

Abstract
The abundance and inherent potential for variations in simple sequence repeats (SSRs) or microsatellites resulted in valuable source for genetic markers in eukaryotes. We describe the organization and abundance of SSRs in fungus Fusarium graminearum (causative agent for Fusarium head blight or head scab of wheat). We identified 1705 SSRs of various nucleotide repeat motifs in the sequence database of F. graminearum. It is observed that mononucleotide repeats (62%) were most abundant followed by di- (20%) and trinucleotide repeats (14%). It is noted that tetra-, penta- and hexanucleotide repeats accounted for only 4% of SSRs. The estimated frequency of Class I SSRs (perfect repeats ≥20 nucleotides) was one SSR per 124.5 kb, whereas the frequency of Class II (perfect repeats >10 nucleotides and ≫20 nucleotides) was one SSR per 25.6 kb. The dynamics of SSRs will be a powerful tool for taxonomic, phylogenetic, genome mapping and population genetic studies as SSR based markers show high levels of allelic variation, codominant inheritance and ease of analysis.
Keywords: SSR, microsatellites, Fusarium graminearum, EST
Background
Fusarium head blight or head scab of wheat is a global problem in the humid and subhumid wheat growing areas of world [1]. Fusarium head blight has been associated with upto 17 organisms, of which Fusarium graminearum is the principle pathogen responsible for head blight in many countries including India [2]. In addition to causing yield losses, strains of F. graminearum are known to produce trichothecene mycotoxins which pose a serious threat to human and animal health and food safety [3].
Simple sequence repeats (SSRs) or microsatellites have been proven to be the markers of choice during the last decade in plant research because of their hypervariability and ease of detection. SSR markers have been developed for many species of plants, animals and fungi from genomic DNA through the construction of SSR enriched libraries. This approach is labor intensive and time consuming. However in recent years, with the establishment of several sequencing projects in crop plants, animals and microorganisms resulted in a wealth of DNA sequence information. This sequence data for expressed sequence tags (ESTs), genes and cDNA clones can be downloaded from various databases in public domain and by using computer programs these can be scanned for identification of SSRs, referred as EST-SSRs or genic microsatellites. Microsatellite sequences obtained through in silico mining have more or less the same utility and potential comparative with those derived from a genomic library. However, the negligible cost of in silico mining and high abundance of microsatellites in different sequence resources make this approach extremely attractive for the generation of microsatellite markers. SSR provides a powerful tool for taxonomic, phylogenetic and population genetic studies because of its highly polymorphic nature. The polymorphism in SSRs is generally believed to be the result of DNA polymerase slippage and unequal recombination [4]. The information on abundance and distribution of SSRs may also help in understanding their relevance in gene function or genome evolution. The main objective of this study was to analyze the abundance and distribution of different classes of SSRs in the EST database Fusarium graminearum, which may help in understanding the evolution and diversity analysis.
Methodology
Dataset
The genomic sequences (433 contigs of 36.22 Mb) of Fusarium graminearum available in Fusarium comparative database of Broad Institute of MIT and Harvard, Cambridge (http://www.broadinstitute.org/ annotation/genome/fusarium_graminearum/) were used for the study.
SSR analysis
Perfect mono-, di-, tri-, tetra-, penta-, and hexanucleotide motifs with a repeat of ≥6 times were identified using the software WebSat (SSR finder program)[5]. The sequences from each contigs were downloaded from Fusarium comparative database and entered in the WebSat software. As the program can process 150,000 characters, the longer sequences were divided into two or more parts and then processed for SSR analysis. The output generated by the program highlight the SSR sequences in yellow color.
Discussion
Genomic sequence data of 36.22 Mb size of F. graminearum assembled into 433 contigs and further assembled into 31 scaffolds or supercontigs from Broad Institute (http://www.broadinstitute.org/) was used to search for mono-, di-, tri-, tetra-, penta- and hexanucleotide motifs with a repeat of ≥6 times. A total number of 1705 SSRs were identified from the EST database >Fusarium graminearum (Table 1 see Table 1). Chromosome 1 possessed highest number of SSRs (611) and chromosome 3 had the least number of SSRs (318). Ten SSRs were identified in the contigs not mapped to any chromosome. Mononucleotides repeats were the most abundant (1063) repeats in all the chromosomes accounting 62% of SSRs. Next to mononucleotides, dinucleotides (20%) were predominant followed by trinucleotides (14%). Tetra-, penta- and hexanucleotide repeats were the least frequent repeats accounting 4% of SSRs. The density of SSRs was found to be one SSR per 21.2 kb.
Among the mononucleotides, polyA and polyT were more abundant repeats with a frequency of 492 and 439 (Table 2 see Table 2), respectively. PolyG and polyC repeats were rare representing 5.9% and 6.5% of mononucleotide repeats. The number of repeat units ranged from 10 to 41 among mononucleotides, but majority of repeats had 10-12 repeat units. Twelve types of dinucleotide repeat motifs (Table 3 see Table 3) were found in the genome. The AT/TA dinucleotide repeat motif was the most predominant while the CG/GC repeat motif was rare. Among trinucleotide repeats, 53 different types of repeat motifs were identified and the CTT repeat motif was predominant in Fusarium graminearum genome. Tetra-, penta - and hexanucleotide repeats were least frequent repeats in the genome, tetranucleotide repeats occur more in number (34) followed by penta- (21) and hexanucleotide repeats (14). The genome possessed 29 different types of tetra-, 20 types of penta- and 14 types of hexanucleotide repeats. The number of repeat units in di-, tri-, tetra-, penta- and hexanucleotides ranged from 6 to 46, but the majority of SSRs (70%) had six to seven repeat units. Some of the highly repeated sequences identified were (AG)28, (AAG)31, (GAA)46, (GTATG)18, (GAAGAG)21, (TGAAGA)22 and (CCCTAA)23.
SSRs were categorized into two groups based on length of SSR tracts and their potential as informative genetic markers: Class I SSRs contain perfect repeats ≥20 nucleotides in length and Class II contain perfect repeats >10 nucleotides and <20 nucleotides in length. Out of 1705 SSRs, 291 repeats were categorized as Class I SSRs. 54% of trinucleotide repeats were Class I SSRs, followed by dinucleotide (14%) and mononucleotide repeats (4.5%) (Figure 1). All tetra- , penta - and hexanucleotide repeats were Class I SSRs. The estimated frequency of Class I SSRs was one SSR per 124.5 kb, whereas the frequency of Class II was one SSR per 25.6 kb.
Figure 1.
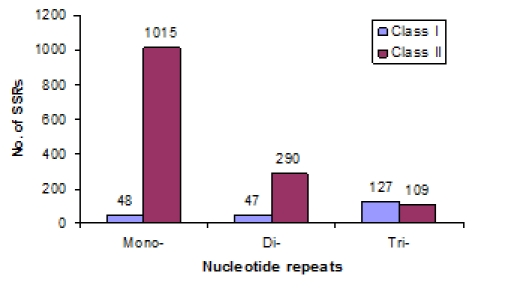
Frequency of Class I and Class II SSRs of mono-, di-and trinucleotide repeats.
The in silico mining of EST database of F. graminearum provided a rich source of SSRs which can be used for taxonomic, evolutionary and population genetic studies. Role of microsatellites in regulation of gene expression and in the evolution of gene regulation [6, 7] are well documented. The implications of excess numbers of short iterated repeats could be extremely important not only for genomic stability, but also for the evolution of additional genomic features such as codon usage [8].
In general, microsatellites show a decrease in abundance with increasing repeat length [9]. However, more than the expected number of long microsatellite repeats were also reported [10]. The rationale for Class I and Class II categories of SSRs is that longer perfect repeats (Class I) are highly polymorphic as evidenced by the experimental data originally reported from human [11] and then confirmed by studies in many other organisms, including rice [12]. Microsatellites in Class II tended to be less variable, representing sites where SSR expansion may occasionally occur but its probability is limited due to a smaller chance of slipped-strand impairing over the shorter SSR template [11, 13]. The microsatellites identified in this study could be used for the development of genome specific markers for evolutionary studies in F. graminearum.
Supplementary material
Acknowledgments
The financial support for Agri Bioinformatics Promotion Program provided by Bioinformatics Initiative Division, Department of Information Technology, Ministry of Communications & Information Technology, Government of India, New Delhi is gratefully acknowledged. The authors are thankful to Project Director, DWR, Karnal for providing facilities for this work.
Footnotes
Citation:Singh et al, Bioinformation 5(10): 402-404 (2011)
References
- 1.Gilbert J, Tekauz A. Can J Plant Pathol. 2000;22:1. [Google Scholar]
- 2.Saharan MS, et al. Proc Indian Natl Sci Acad. 2004;B70:255. [PMC free article] [PubMed] [Google Scholar]
- 3.Bottalico A, Perrone G. Eur J Plant Pathol. 2002;108:611. [Google Scholar]
- 4.Li YC, et al. Mol Ecol. 2002;11:2453. doi: 10.1046/j.1365-294x.2002.01643.x. [DOI] [PubMed] [Google Scholar]
- 5.Martins WS, et al. Bioinformation. 2009;3:282. doi: 10.6026/97320630003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li YC, et al. Mol Biol Evol. 2004;21:991. doi: 10.1093/molbev/msh073. [DOI] [PubMed] [Google Scholar]
- 7.Huang TS, et al. Biochem Biophys Res Commun. 2003;300:901. doi: 10.1016/s0006-291x(02)02962-5. [DOI] [PubMed] [Google Scholar]
- 8.Field D, Wills C. Proc Nat Acad Sci.USA. 1998;95:1947. doi: 10.1073/pnas.95.4.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grover A, et al. Mol Genet Genomics. 2007;277:469. doi: 10.1007/s00438-006-0204-y. [DOI] [PubMed] [Google Scholar]
- 10.Dieringer D, Schlotterer C. Genome Res. 2003;13:2242. doi: 10.1101/gr.1416703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber JL. Genomics. 1990;7:524. doi: 10.1016/0888-7543(90)90195-z. [DOI] [PubMed] [Google Scholar]
- 12.Cho YG, et al. Theor Appl Genet. 2000;95:713. [Google Scholar]
- 13.Temnykh S, et al. Genome Res. 2001;11:1441. doi: 10.1101/gr.184001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.