

Abstract
Phytochrome photoreceptors mediate light responses in plants and in many microorganisms. Here we report studies using 1H–13C magic-angle spinning NMR spectroscopy of the sensor module of cyanobacterial phytochrome Cph1. Two isoforms of the red-light absorbing Pr ground state are identified. Conclusive evidence that photoisomerization occurs at the C15-methine bridge leading to a β-facial disposition of the ring D is presented. In the far-red-light absorbing Pfr state, strong hydrogen-bonding interactions of the D-ring carbonyl group to Tyr-263 and of N24 to Asp-207 hold the chromophore in a tensed conformation. Signaling is triggered when Asp-207 is released from its salt bridge to Arg-472, probably inducing conformational changes in the tongue region. A second signal route is initiated by partner swapping of the B-ring propionate between Arg-254 and Arg-222.
Keywords: chromophore-protein interaction, signal transduction, solid-state NMR, photomorphogenesis
Phytochrome was first demonstrated in plants as a red-light-dependent photoreceptor regulating numerous photomorphogenic processes (1, 2). Phytochromes are, however, also now known in photosynthetic prokaryotes including cyanobacteria (3, 4), nonphotosynthetic bacteria (5), and fungi (6). Generally, the phytochrome apoprotein binds an open-chain tetrapyrrole as a chromophore (7, 8) to form the red-light absorbing Pr ground state (λmax ≈ 658 nm in case of cyanobacterial phytchrome Cph1 from Synechocystis 6803). Red light absorption photoactivates the molecule to form the photoactivated far-red-light absorbing Pfr state (λmax ≈ 702 nm for Cph1) via a series of intermediates (8–10). Photoactivation is thought to be initiated by a double bond isomerization of the chromophore (10, 11). Early NMR spectroscopic studies on proteolytic phytochrome fragments (12, 13) indicated that this isomerization occurs at the C15═C16 double bond (for numbering, see Fig. 1A), a geometrical change in line with vibrational spectroscopic investigations (14–16) and results from recent 13C solid-state NMR (17, 18) in which the most significant changes during the light-triggered conversions are confined to rings C and D. Exact geometries of the chromophore in the Pr state have been resolved as periplanar ZZZssa configurations in bacteriophytochromes from Deinococcus radiodurans (19) and Rhodopseudomonas palustris (20) as well as in the more plant-phytochrome-like Cph1 from the cyanobacterium Synechocystis 6803 (21). On the other hand, the crystal structure of the unusual bacteriophytochrome PaBphP Pseudomonas aeruginosa (22) whose ground state is Pfr shows a ZZEssa conformation, consistent with the expected primary photochemistry at the C15═C16 double bond (Fig. 1 B vs. C). Very recently, however, Ulijasz et al. presented structural simulations based on liquid NMR data of a 20-kDa GAF (cGMP phosphodiesterase/adenylyl cyclase/FhlA) domain fragment of “SyB-Cph1” phytochrome from the thermotolerant cyanobacterium Synechococcus OSB′ (23). Surprisingly, they concluded that photoisomerization occurs at the methine bridge between rings A–B rather than C–D, proposing the former as a general model for phytochrome activation.
Fig. 1.

Interfacial 1H contacts of the PCB chromophore. (A) Schematic of protein-bound PCB shown in the Pr ZZZssa geometry. The tetrapyrrole rings and representative PCB atoms are labeled for reference. (B and C) Structural views showing residue contacts of the chromophore observed in heteronuclear correlation NMR spectra (see also Figs. S1 and S2) as (B) Pr and (C) Pfr. The presentations for Pr (B) and Pfr (C) states are modeled according to the Cph1 2VEA (21) and PaBphP 3C2W crystal structures (22), respectively. Water locations (W1–W7) are marked as red spheres and numbered (42). (D–G) Close-up views of the 1H contacts of the two D-ring carbons, C19 and C171, as Pr (D and F) and Pfr (E and G). The dashed lines highlight the 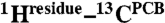 interfacial correlations during Pr → Pfr photoconversion. All 1H contacts of the chromophore are summarized in Datasets S3 and S4.
interfacial correlations during Pr → Pfr photoconversion. All 1H contacts of the chromophore are summarized in Datasets S3 and S4.
Here we investigate the photochemistry of the chromophore within the sensory module of Cph1 phytochrome (residues 1–514, Cph1Δ2) in greater detail. This includes the N-terminal PAS (PER-ARNT-SIM) domain, the chromophore-binding GAF domain, and the PHY (phytochrome-associated) domain that is generally essential for stabilization of the Pfr state. An extended hairpin loop of the PHY domain (the tongue) closes the chromophore pocket, rendering it inaccessible to the solvent. We represent 1H sensitivity-enhanced heteronuclear NMR correlation experiment (MELODI–HETCOR) for selectively recognizing 1H contacts of the uniformly 13C/15N-labeled phycocyanobilin (PCB) chromophore in both Pr and Pfr states at 100% occupancy. The MELODI filter effectively silences magnetization of protons directly bonded to the chromophore, while emphasizing its medium- and long-range 1H contacts (24, 25). The sensitivity enhancement of 1H indirect dimension was achieved through the use of windowed phase-modulated Lee–Goldburg (wPMLG) homonuclear decoupling scheme (26–28) over the 1H evolution period (t1), in our case, wPMLG3+. Our previous 2D 13C–13C dipolar-assisted rotational resonance (DARR) NMR studies on various states of this protein provided full sets of 13C chemical shifts of the chromophore (18) and hence the basis for the interpretation of the heteronuclear spectra presented in Fig. 2 as well as Figs. S1 and S2. The experiments reported here represent an application of the MELODI–HETCOR method to a particularly large protein (approximately 58 kDa).
Fig. 2.

Light-induced changes in PCB chromophore geometry. The contour plot of the 2D 1H–13C correlation spectra of u-[13C,15N]-PCB-Cph1Δ2 as Pr (red) and Pfr (purple) reveals the intramolecular proximity between protons bound to tetrapyrrole nitrogens (HN21–N24) and chromophore carbons (Dataset S1). Chemical shifts of 13C (as indicated by dashed vertical lines, Pr in red, Pfr in purple) and HN (indicated by solid horizontal lines, Pr in red, Pfr in purple) are in agreement with our previous data (18, 33). Several residue correlations in the close proximity to the chromophore (within a region of approximately 3.5 Å) are also resolved (labeled with Latin capitals A to O, listed in Dataset S2). Spectral regions without resonances, 15–35, 57–88, and 95–109 ppm in the ω2-dimension are omitted. For 1H spectra in the ω1-dimension, only the characteristic spectral region of 8–14 ppm is shown.
Results and Discussion
We first recorded the MELODI–HETCOR NMR spectra for Cph1Δ2 in both parent states at 100% occupancy [Fig. 2, Pr (red) and Pfr (purple)] with a 900-μs Lee–Goldburg cross-polarization (LG–CP) contact time to build up intramolecular heteronuclear correlations (i.e., a cutoff distance of approximately 3.5 Å) between the retained NH (HN21–N24) and OH protons with the chromophore. In both states, four protons bound to the tetrapyrrole nitrogens are well resolved and can be assigned straightforwardly according to our previous studies (18) (Datasets S1 and S2). The absence of cross-peaks involving carboxylate protons (C83 and C123) in our data indicates that these side chains are deprotonated, implicated by earlier studies (19–21, 29).
To explore the site of photoisomerization of the chromophore, we focused on its NH contacts. HN24 (9.9 ppm) of ring D correlates in the Pr state (red) with C13 (126.5 ppm), C131 (11.4 ppm), and C14 (146.1 ppm) of ring C, whereas these contacts are lost in Pfr (purple). The disappearance of these signals is most likely driven by the substantial rotation of ring D in which HN24 is moved away from the C-ring carbons, that is, far beyond the magnetization transfer range set (approximately 3.5 Å). Other correlations are preserved upon Pfr formation, implying that the light-induced geometrical changes at those positions are small. In particular, the maintenance of contacts between HN21 (11.6 ppm) of ring A and C6 (149.3 ppm) and C9 (127.9 ppm) of ring B provides unequivocal evidence that C5-methine bridge is essentially unaffected or only transiently affected by Pr → Pfr photoconversion. The site of photoisomerization in Cph1 is also identified by comparing interfacial 1H contacts of the PCB carbons in both states, Pr (spectra shown in Fig. S1, contacts visualized in Fig. 1B) and Pfr (Fig. S2 and Fig. 1C), within an enclosure sphere up to approximately 5.5 Å (SI Text). On photoconversion to Pfr, most of intermolecular 1H contacts of rings A–C are maintained, whereas D-ring carbons, in particular C19 and C171 (Fig. 1 D vs. E, and F vs. G) exhibit entirely different 1H correlations, providing clear evidence that the Pr → Pfr phototransformation is associated with Z-to-E isomerization at the C15═C16 methine bridge (Datasets S3 and S4). Our data thus refute the idea that a photoisomerization between rings A–B constitutes the photoactivation mechanism in all phytochromes (23). The lack of the PHY domain in the SyB-Cph1 (GAF) fragment (23) might explain the apparent discrepancy in the primary motion of the chromophore during photoconversion.
The same spectra also allow investigation of the light-induced changes for amino acids adjacent to the chromophore (Figs. S1 and S2). In most cases, 1H contacts of a specific chromophore carbon have been validated by the 1H contacts of the adjacent carbons. Hence, the assignments in Figs. S1 and S2 represent consistent networks of interfacial 1H–13C correlations within a 5.5-Å sphere. These networks in the Pr state are in harmony with the Cph1 2VEA structure (21). Also, most of the Cph1 Pfr 1H correlations of carbons in rings A–C (Fig. S2 and Dataset S3) agree well with the crystal structure of bacteriophytochrome PaBphP (Protein Data Bank ID 3C2W) (22). However, the 1H correlations of the D-ring carbons (e.g., C171) conflict radically with 3C2W (Fig. 3A). Circular dichroism (CD) studies of Cph1 and plant phytochromes on the one hand and of bacteriophytochromes on the other suggest that the D-ring facial disposition of Pfr is radically different in the two phytochrome types (30). Indeed, modeling of the Cph1 Pfr with a β-facial disposition of ring D (relative to rings B and C) predicts interactions in accord with those measured here (Fig. 3B). We can therefore confirm that the β-facial D-ring disposition in Cph1 Pfr is distinct from that in bacteriophytochromes (α-facial D-ring disposition, Fig. 3A). Because CD spectra for plant phytochrome (31) and Cph1 (30, 32) are similar, plant phytochromes too are likely to have a similar β-facial D-ring disposition. The Cph1 Pfr model requires a movement of the two tyrosine side chains (Tyr-176 and Tyr-203) for the accommodation of its β-facial chromophore ring D, as suggested for PaBphP (22). We do not know the sense of the rotational vector during the ring-D photoflip, although the anticlockwise route from Pr to Pfr is somewhat shorter (150° ∼ 160° instead of 200° ∼ 210° clockwise). Furthermore, with this route, steric hindrance between C131 and C171 might cause the observed Cph1 β-facial disposition of ring D in the Pfr state (30).
Fig. 3.

D-ring facial disposition in the Pfr state of PaBphP and Cph1. In the PaBphP 3C2W Pfr structure (A) (22), the light-induced photoisomerization tilts the ring D approximately 50° anticlockwise with respect to the PCB plane (rings B and C). Such an α-facial D-ring disposition (D-αf) of PaBphP (indicated by a solid arrow; ref. 30) leads to a poor agreement with our observed NMR contacts for Cph1, in particular D-ring carbons (e.g., C171, Datasets S3 and S4). Some 1H contacts of the C171 within the detection range of C–H distance (approximately 5.5 Å) are not resolved (red dashed lines, A), others appear for the C171 despite a distance much beyond the 5.5-Å prediction (blue dashed lines, A). If, on the other hand, the chromophore adopts β-facial disposition for its ring D (D-βf) relative to the rings B and C (B), the distance constraints can match our observed NMR contacts for Cph1 very well, as shown for the C171 (blue dashed lines, B). The residues codes for Cph1 sequence are given in parentheses following the equivalent codes for PaBphP sequence (see Dataset S5).
In the Pr state, ring D forms a conserved hydrogen bond with His-290 via its carbonyl group (21) (Fig. 1D). The side chain of acidic Asp-207, pointing toward Arg-472, is negatively charged in both parent states, indicative from the absence of 1H correlations with the chromophore (an Oδ2⋯C171 distance of 3.55 Å extracted from the 2VEA Pr structure) (21). The carboxylic group of Asp-207 in the Pr state interacts with the guanidium moiety (Nη1/Nη2) of Arg-472 to form a conserved salt bridge. The Pr → Pfr photoflip breaks the interaction between ring D and His-290 and leads to the formation of hydrogen-bonding interactions of the D-ring carbonyl group to Tyr-263 and of N24 to Asp-207 (Figs. 1E and 4C), apparently stabilizing the Pfr chromophore in a more tensed conformation (33), as also implied by the PaBphP 3C2W Pfr structure (22). The newly formed Pfr interactions (Tyr-263⋯C19 and Asp-207⋯N24) require disruption of the Asp-207/Arg-472 salt bridge (Fig. 4 B vs. C) as supported by the remarkable 1H up-field shift of approximately 3.7 ppm for the correlations involving HNη1 of Arg-472 (Dataset S4). The origin for this dramatic 1H change occurring at the guanidium moiety either might be a deprotonation, induced by a sudden increase in hydrophobicity of the local environment as a result of the closer placement of the D-ring carbonyl (14) or a positive charge redistribution among its three nitrogens. In any case, Arg-472 of the tongue is the most prominent site of 1H NMR chemical shift change surrounding the chromophore; as we show below, this change is particularly important for signal transduction.
Fig. 4.

Hydrogen-bonding networks and charge distribution inside the PCB-binding pocket. (A–C) Region surrounding the PCB chromophore for the two Pr isoforms (Pr-I, A and Pr-II, B) as well as for Pfr (C). Dashed lines highlight the hydrogen-bonding networks in the tetrapyrrole cavity. The + (blue) and − (red) signs represent the positive and negative charges, respectively. The conjugation pattern of the chromophore is colored in red for Pr (A and B) and purple for Pfr (C) (18).
The Za → Ea photoisomerization of ring D in Cph1 leads to a shift of the two inner rings B and C within the pocket by a clockwise in-plane rotation (as viewed from the α-face of the bilin ring system) as proposed as the “rotate” step in the “flip-and-rotate” model by Yang et al. (22, 34). A bidentate salt bridge between the B-ring propionate and Arg-254 (two O⋯N distances of 2.67 and 3.33 Å, respectively) (Fig. 4 A and B) in the Pr state is seen in the Cph1 2VEA structure (21) and is clear from our NMR data (Fig. S1 and Dataset S3). Photoconversion to Pfr is associated with the partner swap of the B-ring propionate from Arg-254 to Arg-222, as evidenced by the newly formed Pfr correlations involving C83 and the guanidium moiety (HNη1 and HNη2) of Arg-222 (Fig. 4C and Dataset S3). The partner swap is also supported by published structure and mutational studies of different phytochromes, such as Arg-254 in DrBphP (35) and Arg-101/Arg-133 in SyB-Cph1 (36). This reassociation implies an inward movement of the side chain of Arg-222 toward the GAF domain and is probably important in signal transduction (see below). The positional change of pyrrole rings B and C is also detected by the conserved Ile-208, located on the β-face of the chromophore ring system, via its long-range correlations involving the side chain hydrogens (i.e., HCγ1 and HCδ1) (Dataset S4).
A structural heterogeneity of the chromophore in the Pr state was detected in phyA from oat (Avena sativa L.) (37) as well as in D. radiodurans DrBphP and Agrobacterium Agp1 by optical spectroscopies (35, 38). Also, ultrafast absorption spectroscopy on wild-type and mutants of Rp. palustris RpBphP2 showed two kinetic components (39). Discrepancies between Pr fluorescence excitation and absorbance spectra of Cph1Δ2 have also been noted and associated with Pr heterogeneity (40). Local doubling of 13C signals was also detected in our previous DARR NMR experiments on Synechocystis Cph1Δ2 and a recombinant N-terminal fragment of phytochrome A oat (65 kDa, phyA65), implying the coexistence of two Pr isoforms in solution (18). Our current NMR data allow for exact characterization of these, here termed Pr-I and Pr-II (Fig. 4 A and B, respectively). They are distinguished specifically by their hydrogen-bonding networks and the charge distribution in the tetrapyrrole cavity. The Cph1 Pr correlation spectrum in Fig. S1 reveals a weak proton contact (σH of 8.9 ppm in the ω1-dimension) at 13C frequencies of several carbons of rings C (e.g., C122, C123, and C131) and D (e.g., C16 and C19) (Dataset S4) which is not correlated with any amino acid in the proximity of the chromophore and the conserved water molecules in the binding pocket. We attribute these signals to a hydrated hydroxide cluster in the form of a 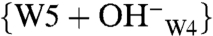 species (41). As shown in Fig. 4B, W4 is closely surrounded by formal charges, for example Nϵ2 of His-260 (distance of 3.19 Å, 2VEA), Oγ of Ser-272 (2.56 Å), W5 (2.73 Å), and the carboxylate group of the C12 propionate side chain (2.43 Å). The proton freed from the highly polarized W4 might cause the observed protonation of His-260, and the remaining hydroxide ion binds strongly with W5 to form the negatively charged hydrated cluster (Fig. 4A).
species (41). As shown in Fig. 4B, W4 is closely surrounded by formal charges, for example Nϵ2 of His-260 (distance of 3.19 Å, 2VEA), Oγ of Ser-272 (2.56 Å), W5 (2.73 Å), and the carboxylate group of the C12 propionate side chain (2.43 Å). The proton freed from the highly polarized W4 might cause the observed protonation of His-260, and the remaining hydroxide ion binds strongly with W5 to form the negatively charged hydrated cluster (Fig. 4A).
Also His-260 is distinguished in the two Pr isoforms. 1H correlations in the spectral region of 10–18 ppm (in ω1-dimension, Fig. S1) identify a cationic and a neutral form of the imidazole side chain. The resonances of HCδ2 (labeled as H260Cδ2-I) and HNϵ2 (H260Nϵ2-I) of the cationic imidazolium (Pr-I, Fig. 4A) are centered at 15.4 and 17.9 ppm, respectively. Striking 1H up-field shifts of 4.0 ppm of HCδ2 (to 11.4 ppm, H260Cδ2-II) and 3.9 ppm of HNϵ2 (to 14.0 ppm, H260Nϵ2-II) are observed in the neutral isoform (Pr-II, Fig. 4B). Also for the highly conserved His-290, the coexistence of two neutral imidazole tautomers Nδ1-H (σH of 11.7 ppm, Fig. 4A and Dataset S4) and Nϵ2-H (σH of 12.7 ppm, Fig. 4B and Dataset S4) is observed. As indicated by the 2VEA structure (21), C182 of D-ring ethyl side chain (172.7 ppm in ω1-dimension, Fig. S1) is at almost equal distance (approximately 4.3 Å) from both imidazole nitrogens, whereas the intensity of the C182⋯N δ1-H signal is approximately 35% of the signal strength of the correlation with the Nϵ2-H tautomer (Fig. S1), suggesting that the Nϵ2-H tautomeric form of His-290 (in Pr-II) is favored. Additional support for the proposal of the coexistence of two Pr conformers in the PCB-binding pocket is obtained from the 1H–13C correlations involving the pyrrole water (W1) (for example, C10 slice centered at 112.8 ppm in the ω2-dimension), which reveals a small but resolved doubling of the W1 signal with two resonances separated by approximately 0.6 ppm in the ω1-dimension (W1-I vs. W1-II). We note that the 2VEA Pr structure (21) represents the Pr-II isoform only. The homogeneity of the crystals might be due to the fact that Pr-II contains one charge-separated ion pair less than Pr-I and is therefore expected to be more stable. In the Pfr state, there is no hint of multiple isoforms; on the contrary, most cross-peaks are remarkably sharp, also suggesting a well-defined structure (Fig. S1 vs. S2).
QM/MM (quantum mechanics/molecular mechanics) calculations of the Pr state of Cph1 (42) suggest that the change of protonation state of His-260 induces a significant change of orientation of its imidazole ring in respect to the chromophore plane (Fig. 4 A and B). Our data indeed show that the overall position of the histidine residue is affected. As shown in Fig. S1, a correlation signal reflecting the contact between HNδ1 of His-260 and C131 of ring C (Fig. S3A) is fully resolved in the Pr-I isoform. This distance of 6.03 Å (2VEA) is well beyond the effective maximum transfer range with a 2.3-ms LG–CP contact time (a cutoff distance of approximately 5.5 Å; SI Text). Similarly, the comparison of heteronuclear correlations involving the HNε2 of His-260 in the two Pr isoforms clearly confirms its shift (Fig. S3 B and C). The movement of the cationic His-260 toward the anionic C-ring propionate is because of their electrostatic interactions in the Pr-I isoform (Fig. 4A).
In both Cph1Δ2 and phyA65, all four chromophore nitrogens are protonated in both parent states (18). The counterions, however, have not yet been identified. Besides the additional charge separation between His-260 and 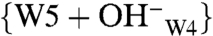 in Pr-I (Fig. 4A), there are no significant differences in the counterionic patterns of the two Pr isoforms. As depicted in Fig. 4 A and B, the C-ring propionate itself might act as a counterion for the protonated tetrapyrrole system. Upon photoconversion to the Pfr state, the carboxylate side chain of the conserved Asp-207 serves as counterion to the positively charged chromophore (Fig. 4C). Hence, the chain of cations and anions forming pairs of counterions is reorganized. In the Pr state, a substantial portion of the net positive charge is delocalized on the two inner rings B and C (43) (Fig. 4 A and B), whereas in the Pfr state, ring D carries the partial positive charge (16) (Fig. 4C). This “soft cation” on the chromophore in Pr could be effectively neutralized by a “soft counterion,” thus partial charges could be involved and the countercharge of the chromophore might be spread over several groups around the C-ring propionate side chain including His-260. The H260Q mutant clearly shows that this residue is critical for chromophore protonation (44). On the other hand, Asp-207, the likely Pfr-state counterion is approximately 2.60 Å from HN24 in PaBphP 3C2W, localizing this electrostatic interaction and conjugation defects at ring D. Moreover, this aspartate residue provides sufficient tension on the ring D to stabilize the chromophore in a more tensed shape (18, 45).
in Pr-I (Fig. 4A), there are no significant differences in the counterionic patterns of the two Pr isoforms. As depicted in Fig. 4 A and B, the C-ring propionate itself might act as a counterion for the protonated tetrapyrrole system. Upon photoconversion to the Pfr state, the carboxylate side chain of the conserved Asp-207 serves as counterion to the positively charged chromophore (Fig. 4C). Hence, the chain of cations and anions forming pairs of counterions is reorganized. In the Pr state, a substantial portion of the net positive charge is delocalized on the two inner rings B and C (43) (Fig. 4 A and B), whereas in the Pfr state, ring D carries the partial positive charge (16) (Fig. 4C). This “soft cation” on the chromophore in Pr could be effectively neutralized by a “soft counterion,” thus partial charges could be involved and the countercharge of the chromophore might be spread over several groups around the C-ring propionate side chain including His-260. The H260Q mutant clearly shows that this residue is critical for chromophore protonation (44). On the other hand, Asp-207, the likely Pfr-state counterion is approximately 2.60 Å from HN24 in PaBphP 3C2W, localizing this electrostatic interaction and conjugation defects at ring D. Moreover, this aspartate residue provides sufficient tension on the ring D to stabilize the chromophore in a more tensed shape (18, 45).
The question rises along which pathway the signal is transduced from the chromophore to the rest of the molecule. The observed changes in the 1H NMR chemical shifts for amino acids surrounding the chromophore during Pr → Pfr photoconversion (Fig. S4) have widespread effects on the protein pocket. A concerted matrix reaction is seen in the region located on α-face of the chromophore ring system undergoing up-field 1H shifts (represented as blue spheres). Interestingly, this region contains the side chain of Arg-472 which participates in a salt bridge with Asp-207 that is broken upon phototransformation (10, 33) (see above). In particular, a dramatic 3.7-ppm 1H up-field shift of tongue residue Arg-472 is observed (Dataset S4). Indeed, proteolytic studies with Pr and Pfr imply different accessibilities of the tongue region (45). Arg-472 is likely to play a major role in determining the structure of this region because it hydrogen bonds via its Nϵ group to Gly-451 (β17 helix) which creates the kink in the tongue. Along the GAF–tongue interface, further hydrogen bondings with conserved tongue residues in the kink region involving Trp-478 and Glu-480 (β20 helix) are likely to play a role in signal transduction. Although mutations at Asp-207 and homologous residues destabilize or prevent the formation of Pfr (44–46), mutation of its salt-bridge partner Arg-472 in Cph1 has little effect on Pr/Pfr absorbance properties and Pfr stability, even though this residue too is conserved. Its likely critical function is in initiating signal transduction, as implied by the loss of Pr/Pfr autokinase regulation in the full-length Arg-472-Ala holoprotein mutant (Fig. S5B) and state-dependent surface changes of the Cph1Δ2 sensory module (21). Although we can rule out a C5-isomerization in Cph1, global changes in the pocket apparently lead to a partner swap between the B-ring propionate (C83) from Arg-254 in Pr to Arg-222 in Pfr (see above). Interestingly, the FixL “two-component” redox sensor might also signal via an analogous tetrapyrrole propionate-Arg partner swap (47). Like Arg-472, although Arg-254 is conserved, R254A has little effect photochemically or on Pfr stability. It is thus likely to be important in intramolecular signal transduction, as implied by the effects of Arg-254-Ala on state-dependent surface changes in Cph1Δ2 (21) and in vivo signaling in the Cph8 chimera (48) (Fig. S5A). Furthermore, the Arg-254-homologous residue in plant phytochrome B is also important in signal transduction via PIF3 (49, 50). Although the salt bridges might have other functions, such as stabilizing Pfr, arginine-to-alanine substitutions at positions 222, 254, and 472 are not associated with rapid Pfr dark reversion in Cph1. Thus it appears that signal transduction is triggered not only via the Asp-207/Arg-472 salt-bridge pathway to the tongue and perhaps the N terminus but also via a salt-bridge partner swap between the B-ring propionate and Arg-254/Arg-222 on the opposite side of the chromophore pocket.
In summary, we have been able to exploit the MELODI–HETCOR NMR technique to investigate the protonic structure and charge distributions of both Pr and Pfr states of the canonical phytochrome Cph1. Our data reveal the simultaneous presence of two Pr isoforms (Fig. 4 A and B) with Pr-II closely matching the 2VEA Pr structure. The Pfr NMR data (Dataset S4) agree with the PaBphP Pfr 3C2W structure except for the D-ring region, due to the ring D in Cph1 adopting an opposite facial disposition (Fig. 3), consistent with CD spectroscopy (30). We also can rule out a C5 photoisomerization between rings A–B in Cph1 as proposed by Ulijasz et al. (23). The photoflip activates two intramolecular signal transduction routes via the salt bridges on either side of the chromophore.
Materials and Methods
Sample Preparation.
Preparation of uniformly 13C- and 15N-labeled PCB chromophore, Cph1Δ2 apo- and holoproteins is described in the SI Text. For NMR measurements of Pr-state Cph1Δ2 phytochrome, the sample was preirradiated with light from an array of far-red light-emitting diodes (λmax ≈ 730 nm, 20 nm FWHM) surrounding a 1-mm bore glass capillary syringe. Pfr Cph1Δ2 phytochrome at 100% occupancy was obtained by size-exclusion chromatography of the 70/30 Pfr/Pr photoequilibrium mixture using Superdex 200 (Pharmacia/GE) (51). Greater than 98% Pfr occupancy was confirmed by UV-visible absorbance spectroscopy. Mutagenesis and assays of LacZ and the control of Cph8 (Cph1:EnvZ chimeric phytochrome, see ref. 48) are given in the SI Text.
Pulse Sequence.
The 2D NMR experiment combines MELODI filter (24, 25), LG–CP block with wPMLG3+ decoupling (26–28) to yield medium- and long-range 1H–13C correlation spectra with 1H-enhanced sensitivity. The wPMLG3+ decoupling efficiency correlates to several experimental parameters, like rf field power (ν1), unit pulse length (Δt), acquisition windows (τw), rotation frequency (νr), and 1H rf offset frequency (Δν). The optimizations were carried out by acquiring 1D 1H wPMLG3+ magic-angle spinning (MAS) spectra on glycine through monitoring the splitting between α-protons lines (28). To optimize performance for 2D fashion, ν1 and Δt were further examined in the indirect dimension of 2D 1H{wPMLG3+}–1H{wPMLG3+} homonuclear correlation spectra of the same sample (52). Optimum values were obtained using a pulse length Δt of 2.1 μs and rf amplitude (ν1) of 78 kHz. In addition, the acquisition window τw was set to 6.5 μs, slightly higher than the calibrated value, for compensation of the spectral quality loss caused by any mistuned wobble curve (52). The PMLG3 segment was composed of six rf pulses (Δt = 2.1 μs) with a delay τx of 0.1 μs (28) between two pulses to stabilize their phases, thus τPMLG3 = 13.2 μs. The overall length of decoupling cycle time (τc) of wPMLG3+ pulse unit was equal to 19.7 μs (τc = τPMLG3 + τw), thus the characteristic frequency (νc) of the wPMLG3+ CRAMPS (combined rotation and multiple-pulse spectroscopy) cycle was 50.76 kHz. To avoid an undesirable destructive interference between MAS and spin space modulation, the rotor period (τr) was chosen to meet the condition of 3.5 < τr/τc < 5, and τr ≠ 4τc (27, 53, 54). For rf power levels available in our spectrometer using 4-mm triple-resonance MAS probe, 10–13.5-kHz spinning speed is commonly used. The rotation frequency (νr) of 11.778 kHz (τr = 84.90 µs, thus τr/τc ≈ 4.31) was found to be optimal for the spectral resolution with 1H rf field strength (ν1) of 78 kHz and to be sufficiently distant from obvious degeneracy number, τr/τc = 4. Moreover, 1H rf offset frequency (Δν) of -6,000 Hz was set to avoid spiky resonance lines from falling over the spectrum (28).
MAS NMR Data Collection.
To maximize the overall NMR signal intensity of each Cph1Δ2 phytochrome sample, approximately 15 mg of protein were loaded into a 4-mm zirconia MAS rotor. All 2D 1H–13C interfacial correlation spectra were acquired at 233 ± 0.3 K on a Bruker AV-750 WB spectrometer. The spinning frequency was 11,778 ± 4 Hz, regulated by a pneumatic control unit. LG–CP contact times of 900 μs and 2.3 ms were used for both Pr and Pfr states (Fig. 2 as well as Figs. S1 and S2). The dipolar filter period of 38 μs was set to suppress the 1H–1H homonuclear dipolar interaction effectively (24, 25). The 1H power was ramped 80–100% during cross-polarization, and 1H rf field strength for TPPM (two-pulse phase modulation) decoupling was about 80 kHz. Typical 1H 90° and 13C 180° pulse lengths were 3.1 and 5.2 μs, respectively. The spectrum was recorded with 3,072 transients with a relaxation delay of 1.536 s for each t1 slice, and a total of 128 slices combined in the indirect dimension, leading to a total experimental time of ca 138 h. The t1 data were recorded in an off-resonance manner and linear predicted by 32 points using 40 linear predication coefficients. A 90° shifted squared sine bell window function was applied and zero-filled to 2,048 points prior to Fourier transformation. The t2 data were zero-filled to 4,096 points and 50 Hz line broadening for exponential multiplication.
13C resonances were externally referenced to the down-field resonance of solid glycine hydrochloride at 176.04 ppm, and 1H solid-state shifts were calibrated by running a conventional 1H–13C HETCOR experiment on the tyrosine hydrochloride through its HCβ and HO peaks centered at 2.1 and 13.1 ppm, respectively. The 1H datasets of Arg, His, and Tyr are experimentally acquired in our lab by employing the conventional 1H–13C/15N HETCORs on those amino acids, and the other detected residues (Ala, Asp, Cys, Ile, Leu, Met, Phe, Pro, Ser, Thr, and Val) are all referenced to the standard values reported in liquid- and solid-state NMR spectroscopy (25, 55, 56). All amino acids used in this study were obtained from Sigma-Aldrich. The NMR data were processed with Bruker Topspin 2.1 and further analyzed by using the Sparky 3.114 (T. D. Goddard and D. G. Kneller, SPARKY 3. University of California, San Francisco, CA). The figures were produced using the PyMOL 1.2 (DeLano Scientific) (57) and Adobe Illustrator CS4 (Adobe) software. For resonance assignments, please see SI Text.
Supplementary Material
Acknowledgments.
The authors thank Prof. S. Vega (Weizmann Institute of Science, van Arkel guest Professor 2010 at Universiteit Leiden), Prof. J. Lugtenburg, Dr. J. Neugebauer (Leiden Institute of Chemistry), and Dr. T. Rohmer for fruitful discussions. Support from F. Lefeber, K.B. Sai Sankar Gupta, and K. Erkelens (Leiden) as well as L. Teufel (Giessen) during various stages of the experiments is gratefully acknowledged. We thank Prof. H.J.M. de Groot for his continuing interest and support. Funding by the Volkswagen-Stiftung (I/82628) is gratefully acknowledged.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013377108/-/DCSupplemental.
References
- 1.Butler WL, Norris KH, Siegelman HW, Hendricks SB. Detection, assay, and preliminary purification of the pigment controlling photoresponsive development of plants. Proc Natl Acad Sci USA. 1959;45:1703–1708. doi: 10.1073/pnas.45.12.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith H. Phytochromes and light signal perception by plants—an emerging synthesis. Nature. 2000;407:585–591. doi: 10.1038/35036500. [DOI] [PubMed] [Google Scholar]
- 3.Hughes J, et al. A prokaryotic phytochrome. Nature. 1997;386:663. doi: 10.1038/386663a0. [DOI] [PubMed] [Google Scholar]
- 4.Yeh K-C, Wu S-H, Murphy JT, Lagarias JC. A cyanobacterial phytochrome two-component light sensory system. Science. 1997;277:1505–1508. doi: 10.1126/science.277.5331.1505. [DOI] [PubMed] [Google Scholar]
- 5.Davis SJ, Vener AV, Vierstra RD. Bacteriophytochromes: Phytochrome-like photoreceptors from nonphotosynthetic eubacteria. Science. 1999;286:2517–2520. doi: 10.1126/science.286.5449.2517. [DOI] [PubMed] [Google Scholar]
- 6.Blumenstein A, et al. The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Curr Biol. 2005;15:1833–1838. doi: 10.1016/j.cub.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 7.Lagarias JC, Lagarias DM. Self-assembly of synthetic phytochrome holoprotein in vitro. Proc Natl Acad Sci USA. 1989;86:5778–5780. doi: 10.1073/pnas.86.15.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borucki B, et al. Light-induced proton release of phytochrome is coupled to the transient deprotonation of the tetrapyrrole chromophore. J Biol Chem. 2005;280:34358–34364. doi: 10.1074/jbc.M505493200. [DOI] [PubMed] [Google Scholar]
- 9.Remberg A, et al. Raman spectroscopic and light-induced kinetic characterization of a recombinant phytochrome of the cyanobacterium Synechocystis. Biochemistry. 1997;36:13389–13395. doi: 10.1021/bi971563z. [DOI] [PubMed] [Google Scholar]
- 10.Heyne K, et al. Ultrafast dynamics of phytochrome from the cyanobacterium Synechocystis, reconstituted with phycocyanobilin and phycoerythrobilin. Biophys J. 2002;82:1004–1016. doi: 10.1016/S0006-3495(02)75460-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braslavsky SE, Gärtner W, Schaffner K. Phytochrome photoconversion. Plant Cell Environ. 1997;20:700–706. [Google Scholar]
- 12.Rüdiger W, Thümmler F, Cmiel E, Schneider S. Chromophore structure of the physiologically active form (Pfr) of phytochrome. Proc Natl Acad Sci USA. 1983;80:6244–6248. doi: 10.1073/pnas.80.20.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagarias JC, Rapoport H. Chromopeptides from phytochrome. The structure and linkage of the Pr form of the phytochrome chromophore. J Am Chem Soc. 1980;102:4821–4828. [Google Scholar]
- 14.Foerstendorf H, et al. FTIR studies of phytochrome photoreactions reveal the C = O bands of the chromophore: Consequences for its protonation states, conformation, and protein interaction. Biochemistry. 2001;40:14952–14959. doi: 10.1021/bi0156916. [DOI] [PubMed] [Google Scholar]
- 15.Matysik J, Hildebrandt P, Schlamann W, Braslavsky SE, Schaffner K. Fourier-transform resonance Raman spectroscopy of intermediates of the phytochrome photocycle. Biochemistry. 1995;34:10497–10507. doi: 10.1021/bi00033a023. [DOI] [PubMed] [Google Scholar]
- 16.Dasgupta J, Frontiera RR, Taylor KC, Lagarias JC, Mathies RA. Ultrafast excited-state isomerization in phytochrome revealed by femtosecond stimulated Raman spectroscopy. Proc Natl Acad Sci USA. 2009;106:1784–1789. doi: 10.1073/pnas.0812056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohmer T, et al. 15N MAS NMR studies of Cph1 phytochrome: Chromophore dynamics and intramolecular signal transduction. J Phys Chem B. 2006;110:20580–20585. doi: 10.1021/jp062454+. [DOI] [PubMed] [Google Scholar]
- 18.Rohmer T, et al. Light-induced chromophore activity and signal transduction in phytochromes observed by 13C and 15N magic-angle spinning NMR. Proc Natl Acad Sci USA. 2008;105:15229–15234. doi: 10.1073/pnas.0805696105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner JR, Zhang J, Brunzelle JS, Vierstra RD, Forest KT. High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J Biol Chem. 2007;282:12298–12309. doi: 10.1074/jbc.M611824200. [DOI] [PubMed] [Google Scholar]
- 20.Yang X, Stojković EA, Kuk J, Moffat K. Crystal structure of the chromophore binding domain of an unusual bacteriophytochrome, RpBphP3, reveals residues that modulate photoconversion. Proc Natl Acad Sci USA. 2007;104:12571–12576. doi: 10.1073/pnas.0701737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Essen L-O, Mailliet J, Hughes J. The structure of a complete phytochrome sensory module in the Pr ground state. Proc Natl Acad Sci USA. 2008;105:14709–14714. doi: 10.1073/pnas.0806477105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Kuk J, Moffat K. Crystal structure of Pseudomonas aeruginosa bacteriophytochrome: Photoconversion and signal transduction. Proc Natl Acad Sci USA. 2008;105:14715–14720. doi: 10.1073/pnas.0806718105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ulijasz AT, et al. Structural basis for the photoconversion of a phytochrome to the activated Pfr form. Nature. 2010;463:250–254. doi: 10.1038/nature08671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao XL, Schmidt-Rohr K, Hong M. Medium- and long-distance 1H–13C heteronuclear correlation NMR in solids. J Magn Reson. 2001;149:139–143. [Google Scholar]
- 25.Yao XL, Hong M. Dipolar filtered 1H–13C heteronuclear correlation spectroscopy for resonance assignment of proteins. J Biomol NMR. 2001;20:263–274. doi: 10.1023/a:1011251924874. [DOI] [PubMed] [Google Scholar]
- 26.Bosman L, Madhu PK, Vega S, Vinogradov E. Improvement of homonuclear dipolar decoupling sequences in solid-state nuclear magnetic resonance utilising radiofrequency imperfections. J Magn Reson. 2004;169:39–48. doi: 10.1016/j.jmr.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Vinogradov E, Madhu PK, Vega S. Proton spectroscopy in solid state nuclear magnetic resonance with windowed phase modulated Lee–Goldburg decoupling sequences. Chem Phys Lett. 2002;354:193–202. [Google Scholar]
- 28.Leskes M, Madhu PK, Vega S. Proton line narrowing in solid-state nuclear magnetic resonance: New insights from windowed phase-modulated Lee–Goldburg sequence. J Chem Phys. 2006;125:124506. doi: 10.1063/1.2352737. [DOI] [PubMed] [Google Scholar]
- 29.Bhoo SE, et al. Phytochrome photochromism probed by site-directed mutations and chromophore esterification. J Am Chem Soc. 1997;119:11717–11718. [Google Scholar]
- 30.Rockwell NC, Shang L, Martin SS, Lagarias JC. Distinct classes of red/far-red photochemistry within the phytochrome superfamily. Proc Natl Acad Sci USA. 2009;106:6123–6127. doi: 10.1073/pnas.0902370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Litts JC, Kelly JM, Lagarias JC. Structure-function studies on phytochrome: Preliminary characterization of highly purified phytochrome from Avena sativa enriched in the 124-kilodalton species. J Biol Chem. 1983;258:11025–11031. [PubMed] [Google Scholar]
- 32.Borucki B, et al. Mechanism of Cph1 phytochrome assembly from stopped-flow kinetics and circular dichroism. Biochemistry. 2003;42:13684–13697. doi: 10.1021/bi035511n. [DOI] [PubMed] [Google Scholar]
- 33.Rohmer T, et al. Phytochrome as molecular machine: Revealing chromophore action during the Pfr → Pr photoconversion by magic-angle spinning NMR spectroscopy. J Am Chem Soc. 2010;132:4431–4437. doi: 10.1021/ja9108616. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Kuk J, Moffat K. Conformational differences between the Pfr and Pr states in Pseudomonas aeruginosa bacteriophytochrome. Proc Natl Acad Sci USA. 2009;106:15639–15644. doi: 10.1073/pnas.0902178106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner JR, et al. Mutational analysis of Deinococcus radiodurans bacteriophytochrome reveals key amino acids necessary for the phytochromicity and proton exchange cycle of phytochromes. J Biol Chem. 2008;283:12212–12226. doi: 10.1074/jbc.M709355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornilescu G, Ulijasz AT, Cornilescu CC, Markley JL, Vierstra RD. Solution structure of a cyanobacterial phytochrome GAF domain in the red-light-absorbing ground state. J Mol Biol. 2008;383:403–413. doi: 10.1016/j.jmb.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt P, et al. The complexity of the Pr to Pfr phototransformation kinetics is an intrinsic property of native phytochrome. Photochem Photobiol. 1998;68:754–761. [Google Scholar]
- 38.von Stetten D, et al. Chromophore heterogeneity and photoconversion in phytochrome crystals and solutions studied by resonance Raman spectroscopy. Angew Chem Int Ed. 2008;47:4753–4755. doi: 10.1002/anie.200705716. [DOI] [PubMed] [Google Scholar]
- 39.Toh KC, Stojković EA, van Stokkum IHM, Moffat K, Kennis JTM. Proton-transfer and hydrogen-bond interactions determine fluorescence quantum yield and photochemical efficiency of bacteriophytochrome. Proc Natl Acad Sci USA. 2010;107:9170–9175. doi: 10.1073/pnas.0911535107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sineshchekov V, Koppel L, Esteban B, Hughes J, Lamparter T. Fluorescence investigation of the recombinant cyanobacterial phytochrome (Cph1) and its C-terminally truncated monomeric species (Cph1Δ2): Implication for holoprotein assembly, chromophore–apoprotein interaction and photochemistry. J Photochem Photobiol, B. 2002;67:39–50. doi: 10.1016/s1011-1344(02)00282-8. [DOI] [PubMed] [Google Scholar]
-
41.Diken EG, et al. Fundamental excitations of the shared proton in the
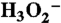 and
and 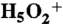 complexes. J Phys Chem A. 2005;109:1487–1490. doi: 10.1021/jp044155v. [DOI] [PubMed] [Google Scholar]
complexes. J Phys Chem A. 2005;109:1487–1490. doi: 10.1021/jp044155v. [DOI] [PubMed] [Google Scholar] - 42.Mroginski MA, et al. Chromophore structure of cyanobacterial phytochrome Cph1 in the Pr state: Reconciling structural and spectroscopic data by QM/MM calculations. Biophys J. 2009;96:4153–4163. doi: 10.1016/j.bpj.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borg OA, Durbeej B. Which factors determine the acidity of the phytochromobilin chromophore of plant phytochrome? Phys Chem Chem Phys. 2008;10:2528–2537. doi: 10.1039/b719190a. [DOI] [PubMed] [Google Scholar]
- 44.Hahn J, et al. Probing protein–chromophore interactions in Cph1 phytochrome by mutagenesis. FEBS J. 2006;273:1415–1429. doi: 10.1111/j.1742-4658.2006.05164.x. [DOI] [PubMed] [Google Scholar]
- 45.Esteban B, Carrascal M, Abian J, Lamparter T. Light-induced conformational changes of cyanobacterial phytochrome Cph1 probed by limited proteolysis and autophosphorylation. Biochemistry. 2005;44:450–461. doi: 10.1021/bi0484365. [DOI] [PubMed] [Google Scholar]
- 46.von Stetten D, et al. Highly conserved residues Asp-197 and His-250 in Agp1 phytochrome control the proton affinity of the chromophore and Pfr formation. J Biol Chem. 2007;282:2116–2123. doi: 10.1074/jbc.M608878200. [DOI] [PubMed] [Google Scholar]
- 47.Gong W, Hao B, Chan MK. New mechanistic insights from structural studies of the oxygen-sensing domain of Bradyrhizobium japonicum FixL. Biochemistry. 2000;39:3955–3962. doi: 10.1021/bi992346w. [DOI] [PubMed] [Google Scholar]
- 48.Levskaya A, et al. Synthetic biology: Engineering Escherichia coli to see light. Nature. 2005;438:441–442. doi: 10.1038/nature04405. [DOI] [PubMed] [Google Scholar]
- 49.Oka Y, et al. Mutant screen distinguishes between residues necessary for light-signal perception and signal transfer by phytochrome B. PLoS Genet. 2008;4:e1000158. doi: 10.1371/journal.pgen.1000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kikis EA, Oka Y, Hudson ME, Nagatani A, Quail PH. Residues clustered in the light-sensing knot of phytochrome B are necessary for conformer-specific binding to signalling partner PIF3. PLoS Genet. 2009;5:e1000352. doi: 10.1371/journal.pgen.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strauss HM, Schmieder P, Hughes J. Light-dependent dimerisation in the N-terminal sensory module of cyanobacterial phytochrome 1. FEBS Lett. 2005;579:3970–3974. doi: 10.1016/j.febslet.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 52.Coelho C, Rocha J, Madhu PK, Mafra L. Practical aspects of Lee–Goldburg based CRAMPS techniques for high-resolution 1H NMR spectroscopy in solids: Implementation and applications. J Magn Reson. 2008;194:264–282. doi: 10.1016/j.jmr.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 53.Zorin VE, Ernst M, Brown SP, Hodgkinson P. Insights into homonuclear decoupling from efficient numerical simulation: Techniques and examples. J Magn Reson. 2008;192:183–196. doi: 10.1016/j.jmr.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 54.Mafra L, Coelho C, Siegel R, Rocha J. Assessing the performance of windowed 1H CRAMPS methods, on biological solids, at high-field and MAS up to 35 kHz. J Magn Reson. 2009;197:20–27. doi: 10.1016/j.jmr.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Gu Z, Ridenour CF, Bronnimann CE, Iwashita T, McDermott A. Hydrogen bonding and distance studies of amino acids and peptides using solid state 2D 1H–13C heteronuclear correlation spectra. J Am Chem Soc. 1996;118:822–829. [Google Scholar]
- 56.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 57.DeLano WL. The PyMOL Molecular Graphic System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.