNFα (797.5 ± 266.1 pg/mL) and IL-6 (21703 ± 7185 pg/mL) were significantly increased at day three in WT mice compared to ETOH-treated controls (196.7 ± 53.9 pg/mL and 1602 ± 445.9 pg/mL, respectively, p<0.05 and <0.01, see Figs. 4B and 4C). Neither cytokine was significantly increased at day three in TLR4 deficient mice, and both were significantly lower than WT controls at this time point (p<0.01). Weight loss following TNBS administration has been linked to activation of systemic immunity and central anorexia leading to reduced chow intake. Both WT and TLR4 deficient mice experienced maximal weight loss by day 3, and had recovered weight by day 6 (see Fig. 4D). However, maximal weight loss was substantially lower in the TLR4 deficient mice compared to the WT controls, consistent with the reduction in serum LBP, TNFα, and IL-6 production.
Figure 4. Circulating LBP, Pro-Inflammatory Cytokines, and Weight Loss Following TNBS Administration to WT and TLR4 deficient mice.
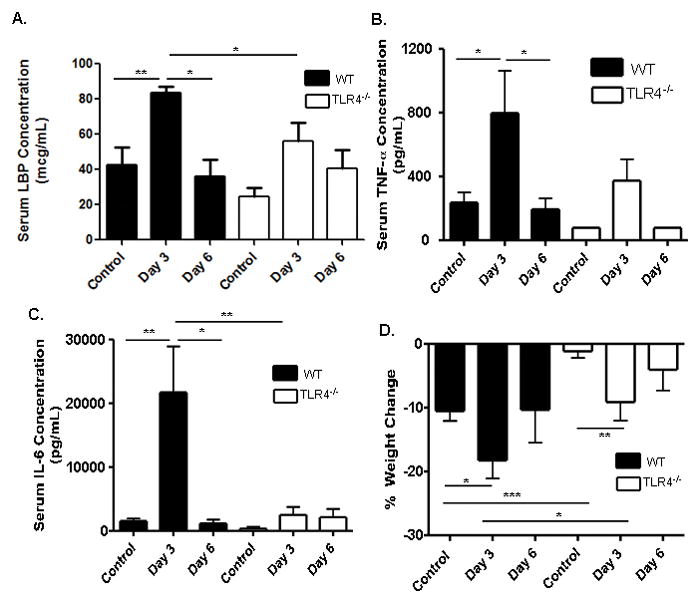
(A) Serum LBP concentration was measured at day 3 in WT (n=4) and TLR4 deficient (TLR4−/−) (n=6) mice, as well as WT (n=6) and TLR4−/− (n=3) control mice and at day 6 in WT (n=4) and TLR4−/− (n=3) mice by ELISA. (B) TNF-α was measured at day 3 in WT (n=4) and TLR4−/− (n=5) mice, as well as WT (n=13) and TLR4−/− (n=4) control mice and at day 6 in WT (n=4) and TLR4−/− (n=6) mice by ELISA. (C) IL-6 was measured at day 3 in WT (n=4) and TLR4−/− (n=7) mice, as well as WT (n=13) and TLR4−/− (n=4) control mice and at day 6 in WT (n=4) and TLR4−/− (n=6) mice by ELISA. (D) Weight change from baseline was recorded for WT (n=11) and TLR4 −/− (n=11) mice. Data are shown as the mean (SEM). *p < 0.05, **p < 0.01, ***p < 0.001, by unpaired t test or Mann-Whitney test.
Serum IL-10 and IFNγ Production and Severity of Colitis following TNBS Administration
Recent studies have linked TLR4 signaling to both counter-regulatory and pro-inflammatory T cell responses. We found that the mean (SEM) serum IL-10 concentration increased significantly in WT mice (898 ± 119.7 pg/mL) and, though not significant, also increased in TLR4 deficient mice (873 ± 248.6 pg/mL) at day three, compared to 219.5 ± 61.45 pg/mL and 183.4 ± 87.1 pg/mL, respectively, in ETOH-treated controls (see Fig. 5A). The increase in serum IL-10 persisted at day six in WT mice, but returned to basal levels at this time point in TLR4 deficient mice. The converse was observed for IFNγ, which increased from 137 ± 59 pg/mL in ETOH-treated controls to 16018 ± 8964 pg/mL at day three in TLR4 deficient mice (see Fig. 5B, p <0.01), and exhibited only a modest increase, to 3298 ± 1694 pg/mL, in WT mice. We then asked whether these differences in circulating IL-10 and IFNγ would be associated with a difference in mortality or colon histology. We found that mortality did not vary between the WT and TLR4 deficient mice (data not shown). Neither the WT nor TLR4 deficient mice exhibited a significant increase in histological injury three days after low dose TNBS administration, compared to controls which received ETOH alone (see Fig. 5C). However, by day six after TNBS administration, the mean (SEM) histological score was significantly increased in the TLR4 deficient mice, to 1.8 ± 0.3, compared to 0.66 ± 0.15 at day three and 0.78 ± 0.08 in controls who received ETOH alone (see Fig. 5C, p <0.01 and <0.05 respectively). The histological abnormality in the TLR4 deficient mice at day six included crypt hyperplasia, mucin depletion, and a mononuclear cell infiltrate.
Figure 5. Circulating IL-10 and IFNγ and Colon Inflammation Following TNBS Administration to WT and TLR4 −/− mice.
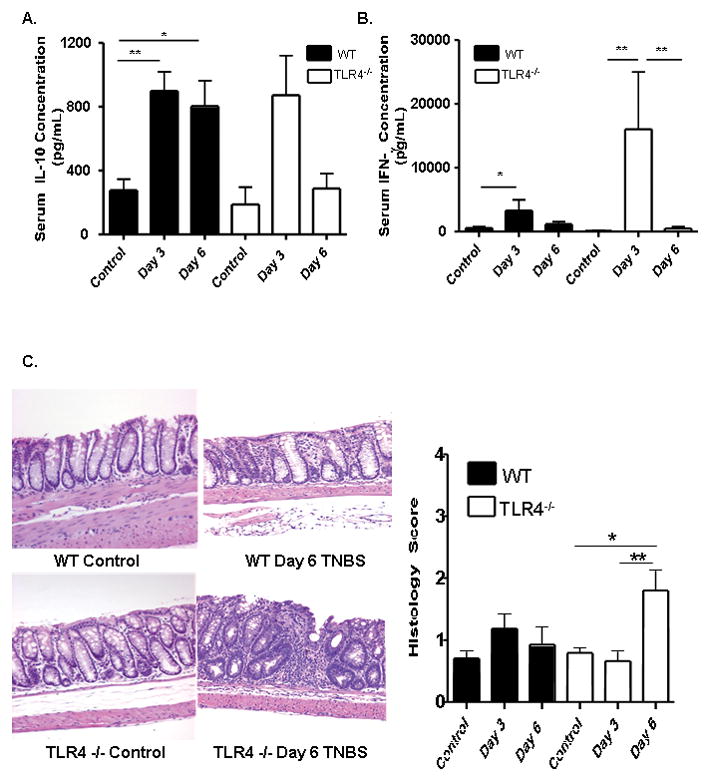
TNBS (50mg/kg) was administered to WT and TLR4 deficient mice at day 0. (A) IL-10 was measured at day 3 in WT (n=4) and TLR4−/− (n=6) mice, as well as WT (n=13) and TLR4−/− (n=4) control mice and at day 6 in WT (n=4) and TLR4−/− (n=6) mice by ELISA. (B) IFN-γ was measured at day 3 in WT (n=4) and TLR4−/− (n=6) mice, as well as WT (n=13) and TLR4−/− (n=4) control mice and at day 6 in WT (n=4) and TLR4−/− (n=6) mice by ELISA. (C) Representative distal colon sections from WT (n= 5) and TLR4 −/− (n=6) mice at day 6 are shown. Histologic scores of WT (n=6) and TLR4−/− (n=9) mice harvested at day 3 and WT (n= 5) and TLR4 −/− (n=6) mice harvested at day 6 as well as WT (n=20) and TLR4 −/− (n=23) controls were recorded.*p <0 .05, **p < 0.01, by unpaired t-test. Data are shown as the mean (SEM).
T-Lymphocyte Responses in WT and TLR4 −/− mice following TNBS Administration
We then asked whether these differences in circulating IL-10 and IFNγ would be associated with a difference in the frequency of regulatory and activated/memory T cells in the draining mesenteric lymph nodes (MLN) or spleen. Three and six days after low dose TNBS administration, we observed a modest increase in the frequency of CD4+CD44+CD62Llo activated/memory T cells in the MLN of both WT and TLR4 deficient mice which did not reach statistical significance (see Fig. 6A). By day six, the frequency of splenic CD4+CD44+CD62Llo activated/memory T cells had increased in both groups. By comparison, the frequency of CD4+FOXP3+ regulatory T cells was significantly increased in the MLN of both groups at days three and six (p < 0.001 and p < 0.01, respectively; see Fig. 6B). This included an increase in both the CD25+ and CD25− populations. However, there was a greater relative increase (approximately two-fold higher) in this population in the WT mice compared to the TLR4 deficient mice at day three (p < 0.01). Therefore, the overall MLN Treg/Teff ratio was significantly higher in the WT mice compared to the TLR4 deficient mice at this timepoint (p < 0.05; see Fig. 6C). The splenic Treg response did not differ between the groups. Taken together, these data suggested that TLR4 signaling was required for a counter-regulatory response to a mild TNBS-induced antigenic exposure in the gut, which involved IL-10 production and expansion of FOXP3+ T cells in the MLN. When this response was reduced by TLR4 deficiency, a mild colitis ensued.
Figure 6. TLR4 Dependent T Lymphocyte Responses to TNBS Administration.
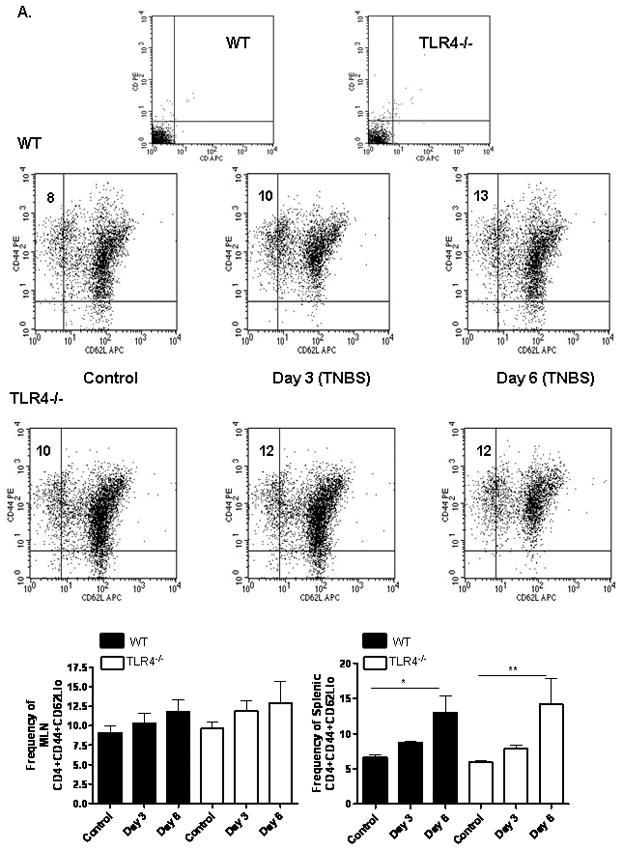
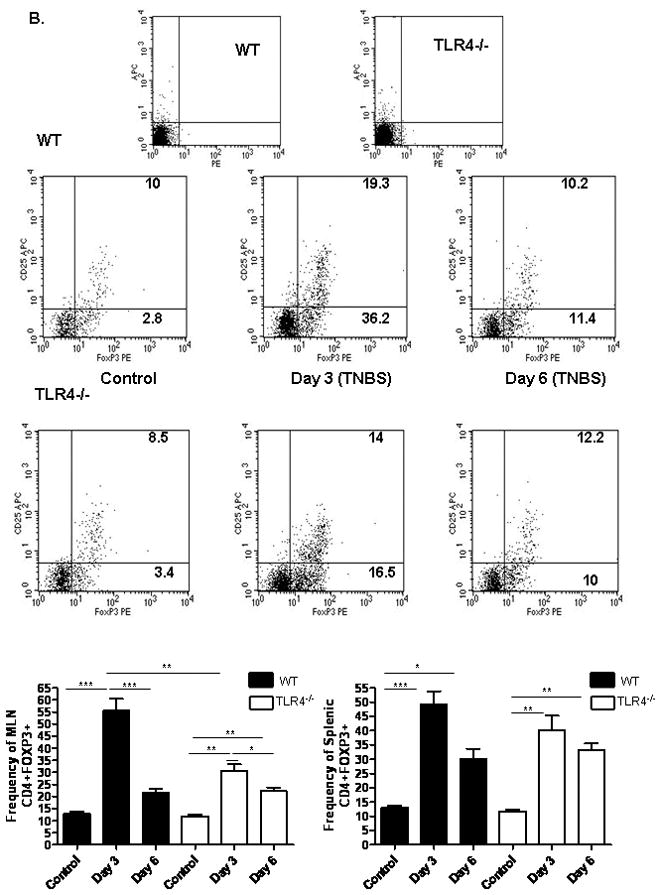
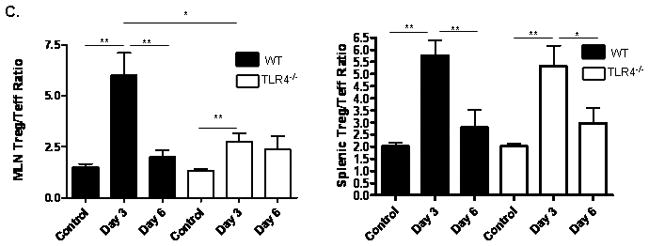
(A) The effect of TNBS administration upon the frequency of mesenteric lymph node (MLN) or splenic CD44+CD62Llo activated/memory T lymphocytes was determined by flow cytometry at days 3 and 6 from WT (n=6) and TLR4 −/− (n=6) mice, as well as WT (n=8) and TLR4 −/− (n=10) controls. Representative scatter graphs from MLN for each group are shown, as well as the mean (SEM). The first two scatter graphs are for the isotype controls used to set the gates. (B) The frequency of MLN or splenic CD4+foxp3+ regulatory T lymphocytes was determined by flow cytometry at days 3 and 6 from WT (n=5) and TLR4 −/− (n=6) mice, as well as WT (n=8) and TLR4 −/− (n=10) controls. Representative scatter graphs from MLN for each group are shown, as well as the mean (SEM). The first two scatter graphs are for the isotype controls used to set the gates. (C) The Treg/Teff ratio was calculated for each mouse from the MLN or splenic CD4+foxp3+ regulatory T lymphocyte and CD44+CD62L− activated/memory T lymphocyte frequencies. The mean (SEM) for each group was as shown. *p < 0.05, **p < 0.01, ***p < 0.001 by ANOVA with Bonferroni’s multiple correction test.
DISCUSSION
Recent studies in adult-onset IBD have linked susceptibility in part to genotypic variation in TLR4, and have described increased circulating levels of endotoxin and markers of endotoxin exposure in patients with active disease (7, 8, 27). Whether this would also be the case in pediatric-onset IBD was not known. We found that pediatric patients with IBD exhibit increased systemic endotoxin responses which are greater for those with CD compared to UC. Surprisingly, these endotoxin responses persisted despite clinical remission, and were independent of disease location or behavior. Recent studies in patients with HIV infection have characterized a defect in gut permeability in patients with progressive disease, leading to increased bacterial translocation and chronic immune activation (28). Considering a similar barrier defect occurs in CD, it is conceivable that similar effects on immune activation exist. Taken together, our data suggest that endotoxin may therefore be a key contributor to both chronic inflammation and extra-intestinal complications including growth failure in pediatric IBD.
Linear growth failure is common at diagnosis of CD, and often persists despite normalization of nutritional status. This has suggested the presence of an inflammatory growth hormone (GH) resistance. We have recently determined that endotoxin will induce GH resistance in mice via proteolytic degradation of the GH receptor (GHR), and have previously reported that TNFα neutralization will rapidly restore GH signaling in murine colitis, prior to resolution of mucosal inflammation (18, 29). We found that linear growth retardation was more common in CD patients with evidence of endotoxin exposure, as suggested by elevated serum EndoCAb IgA, and an increased APR, as demonstrated by elevated serum LBP. Importantly, weight z scores did not differ between these groups, suggesting that linear growth failure was not simply due to malnutrition. Whether endotoxin directly induces GH resistance via GHR proteolysis in CD patients is not known, and is the subject of our ongoing studies. However, it would appear likely that the subset of CD patients with persistent endotoxin exposure and a chronically activated APR are most likely to experience permanent growth failure, and could benefit from therapies targeting this pathway. Persistent elevation of serum LBP may therefore serve as a useful biomarker for identifying pediatric CD patients with the highest likelihood of experiencing refractory growth failure. As LBP is induced by both LPS and inflammatory cytokines such as IL-6, it will be important in future studies to confirm that these are in fact chronically increased in children with CD and growth failure.
As endotoxin signals through TLR4, we sought to characterize its role in the initiation of the systemic APR utilizing an experimental colitis model which encompasses both innate and adaptive features. The TNBS model was selected because of the T-cell dependency of the ensuing colitis, and because the role of TLR4 signaling in this model has not previously been explored. In addition, this model has been shown by gene array to mimic human disease (30). We found that markers of endotoxin exposure and the APR persisted at high levels in pediatric CD patients despite clinical remission. We therefore chose to utilize a low dose of TNBS based upon prior studies by our group and others, and carefully accounted for the weight of the mice (31, 32). By doing so, we sought to test the impact of intact TLR signaling upon the systemic APR and weight loss following minor colonic epithelial injury and antigenic exposure.
As a means of confirming the utility of this model in investigating the endotoxin dependent APR and for comparison with our patient-based studies, LBP was measured. To our knowledge, this has not previously been measured in a murine model of colitis. With striking similarity to the IBD patients, WT mice exhibited elevated circulating levels of LBP three days after low dose TNBS administration. LBP returned to basal levels by day 6. We then utilized a novel technique to test for TLR4 dependent differences in the associated APR. The Cincinnati capture assay allows quantification of integrated cytokine production over a 24 hour period. Biotinylated cytokine-specific antibodies are administered to mice 24 hours prior to sacrifice, and “capture” all of the cytokine which is released into the serum over that time period (26). Our data support a critical role for TLR4 in the APR to TNBS administration. We have identified for the first time a TLR4-dependent increase in both LBP and the pro-inflammatory cytokines TNFα and IL-6 at day three, which returned to basal levels by day six. This was associated with a substantial reduction in weight loss in TLR4 deficient mice compared to WT controls. Collectively, these studies have confirmed that intact TLR4 signaling is required for systemic induction of pro-inflammatory cytokines and LBP expression, and associated weight loss, following minor colonic injury. This raises the intriguing possibility that the reduction in linear growth observed in association with increased markers of endotoxin exposure and the APR in children with CD is also dependent upon intact TLR4 signaling.
Conversely, we found that the Th1 cytokine IFNγ was induced in TLR4-deficient mice following TNBS administration, relative to a more modest up-regulation in WT controls. We therefore asked whether there might also be a TLR4 dependent difference in the magnitude or kinetics of counter-regulatory IL-10 production. We found that TNBS administration led to a robust increase in IL-10 production at day three in both WT and TLR4 deficient mice. This persisted at day six in WT mice, but returned to basal levels in TLR4 deficient mice, coincident with the development of mild colitis. These data are consistent with recent studies in transfer models which have demonstrated that TLR signaling via myd88 may regulate T cell responses in colitis (22).
Considering the differences found in IFNγ and IL-10 production, we then focused upon phenotyping the MLN T cells. A modest increase in the frequency of MLN activated/memory T cells which did not reach significance was noted in both TLR4 deficient and WT mice. A more significant increase in the frequency of MLN FOXPP3+ regulatory T cells was noted in WT mice, relative to a blunted response in TLR4 deficient mice. When considering the ratio of MLN regulatory to activated/memory T cells, this was significantly increased in WT mice, and abrogated in TLR4 deficient mice. Moreover, as the activated/memory and regulatory T cell response to TNBS in the spleen did not vary by TLR4 genotype, these differences appeared to be specific for local MLN responses. This would be consistent with studies showing that TLR4 is required for the development of tolerance to food antigens in the gut (33), and that minor non-specific epithelial injury in the colon can trigger an antigen-specific regulatory T cell response (16). Future studies will define the functional properties of the foxp3+ T cells isolated from WT and TLR4 deficient mice.
Taken together, our results support a role for endotoxin and intact TLR4 signaling in both pediatric and murine colitis. Our patient-based studies have for the first time identified an association between endotoxin exposure, activation of the APR, and linear growth failure which persists during therapy of pediatric CD. Our data in the animal model have confirmed that intact TLR4 signaling is required for robust up-regulation of TNFα, IL-6, and LBP following minor colonic injury. However, our experimental data also suggest that TLR4 may exert an acute protective effect in the gut related to its role in IL-10 production and counter-regulatory T cell responses. These divergent effects of TLR4 signaling will need to be accounted for as therapies are developed to more specifically target the endotoxin:TLR4 pathway in IBD.
Supplementary Material
Linear Growth and Age, Gender, Disease Activity, and Biologic Therapy.
(A) Height z score at follow-up is shown relative to age at diagnosis. (B) Height z score at follow-up is shown stratified by gender. (C) Height z score at follow-up is shown relative to the frequency of moderate-to-severe disease activity during follow-up (0.0 to 1.0), and stratified by whether a patient experienced moderate-to-severe disease activity at any point during follow-up (mild versus moderate-to-severe). (D) Height z score at follow-up is shown stratified by exposure to infliximab therapy at any time during follow-up.
Acknowledgments
This work was supported by the Pediatric Scientist Development Program, NICHD Grant Award K12-HD00850 (BP & CK), NIH grant R01 AI075159 (CK), NIH grant R01 DK058259 (LD), the Crohn’s and Colitis Foundation of America (LD), and the Cincinnati Children’s Hospital Research Foundation Digestive Health Center Integrative Morphology Core (PHS Grant P30 DK0789392). Ramona Bezold and Kathleen Lake coordinated the patient-based studies, and Anne Ryan provided outstanding technical support.
This work was supported by the Pediatric Scientist Development Program, NICHD Grant Award K12-HD00850 (BP & CK), NIH grant R01 AI075159 (CK), NIH grant R01 DK058259 (LD), the Crohn’s and Colitis Foundation of America (LD), and the Cincinnati Children’s Hospital Research Foundation Digestive Health Center Integrative Morphology Core (PHS Grant P30 DK0789392).
Footnotes
Conflict of Interest Disclosure: The authors do not have any conflict of interest to declare; they do not have any financial interests or affiliations with institutions, organizations, or companies that are mentioned in the manuscript or whose products or services are discussed.
References
- 1.Packey CD, Sartor RB. Interplay of commensal and pathogenic bacteria, genetic mutations, and immunoregulatory defects in the pathogenesis of inflammatory bowel diseases. J Intern Med. 2008;263:597–606. doi: 10.1111/j.1365-2796.2008.01962.x. [DOI] [PubMed] [Google Scholar]
- 2.Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 4.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubinsky MC, Kugathasan S, Mei L, et al. Increased immune reactivity predicts aggressive complicating Crohn’s disease in children. Clin Gastroenterol Hepatol. 2008;6:1105–1111. doi: 10.1016/j.cgh.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollander D, Vadheim CM, Brettholz E, et al. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 7.Gardiner KR, Halliday MI, Barclay GR, et al. Significance of systemic endotoxaemia in inflammatory bowel disease. Gut. 1995;36:897–901. doi: 10.1136/gut.36.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pastor Rojo O, Lopez San Roman A, Albeniz Arbizu E, et al. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:269–277. doi: 10.1002/ibd.20019. [DOI] [PubMed] [Google Scholar]
- 9.Wellmann W, Fink PC, Benner F, et al. Endotoxaemia in active Crohn’s disease. Treatment with whole gut irrigation and 5-aminosalicylic acid. Gut. 1986;27:814–820. doi: 10.1136/gut.27.7.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zweigner J, Schumann RR, Weber JR. The role of lipopolysaccharide-binding protein in modulating the innate immune response. Microbes Infect. 2006;8:946–952. doi: 10.1016/j.micinf.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Currie CG, McCallum K, Poxton IR. Mucosal and systemic antibody responses to the lipopolysaccharide of Escherichia coli O157 in health and disease. J Med Microbiol. 2001;50:345–354. doi: 10.1099/0022-1317-50-4-345. [DOI] [PubMed] [Google Scholar]
- 12.Moretti EW, Newman MF, Muhlbaier LH, et al. Effects of decreased preoperative endotoxin core antibody levels on long-term mortality after coronary artery bypass graft surgery. Arch Surg. 2006;141:637–641. doi: 10.1001/archsurg.141.7.637. discussion 642. [DOI] [PubMed] [Google Scholar]
- 13.Stephens RC, Fidler K, Wilson P, et al. Endotoxin immunity and the development of the systemic inflammatory response syndrome in critically ill children. Intensive Care Med. 2006;32:286–294. doi: 10.1007/s00134-005-0019-z. [DOI] [PubMed] [Google Scholar]
- 14.Kronborg G, Fomsgaard A, Galanos C, et al. Antibody responses to lipid A, core, and O sugars of the Pseudomonas aeruginosa lipopolysaccharide in chronically infected cystic fibrosis patients. J Clin Microbiol. 1992;30:1848–1855. doi: 10.1128/jcm.30.7.1848-1855.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamilton-Davies C, Barclay GR, Murphy WG, et al. Passive immunisation with IgG endotoxin core antibody hyperimmune fresh frozen plasma. Vox Sang. 1996;71:165–169. doi: 10.1046/j.1423-0410.1996.7130165.x. [DOI] [PubMed] [Google Scholar]
- 16.Boirivant M, Amendola A, Butera A, et al. A transient breach in the epithelial barrier leads to regulatory T-cell generation and resistance to experimental colitis. Gastroenterology. 2008;135:1612–1623. e1615. doi: 10.1053/j.gastro.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 17.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Jiang J, Warram J, et al. Endotoxin-induced proteolytic reduction in hepatic growth hormone (GH) receptor: a novel mechanism for GH insensitivity. Mol Endocrinol. 2008;22:1427–1437. doi: 10.1210/me.2007-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomariz RP, Arranz A, Abad C, et al. Time-course expression of Toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of VIP. J Leukoc Biol. 2005;78:491–502. doi: 10.1189/jlb.1004564. [DOI] [PubMed] [Google Scholar]
- 20.Szebeni B, Veres G, Dezsofi A, et al. Increased expression of Toll-like receptor (TLR) 2 and TLR4 in the colonic mucosa of children with inflammatory bowel disease. Clin Exp Immunol. 2008;151:34–41. doi: 10.1111/j.1365-2249.2007.03531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukata M, Michelsen KS, Eri R, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 22.Fukata M, Breglio K, Chen A, et al. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886–1894. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffiths AM, Nguyen P, Smith C, et al. Growth and clinical course of children with Crohn’s disease. Gut. 1993;34:939–943. doi: 10.1136/gut.34.7.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satsangi J, Silverberg MS, Vermeire S, et al. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55:749–753. doi: 10.1136/gut.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finkelman FD, Morris SC. Development of an assay to measure in vivo cytokine production in the mouse. Int Immunol. 1999;11:1811–1818. doi: 10.1093/intimm/11.11.1811. [DOI] [PubMed] [Google Scholar]
- 27.Browning BL, Huebner C, Petermann I, et al. Has toll-like receptor 4 been prematurely dismissed as an inflammatory bowel disease gene? Association study combined with meta-analysis shows strong evidence for association. Am J Gastroenterol. 2007;102:2504–2512. doi: 10.1111/j.1572-0241.2007.01463.x. [DOI] [PubMed] [Google Scholar]
- 28.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 29.DiFedele LM, He J, Bonkowski EL, et al. Tumor necrosis factor alpha blockade restores growth hormone signaling in murine colitis. Gastroenterology. 2005;128:1278–1291. doi: 10.1053/j.gastro.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 30.te Velde AA, de Kort F, Sterrenburg E, et al. Comparative analysis of colonic gene expression of three experimental colitis models mimicking inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:325–330. doi: 10.1002/ibd.20079. [DOI] [PubMed] [Google Scholar]
- 31.Han X, Osuntokun B, Benight N, et al. Signal transducer and activator of transcription 5b promotes mucosal tolerance in pediatric Crohn’s disease and murine colitis. Am J Pathol. 2006;169:1999–2013. doi: 10.2353/ajpath.2006.060186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.te Velde AA, Verstege MI, Hommes DW. Critical appraisal of the current practice in murine TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:995–999. doi: 10.1097/01.mib.0000227817.54969.5e. [DOI] [PubMed] [Google Scholar]
- 33.Berin MC, Zheng Y, Domaradzki M, et al. Role of TLR4 in allergic sensitization to food proteins in mice. Allergy. 2006;61:64–71. doi: 10.1111/j.1398-9995.2006.01012.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Linear Growth and Age, Gender, Disease Activity, and Biologic Therapy.
(A) Height z score at follow-up is shown relative to age at diagnosis. (B) Height z score at follow-up is shown stratified by gender. (C) Height z score at follow-up is shown relative to the frequency of moderate-to-severe disease activity during follow-up (0.0 to 1.0), and stratified by whether a patient experienced moderate-to-severe disease activity at any point during follow-up (mild versus moderate-to-severe). (D) Height z score at follow-up is shown stratified by exposure to infliximab therapy at any time during follow-up.