

In developing and validating the concept of frailty as a geriatric syndrome, it has been necessary to distinguish the clinical expression of frailty from normal age-related changes and from other age-related disease pathologies. The work of Fried et al. has provided a framework for excluding potentially confounding disease, a working clinical tool to diagnose frailty and has shown associations between frailty and other pathophysiologies [1-5]. However, investigating the underlying biological basis for the geriatric syndrome of frailty through the study of basic homeostatic pathways and mechanisms has not proceeded at the same rate. The following chapter provides an overview of the homeostatic pathways emphasized in aging research and how this science may help to stimulate frailty research.
Normal aging
Aging can be defined as the decline and deterioration of functional properties at the cellular, tissue and organ level. This loss of functional properties yields a loss of homeostasis and decreased adaptability to internal and external stress yielding an increased vulnerability to disease and mortality [6]. Aging is a breakdown in maintenance of specific molecular structures and pathways, a loss of homeostasis, a failure in homeodynamics [7]. Homeodynamics refers to biological systems that do not actively mandate stasis, but rather dynamically reorganize and reset points of balance in response to internal and external change in order to maintain the systems' functional capacity across time.
Individuals are extremely heterogeneous in the onset of the aging process, rate at which it progresses, and extent to which it progresses. Differences in the manifestations of aging reflect differences in the functional capacity. Functional capacity is a direct measure of the ability of cells, tissues, and organ systems to operate properly/optimally and is influenced by both genes and environment. Optimal cellular, organ and organism operation reflect homeodynamic mechanisms/maintenance pathways. Mechanisms of maintenance include: DNA repair, synthesis and fidelity surveillance; detection and clearance of defective proteins and lipids; clearance of defective organelles and cells; and defense against pathogens and injury. Many of the physiological theories of aging are direct counterparts to these maintenance mechanisms (e.g. - DNA damage, error catastrophe, free radical, mitochondrial damage and immunosenescence theories). These maintenance mechanisms, in turn, affect homeodynamics through the cellular responses of apoptosis, senescence and repair and the systemic response of immune activation/inflammation. For example, when DNA damage is too great to be repaired, cells undergo apoptosis [8]. Cells can respond to free radical damage to DNA by either inducing senescence [9] or initiating apoptosis [10]. Oxidative damage and apoptosis are correlated negatively with the repair mechanism autophagy [11]. Proteins aberrantly modified with non-enzymatic glycation [12] or free radicals [13] can induce inflammation. Inflammation and immune responses resolve, in part, through targeted immune cell apoptosis [14]. In addition to critical telomere length, senescence can be triggered by oxidative stress [9, 15] and protein glycation/cross-links [16, 17]. Hormones contribute to homeodynamics in part through modulation of apoptosis, senescence and inflammation [18-20].
Frailty
Frailty is a geriatric syndrome characterized by weakness, weight loss, and low activity that is associated with adverse health outcomes. Frailty manifests as an age-related, biological vulnerability to stressors and decreased physiological reserves yielding a limited capacity to maintain homeostasis [3]. The validated and widely utilized five-item frailty criteria for screening: self-reported exhaustion, slowed performance (by walking speed), weakness (by grip strength), unintentional weight loss (10 lbs in past year), and low physical activity [1] are composite outcomes of multiple organ systems. The surrogate endpoint markers: elevated cytokines and chemokines [21-23]; reduced IGF-I, DHEAS and leptin [24]; perturbed neutrophil, monocyte, and white blood cell distribution [25, 26]; indicate multiple systems are dysregulated in frailty.
This definition of frailty (multisystem dysregulation yielding decreased physiological reserves and increased vulnerability to stressors) has commonality to that of aging (loss of molecular/cellular functional properties yielding decreased adaptability to internal/external stress and increased vulnerability to disease and mortality). Both have a basis in loss of homeostasis, though with aging the failure in homeodynamics is global whereas with frailty the failure in homeodynamics cycle around energy metabolism and neuromuscular changes. As researchers have characterized frail elder populations, the observed changes in both functional performance and biomarker distribution are distinct from the corresponding age-related changes observed in the non-frail (normal) individuals[1-4].
Cellular responses to stressors
For both aging and frailty, loss of homeostasis gives rise to an increased vulnerability to stressors. An organism's reaction to stressors involves cellular responses of apoptosis, senescence and repair. At the cellular level, stressors (e.g. - free radicals, DNA damage, cell injury/insult) challenge maintenance mechanisms (Figure 1). The evolved cellular response apoptosis removes damaged/aberrant cells through controlled cell death, senescence alters their phenotype and blocks further proliferation, while repair removes damaged proteins, lipids and organelles; and recycles constituent parts. Failure in these responses gives rise to transformed cells/neoplasms that can radically compromise organ function and survival. Dysregulation of these cellular responses can contribute to tissue pathology when, for example, increased apoptosis leads to tissue/organ atrophy or when expression of the senescent cell phenotype increases pro-inflammatory cytokine release. Each of these cellular responses has been implicated as playing a role in normal aging and are discussed in greater detail below. The balance between apoptosis and senescence or the acceleration of either may well precipitate changes in multiple systems and ultimately frailty and related late-life vulnerability.
Figure 1.
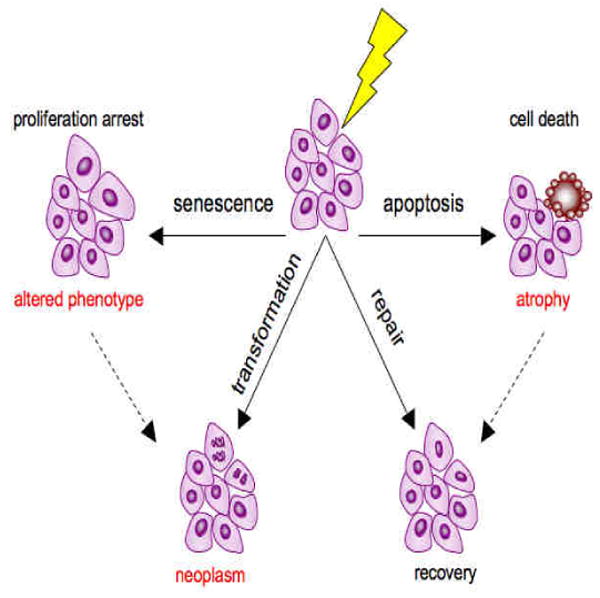
Cellular responses to stressors. Stressors (free radicals, DNA damage, nutrient or oxygen constriction, cell injury; represented by the lightening bolt) challenge cellular homeostasis. The cellular response can be senescence, apoptosis, repair, or neoplastic transformation of the cell. Senescence, a tumor suppressive response is associated with an altered secretory phenotype. The controlled cell death of apoptosis can also be tumor suppressive, though many cells, especially immune cells, normally exit through apoptosis. Apoptosis can, however yield tissue/organ atrophy. Repair enables recovery of homeostasis. In some cases apoptosis is a precursor to repair and recovery (dotted line with arrow). Additionally, the senescent cell phenotype is sometimes a precursor/ contributory to neoplasm formation and cancer progression.
(a) Apoptosis
Apoptosis is an orderly process of cellular self-destruction, a process as crucial for survival of multi-cellular organisms as cell division. Apoptosis is important in embryogenesis: morphogenesis (eliminates excess cells such as webbing between digits in embryos), selection (eliminates non-functional cells, such as neuronal pruning), immunity (eliminates dangerous cells such as self-antigen recognizing cells), and organ size (eliminates excess cells) [27]. Apoptosis is also important in adults in tissue remodeling (eliminates cells no longer needed such as mammary gland involution after lactation), maintains organ size and function [28], and eliminates damaged/dysfunctional cells [29, 30].
Apoptosis can be initiated by external signals that bind to physiological receptors on cell surfaces or by intrinsic damage that propagates cytosolic signals. Both pathways converge with mitochondrial signals that lead to a caspase cleavage cascade that results in the orderly proteolysis of proteins and DNA, the cross-linking of cell corpses and their subsequent engulfment. While death by necrosis and oncoisis (ischemic cell death) invoke major inflammatory responses and collateral damage, apoptosis is a controlled cellular demolition with no inflammatory response. Key apoptosis steps (chromatin condensation, vesicle formation and activity of hydrolytic enzymes) have a high energy demand and apoptosis is, thus, an ATP-dependent process [31]. Thus, at insufficient ATP levels, cells shift from apoptotic to necrotic cell death [32]. Consistent with this, an age-related decline in cellular ATP levels was found to promote necrotic fibroblast cell death over apoptosis in response to oxidative stress [33]. These molecular processes may stimulate chronic inflammation in older adults, which likely facilitates frailty and late-life decline through mechanisms discussed below.
As a sentinel homeodynamic cellular response, apoptosis can have pathophysiological consequences in aging. For example, too much apoptosis can yield tissue degeneration [34], while too little apoptosis allows either dysfunctional cells to accumulate or differentiated immune cells to persist [35]. Evidence suggests that sarcopenia is apoptosis driven [36]. Conversely, failures in apoptosis can contribute to the senescent cell phenotype as well as rogue cell proliferation [37]. It has been shown that apoptosis is an important cellular defense mechanism in maintaining genetic stability, and centenarians who have aged successfully possess cells that are more prone to apoptosis [38]. It is of note that the pro-inflammatory marker interleukin-6 (IL-6) appears to be protective against apoptosis [39], its serum levels are known to increase with increasing age [40] and have an inverse correlation with apoptosis [41].
(b) Senescence
Cellular senescence is a response of normal cells to potentially cancer-causing events. The term replicative senescence identifies the subset of senescent cells whose arrest in proliferation is associated with their high number of cell divisions (the Hayflick limit – typically between 40 And 60 cell division) [42]. The mechanism for replicative senescence involves each division resulting in a shortening of the telomere region of chromosomes such that a cumulative critical length is reached that does not support the DNA replication machinery. Senescent cells exhibit an irreversible arrest of cell proliferation, an altered function and in some cases a resistance to apoptosis. Besides telomere shortening, inducers of senescence include DNA damage, oncogene expression, and supermitogenic signals [43] and telomere-independent pathways which include: cytoskeletal, interferon-related, insulin-like growth factor (IGF)-related, MAP kinase and oxidative stress pathways [44]. The arrest in proliferation is imposed and maintained on cells by the induction of cyclin-dependent kinase inhibitors p16 and p21 that implement cell cycle arrest [45]. Cellular senescence is associated with typical phenotypic changes such as an enlarged morphology, activation of senescence-associated β-galactosidase, elevated expression of proteases, cell cycle inhibitors and pro-inflammatory cytokines [43].
The contribution of cellular (replicative) senescence to organismal aging has been controversial, though increasingly; evidence appears to link cellular senescence and aging [46, 47]. Senescent cells accumulate with age and at sites of age-related pathology [47]. The senescent phenotype (e.g. secretion of IL-6) may be contributory to the pro-inflammatory state observed in normal aging and that is exacerbated in frailty.
(c) Repair
The repair cellular response involves removal of damaged proteins/lipids, organelles and recycling of constituent components via the catabolic/degradative machinery of cells - proteasomes, lysosomes and autophagosomes [48]. Proteasomes are large cellular protein complexes that degrade unneeded or damaged proteins tagged with ubiquitin. Lysosomes are cellular organelles that fuse with vacuoles and dispense enzymes that degrade proteins, polysaccharides, nucleic acids and lipids present in the vacuoles. Autophagosomes are involved in sequestering cytosolic components/organelles through phagophore formation, fusing of the formed autophagosome with lysosomes, and the degrading material by the lysosomal machinery (i.e. – autophagy). Normal cycling (flux) of these cellular catabolic vacuoles prevents the accumulation of damaged/aberrant molecular and cellular components [49].
The removal and recycling phases of repair have gained considerable interest in research on aging. Conditions that modulate lifespan - mutations in the insulin, insulin-like growth factor-I (IGF-I) signaling system, treatments that reduce the expression/activity of the transcription factor mammalian Target of Rapamycin (mTOR) as well as caloric restriction all increase autophagy [49]. Aging cells exhibit an increase in mitochondrial DNA mutations and a decline in mitochondrial function [50]. In addition, free radical generation by damaged mitochondria increases. Autophagy normally maintains homeostasis through mitochondrial turnover. Dysregulation of autophagy, [51], proteasomes [52] and lysosomes [53] have all been observed with increasing age. Thus, functional capacity to remove oxidized, cross-linked, and/or unfolded proteins, nucleic acids lipids and polysaccharides is impaired and these products increase with age. Lipofuscin (“aging pigment”) arises in lysosomes through the oxidation of unsaturated fatty acids through iron-generated free radicals. Lipofuscin is insoluble and refractile to cellular removal and recycling, accumulates with age and may compromise organelle functional capacity by either ‘overstuffing’ organelles or by diverting hydolases and lipases away from their normal substrates.
Systemic responses to stressors: inflammation
Inflammation is a sentinel systemic response to stressors that plays a central role in the aging process in normal, healthy individuals [54]. The classical definition of inflammation refers to five cardinal signs: calor (heat), dolor (pain), rubor (redness), tumor (swelling), and functio laesa (loss of function). Inflammation modulates the cellular responses to stressors and cellular responses can regulate components of the inflammatory response. Immunosenescence is the decline in the function of the adaptive immune system that occurs during aging that is associated with thymic involution, alterations in T cell subsets, and reduction in antibody production. As adaptive immunity declines, innate immunity systems exhibit low-level but chronic activation that with oxidative stress leads to a low-level but chronic pro-inflammatory phenotype. A pro-inflammatory state may underlie a number of pathologies including cancer [55], cardiovascular disease [56, 57], diabetes mellitus [58], osteoporosis [59], rheumatoid arthritis [60], and cognitive disorders such as Alzheimers and Parkinson's disease [61]. It is thought that these altered cytokines, in the local tissue microenvironment, perturb cellular functional capacity facilitating disease progression. ‘Inflammaging’ is the term used to describe this pro-inflammatory state associated with aging [62] which has also been termed ‘molecular inflammation’ [63, 64].
A number of studies have shown significant association of elevated IL-6 levels with frailty in older adults [21, 65, 22, 25, 66]. The altered inflammatory state observed in frailty may contribute to a number of frailty-associated pathologies. For example, pro-inflammatory cytokines affect the growth hormone/IGF-I axis [67]. “Sickness behavior,” (fatigue, malaise, loss of interest in social activities, difficulty concentrating, and changes in sleep patterns) are triggered by the production of pro-inflammatory cytokines by macrophages and other cells of the innate immune system in response to immune challenge [68]. The ‘cytokine hypothesis of depression’ suggests that pro-inflammatory cytokines can act as neuromodulators - key factors in the (central) mediation of the behavioral, neuroendocrine and neurochemical features of depressive disorders [69, 70]. The central action of cytokines may also account for the hypothalamic-pituitary-adrenal (HPA) axis hyperactivity that is frequently observed in a number of age-related disorders, as pro-inflammatory cytokines disturb the negative feedback inhibition of circulating corticosteroids on the HPA axis [71].
Cross talk between cellular responses and inflammation
The tumor suppressors retinoblastoma protein (pRb), p53, and forkhead transcription factor (FOXO) are key modulators of apoptosis, senescence and repair responses. The central transcription factor in inflammatory responses is nuclear factor-kappaB (NF-κB). NF-κB also modulates apoptosis, senescence and repair responses. The proteins p53, pRb, FOXO, and NF-κB are controlled by complex pathways involving upstream regulators, downstream effectors and cytosolic inhibitors (for NF-κB) that regulate expression of other genes, modulate cell cycle progression and are crucial for allowing normal cells to sense and respond to apoptosis and senescence signals. These transcription factors have direct effects on either increasing or decreasing cellular responses (Table 1).
Table 1.
Cellular and systemic response regulation by key transcription factors.
| Transcription Factor | ||||
|---|---|---|---|---|
| Response | p53 | Rb | NF-κB | FOXO |
| Apoptosis | ↑ [72] | ↓ [73] | ↓ [74] | ↑ [75] |
| Senescence | ↑ [76] | ↑ [45] | ↑ [77] | ↑ [78] |
| Repair/autophagy | ↓ [79] | ↓ [80] | ↓ [81] | ↑ [82] |
| Inflammation | ↓ [83] | ↓ [84] | ↑ [85] | ↓ [86] |
The induction (↑) or repression (↓) of cellular and systemic responses by key transcription factors and the references supporting that assessment are given in parenthesis.
Interaction between p53 and pRb is well known in their role in determining whether DNA damage can be repaired or whether apoptosis should occur [87]. Cross talk also occurs between p53 and FOXO [88] and pRb and NF- κB [89]. The communication between response regulators can be multi-leveled. For example, p53 acts in at least two stages of inflammation as a general inhibitor of NF- κB-dependent transcription and, through an unknown mechanism, as a positive regulator of neutrophil clearance by macrophages [90].
Summary
The cellular responses of apoptosis, senescence and repair and the systemic response of immune activation/inflammation have evolving roles in contributing to the aging phenotype. Homeostatic mechanisms effect change through these responses, thus they are likely candidates as pathways that may be contributory to the failure in homeodynamics seen in frailty. Dysregulation of apoptosis may be contributory to the frailty traits of sarcopenia and weakness. Similarly, the cellular senescent phenotype – secreting pro-inflammatory cytokines may be contributory to the dysregulated inflammatory state seen in frailty. One can further speculate that deficits in repair in specific tissues (muscle, nerves, bone, etc) contribute to frailty. Facets of senescence, apoptosis and the repair response have yet to be carefully studied in the setting of frailty. It may be expected that the patterns observed in normal aging between these different cellular and systemic responses would be further perturbed or accelerated in the syndrome of frailty. Future investigations targeting these areas will provide the data necessary to test these hypotheses related to the biology of aging.
Acknowledgments
This work was supported in part by Grant No. AG000120 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146–56. doi: 10.1093/gerona/56.3.m146. [DOI] [PubMed] [Google Scholar]
- 2.Walston J, McBurnie MA, Newman A, et al. Frailty and activation of the inflammation and coagulation systems with and without clinical comorbidities: results from the Cardiovascular Health Study. Arch Intern Med. 2002;162(20):2333–41. doi: 10.1001/archinte.162.20.2333. [DOI] [PubMed] [Google Scholar]
- 3.Fried LP, Ferrucci L, Darer J, et al. Untangling the concepts of disability, frailty, and comorbidity: implications for improved targeting and care. J Gerontol A Biol Sci Med Sci. 2004;59(3):255–63. doi: 10.1093/gerona/59.3.m255. [DOI] [PubMed] [Google Scholar]
- 4.Bandeen-Roche K, Xue QL, Ferrucci L, et al. Phenotype of frailty: characterization in the women's health and aging studies. J Gerontol A Biol Sci Med Sci. 2006;61(3):262–6. doi: 10.1093/gerona/61.3.262. [DOI] [PubMed] [Google Scholar]
- 5.Chang SS, Weiss CO, Xue QL, et al. Patterns of Comorbid Inflammatory Diseases in Frail Older Women: The Women's Health and Aging Studies I and II. J Gerontol A Biol Sci Med Sci. 2009 doi: 10.1093/gerona/glp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holliday R. Developmental and cell biology series. Vol. 30. Cambridge; New York: Cambridge University Press; 1995. Understanding ageing; p. xiv.p. 207. [Google Scholar]
- 7.Lloyd D, Aon MA, Cortassa S. Why homeodynamics, not homeostasis? Scientific World Journal. 2001;1:133–45. doi: 10.1100/tsw.2001.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12(9):440–50. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Fischer A, Reagan JD, et al. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A. 1995;92(10):4337–41. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozawa T. Oxidative damage and fragmentation of mitochondrial DNA in cellular apoptosis. Biosci Rep. 1997;17(3):237–50. doi: 10.1023/a:1027324410022. [DOI] [PubMed] [Google Scholar]
- 11.Wohlgemuth SE, Seo AY, Marzetti E, et al. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp Gerontol. 2009 doi: 10.1016/j.exger.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramasamy R, Vannucci SJ, Yan SS, et al. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15(7):16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- 13.Nabeshi H, Oikawa S, Inoue S, et al. Proteomic analysis for protein carbonyl as an indicator of oxidative damage in senescence-accelerated mice. Free Radic Res. 2006;40(11):1173–81. doi: 10.1080/10715760600847580. [DOI] [PubMed] [Google Scholar]
- 14.Marshall JC, Watson RW. Programmed cell death (apoptosis) and the resolution of systemic inflammation. Can J Surg. 1997;40(3):169–74. [PMC free article] [PubMed] [Google Scholar]
- 15.von Zglinicki T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann N Y Acad Sci. 2000;908:99–110. doi: 10.1111/j.1749-6632.2000.tb06639.x. [DOI] [PubMed] [Google Scholar]
- 16.Nagaraj RH, Sell DR, Prabhakaram M, et al. High correlation between pentosidine protein crosslinks and pigmentation implicates ascorbate oxidation in human lens senescence and cataractogenesis. Proc Natl Acad Sci U S A. 1991;88(22):10257–61. doi: 10.1073/pnas.88.22.10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravelojaona V, Robert AM, Robert L. Expression of senescence-associated beta-galactosidase (SA-beta-Gal) by human skin fibroblasts, effect of advanced glycation end-products and fucose or rhamnose-rich polysaccharides. Arch Gerontol Geriatr. 2008 doi: 10.1016/j.archger.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Evans-Storms RB, Cidlowski JA. Regulation of apoptosis by steroid hormones. J Steroid Biochem Mol Biol. 1995;53(1-6):1–8. doi: 10.1016/0960-0760(95)00034-w. [DOI] [PubMed] [Google Scholar]
- 19.Hertoghe T. The “multiple hormone deficiency” theory of aging: is human senescence caused mainly by multiple hormone deficiencies? Ann N Y Acad Sci. 2005;1057:448–65. doi: 10.1196/annals.1322.035. [DOI] [PubMed] [Google Scholar]
- 20.Roubenoff R. Molecular basis of inflammation: relationships between catabolic cytokines, hormones, energy balance, and muscle. JPEN J Parenter Enteral Nutr. 2008;32(6):630–2. doi: 10.1177/0148607108324875. [DOI] [PubMed] [Google Scholar]
- 21.Leng S, Chaves P, Koenig K, et al. Serum interleukin-6 and hemoglobin as physiological correlates in the geriatric syndrome of frailty: a pilot study. J Am Geriatr Soc. 2002;50(7):1268–71. doi: 10.1046/j.1532-5415.2002.50315.x. [DOI] [PubMed] [Google Scholar]
- 22.De Martinis M, Franceschi C, Monti D, et al. Inflammation markers predicting frailty and mortality in the elderly. Exp Mol Pathol. 2006;80(3):219–27. doi: 10.1016/j.yexmp.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Qu T, Yang H, Walston JD, et al. Upregulated monocytic expression of CXC chemokine ligand 10 (CXCL-10) and its relationship with serum interleukin-6 levels in the syndrome of frailty. Cytokine. 2009;46(3):319–24. doi: 10.1016/j.cyto.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubbard RE, O'Mahony MS, Calver BL, et al. Plasma esterases and inflammation in ageing and frailty. Eur J Clin Pharmacol. 2008;64(9):895–900. doi: 10.1007/s00228-008-0499-1. [DOI] [PubMed] [Google Scholar]
- 25.Leng SX, Xue QL, Tian J, et al. Inflammation and frailty in older women. J Am Geriatr Soc. 2007;55(6):864–71. doi: 10.1111/j.1532-5415.2007.01186.x. [DOI] [PubMed] [Google Scholar]
- 26.Leng SX, Xue QL, Tian J, et al. Associations of neutrophil and monocyte counts with frailty in community-dwelling disabled older women: results from the Women's Health and Aging Studies I. Exp Gerontol. 2009;44(8):511–6. doi: 10.1016/j.exger.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Haanen C, Vermes I. Apoptosis: programmed cell death in fetal development. Eur J Obstet Gynecol Reprod Biol. 1996;64(1):129–33. doi: 10.1016/0301-2115(95)02261-9. [DOI] [PubMed] [Google Scholar]
- 28.Mondello C, Scovassi AI. Apoptosis: a way to maintain healthy individuals. Subcell Biochem. 50:307–23. doi: 10.1007/978-90-481-3471-7_16. [DOI] [PubMed] [Google Scholar]
- 29.Muradian K, Schachtschabel DO. The role of apoptosis in aging and age-related disease: update. Z Gerontol Geriatr. 2001;34(6):441–6. doi: 10.1007/s003910170015. [DOI] [PubMed] [Google Scholar]
- 30.Pollack M, Phaneuf S, Dirks A, et al. The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann N Y Acad Sci. 2002;959:93–107. doi: 10.1111/j.1749-6632.2002.tb02086.x. [DOI] [PubMed] [Google Scholar]
- 31.Kass GE, Eriksson JE, Weis M, et al. Chromatin condensation during apoptosis requires ATP. Biochem J. 1996;318(Pt 3):749–52. doi: 10.1042/bj3180749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richter C, Schweizer M, Cossarizza A, et al. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996;378(2):107–10. doi: 10.1016/0014-5793(95)01431-4. [DOI] [PubMed] [Google Scholar]
- 33.Miyoshi N, Oubrahim H, Chock PB, et al. Age-dependent cell death and the role of ATP in hydrogen peroxide-induced apoptosis and necrosis. Proc Natl Acad Sci U S A. 2006;103(6):1727–31. doi: 10.1073/pnas.0510346103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams JD, Mukherjee SK, Klaidman LK, et al. Apoptosis and oxidative stress in the aging brain. Ann N Y Acad Sci. 1996;786:135–51. doi: 10.1111/j.1749-6632.1996.tb39058.x. [DOI] [PubMed] [Google Scholar]
- 35.Gupta S. Molecular mechanisms of apoptosis in the cells of the immune system in human aging. Immunol Rev. 2005;205:114–29. doi: 10.1111/j.0105-2896.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- 36.Marzetti E, Leeuwenburgh C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp Gerontol. 2006;41(12):1234–8. doi: 10.1016/j.exger.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Hsu HC, Scott DK, Mountz JD. Impaired apoptosis and immune senescence - cause or effect? Immunol Rev. 2005;205:130–46. doi: 10.1111/j.0105-2896.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- 38.Franceschi C, Monti D, Scarfi MR, et al. Genomic instability and aging. Studies in centenarians (successful aging) and in patients with Down's syndrome (accelerated aging) Ann N Y Acad Sci. 1992;663:4–16. doi: 10.1111/j.1749-6632.1992.tb38643.x. [DOI] [PubMed] [Google Scholar]
- 39.Biffl WL, Moore EE, Moore FA, et al. Interleukin-6 delays neutrophil apoptosis. Arch Surg. 1996;131(1):24–9. doi: 10.1001/archsurg.1996.01430130026005. discussion 9-30. [DOI] [PubMed] [Google Scholar]
- 40.Giuliani N, Sansoni P, Girasole G, et al. Serum interleukin-6, soluble interleukin-6 receptor and soluble gp130 exhibit different patterns of age- and menopause-related changes. Exp Gerontol. 2001;36(3):547–57. doi: 10.1016/s0531-5565(00)00220-5. [DOI] [PubMed] [Google Scholar]
- 41.Kovalovich K, Li W, DeAngelis R, et al. Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl-xL. J Biol Chem. 2001;276(28):26605–13. doi: 10.1074/jbc.M100740200. [DOI] [PubMed] [Google Scholar]
- 42.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 43.Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal. 2009;11(1):59–98. doi: 10.1089/ars.2008.2104. [DOI] [PubMed] [Google Scholar]
- 44.Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008;27(46):5975–87. doi: 10.1038/onc.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohtani N, Yamakoshi K, Takahashi A, et al. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest. 2004;51(3-4):146–53. doi: 10.2152/jmi.51.146. [DOI] [PubMed] [Google Scholar]
- 46.Campisi J. Cellular senescence and apoptosis: how cellular responses might influence aging phenotypes. Exp Gerontol. 2003;38(1-2):5–11. doi: 10.1016/s0531-5565(02)00152-3. [DOI] [PubMed] [Google Scholar]
- 47.Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008;129(7-8):467–74. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2009 doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 49.Vellai T, Takacs-Vellai K, Sass M, et al. The regulation of aging: does autophagy underlie longevity? Trends Cell Biol. 2009;19(10):487–94. doi: 10.1016/j.tcb.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuervo AM, Bergamini E, Brunk UT, et al. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1(3):131–40. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 52.Chondrogianni N, Gonos ES. Proteasome dysfunction in mammalian aging: steps and factors involved. Exp Gerontol. 2005;40(12):931–8. doi: 10.1016/j.exger.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 53.Chondrogianni N, Fragoulis EG, Gonos ES. Protein degradation during aging: the lysosome-, the calpain- and the proteasome-dependent cellular proteolytic systems. Biogerontology. 2002;3(1-2):121–3. doi: 10.1023/a:1015236203379. [DOI] [PubMed] [Google Scholar]
- 54.Stout RD, Suttles J. Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunol Rev. 2005;205:60–71. doi: 10.1111/j.0105-2896.2005.00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aggarwal BB, Shishodia S, Sandur SK, et al. Inflammation and cancer: how hot is the link? Biochem Pharmacol. 2006;72(11):1605–21. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 56.Osiecki H. The role of chronic inflammation in cardiovascular disease and its regulation by nutrients. Altern Med Rev. 2004;9(1):32–53. [PubMed] [Google Scholar]
- 57.Diomedi M, Leone G, Renna A. The role of chronic infection and inflammation in the pathogenesis of cardiovascular and cerebrovascular disease. Drugs Today (Barc) 2005;41(11):745–53. doi: 10.1358/dot.2005.41.11.917342. [DOI] [PubMed] [Google Scholar]
- 58.Duncan BB, Schmidt MI. The epidemiology of low-grade chronic systemic inflammation and type 2 diabetes. Diabetes Technol Ther. 2006;8(1):7–17. doi: 10.1089/dia.2006.8.7. [DOI] [PubMed] [Google Scholar]
- 59.Ginaldi L, Di Benedetto MC, De Martinis M. Osteoporosis, inflammation and ageing. Immun Ageing. 2005;2:14. doi: 10.1186/1742-4933-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong SH, Lord JM. Factors underlying chronic inflammation in rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) 2004;52(6):379–88. [PubMed] [Google Scholar]
- 61.McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–16. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- 62.Giunta S. Is inflammaging an auto[innate]immunity subclinical syndrome? Immun Ageing. 2006;3:12. doi: 10.1186/1742-4933-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chung HY, Kim HJ, Kim KW, et al. Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc Res Tech. 2002;59(4):264–72. doi: 10.1002/jemt.10203. [DOI] [PubMed] [Google Scholar]
- 64.Chung HY, Cesari M, Anton S, et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8(1):18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leng SX, Yang H, Walston JD. Decreased cell proliferation and altered cytokine production in frail older adults. Aging Clin Exp Res. 2004;16(3):249–52. doi: 10.1007/BF03327392. [DOI] [PubMed] [Google Scholar]
- 66.Hubbard RE, O'Mahony MS, Calver BL, et al. Nutrition, inflammation, and leptin levels in aging and frailty. J Am Geriatr Soc. 2008;56(2):279–84. doi: 10.1111/j.1532-5415.2007.01548.x. [DOI] [PubMed] [Google Scholar]
- 67.Johnson RW, Arkins S, Dantzer R, et al. Hormones, lymphohemopoietic cytokines and the neuroimmune axis. Comp Biochem Physiol A Physiol. 1997;116(3):183–201. doi: 10.1016/s0300-9629(96)00277-0. [DOI] [PubMed] [Google Scholar]
- 68.Dantzer R, Bluthe RM, Laye S, et al. Cytokines and sickness behavior. Ann N Y Acad Sci. 1998;840:586–90. doi: 10.1111/j.1749-6632.1998.tb09597.x. [DOI] [PubMed] [Google Scholar]
- 69.Myint AM, Kim YK. Cytokine-serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses. 2003;61(5-6):519–25. doi: 10.1016/s0306-9877(03)00207-x. [DOI] [PubMed] [Google Scholar]
- 70.Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):201–17. doi: 10.1016/j.pnpbp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 71.Porter RJ, Gallagher P, O'Brien JT. Effects of rapid tryptophan depletion on salivary cortisol in older people recovered from depression, and the healthy elderly. J Psychopharmacol. 2006 doi: 10.1177/0269881106064341. [DOI] [PubMed] [Google Scholar]
- 72.Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000;2(4):E65–7. doi: 10.1038/35008695. [DOI] [PubMed] [Google Scholar]
- 73.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22(56):9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 74.Cardoso SM, Oliveira CR. Inhibition of NF-kB renders cells more vulnerable to apoptosis induced by amyloid beta peptides. Free Radic Res. 2003;37(9):967–73. [PubMed] [Google Scholar]
- 75.Jagani Z, Singh A, Khosravi-Far R. FoxO tumor suppressors and BCR-ABL-induced leukemia: a matter of evasion of apoptosis. Biochim Biophys Acta. 2008;1785(1):63–84. doi: 10.1016/j.bbcan.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wesierska-Gadek J, Ranftler C, Schmid G. Physiological ageing: role of p53 and PARP-1 tumor suppressors in the regulation of terminal senescence. J Physiol Pharmacol. 2005 2:56. 77–88. [PubMed] [Google Scholar]
- 77.Bernard D, Gosselin K, Monte D, et al. Involvement of Rel/nuclear factor-kappaB transcription factors in keratinocyte senescence. Cancer Res. 2004;64(2):472–81. doi: 10.1158/0008-5472.can-03-0005. [DOI] [PubMed] [Google Scholar]
- 78.Kyoung Kim H, Kyoung Kim Y, Song IH, et al. Down-regulation of a forkhead transcription factor, FOXO3a, accelerates cellular senescence in human dermal fibroblasts. J Gerontol A Biol Sci Med Sci. 2005;60(1):4–9. doi: 10.1093/gerona/60.1.4. [DOI] [PubMed] [Google Scholar]
- 79.Maiuri MC, Galluzzi L, Morselli E, et al. Autophagy regulation by p53. Curr Opin Cell Biol. 2009 doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 80.Tracy K, Dibling BC, Spike BT, et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27(17):6229–42. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Copetti T, Bertoli C, Dalla E, et al. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29(10):2594–608. doi: 10.1128/MCB.01396-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao J, Brault JJ, Schild A, et al. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy. 2008;4(3):378–80. doi: 10.4161/auto.5633. [DOI] [PubMed] [Google Scholar]
- 83.Zheng SJ, Lamhamedi-Cherradi SE, Wang P, et al. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes. 2005;54(5):1423–8. doi: 10.2337/diabetes.54.5.1423. [DOI] [PubMed] [Google Scholar]
- 84.Ying L, Marino J, Hussain SP, et al. Chronic inflammation promotes retinoblastoma protein hyperphosphorylation and E2F1 activation. Cancer Res. 2005;65(20):9132–6. doi: 10.1158/0008-5472.CAN-05-1358. [DOI] [PubMed] [Google Scholar]
- 85.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Snoeks L, Weber CR, Wasland K, et al. Tumor suppressor FOXO3 participates in the regulation of intestinal inflammation. Lab Invest. 2009;89(9):1053–62. doi: 10.1038/labinvest.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hooper ML. The role of the p53 and Rb-1 genes in cancer, development and apoptosis. J Cell Sci Suppl. 1994;18:13–7. doi: 10.1242/jcs.1994.supplement_18.3. [DOI] [PubMed] [Google Scholar]
- 88.You H, Mak TW. Crosstalk between p53 and FOXO transcription factors. Cell Cycle. 2005;4(1):37–8. doi: 10.4161/cc.4.1.1401. [DOI] [PubMed] [Google Scholar]
- 89.Kim YA, Lee WH, Choi TH, et al. Involvement of p21WAF1/CIP1, pRB, Bax and NF-kappaB in induction of growth arrest and apoptosis by resveratrol in human lung carcinoma A549 cells. Int J Oncol. 2003;23(4):1143–9. [PubMed] [Google Scholar]
- 90.Zhou M, Gu L, Zhu N, et al. Transfection of a dominant-negative mutant NF-kB inhibitor (IkBm) represses p53-dependent apoptosis in acute lymphoblastic leukemia cells: interaction of IkBm and p53. Oncogene. 2003;22(50):8137–44. doi: 10.1038/sj.onc.1206911. [DOI] [PubMed] [Google Scholar]