

Abstract
Inherited erythromelalgia (IEM) and paroxysmal extreme pain disorder (PEPD) are inherited pain syndromes predominantly caused by missense mutations in the peripheral neuronal voltage-gated sodium channel (Nav) isoform Nav1.7. While both IEM and PEPD mutations increase neuronal excitability, IEM mutations primarily enhance activation and PEPD mutations impair inactivation. In addition, one PEPD mutation, Nav1.7-I1461T, has been shown to increase resurgent sodium currents in dorsal root ganglion (DRG) neurons. Because resurgent currents have been implicated in increased neuronal excitability, we asked whether (1) additional PEPD mutations located within the putative inactivation gate and docking sites and (2) IEM mutations might also increase these unusual currents. Resurgent currents are generated following open-channel block at positive potentials by an endogenous blocking particle and subsequent expulsion of this blocker upon repolarization to moderately negative potentials. Here we used a mimetic of the putative blocking particle, the Navβ4 peptide, to determine if enhanced resurgent currents are induced by three distinct PEPD mutations and two IEM mutations in stably transfected HEK293 cells. We demonstrate that (1) Nav1.7-mediated resurgent currents are observed in HEK293 cells with the Navβ4 peptide in the recording pipette, (2) while the PEPD mutants M1627K, T1464I and V1299F exhibit enhanced resurgent current amplitudes compared to wild-type, the IEM mutants I848T and L858H do not, and (3) there is a strong correlation between the decay time constant of open-channel fast inactivation and resurgent current amplitude. These data suggest that resurgent currents may play a role in the neuronal hyperexcitability associated with PEPD, but not IEM, mutations.
Non-technical summary
Abnormal pain sensitivity associated with inherited and acquired pain disorders occurs through increased excitability of peripheral sensory neurons in part due to changes in the properties of voltage-gated sodium channels (Navs). Resurgent sodium currents (INaR) are atypical currents believed to be associated with increased excitability of neurons and may have implications in pain. Mutations in Nav1.7 (peripheral Nav isoform) associated with two genetic pain disorders, inherited erythromelalgia (IEM) and paroxysmal extreme pain disorder (PEPD), enhance Nav1.7 function via distinct mechanisms. We show that changes in Nav1.7 function due to mutations associated with PEPD, but not IEM, are important in INaR generation, suggesting that INaR may play a role in pain associated with PEPD. This knowledge provides us with a better understanding of the mechanism of INaR generation and may lead to the development of specialized treatment for pain disorders associated with INaR.
Introduction
Pain hypersensitivity as a result of injury or disease occurs primarily through increased excitability of peripheral sensory neurons. Voltage-gated sodium channels (Nav) are key determinants regulating action-potential generation and propagation (Hodgkin & Huxley, 1952). The sodium channel complex consists of a pore-forming α-subunit (220–260 kDa) and auxiliary β-subunits (32–36 kDa) (Catterall, 2000). To date, nine α-subunits (Nav1.1–1.9) and four β-subunits (β1–4) have been identified in mammals (Goldin et al. 2000). The Nav isoforms exhibit differential distribution (Felts et al. 1997) as well as distinguishing electrophysiological (Catterall et al. 2005) and pharmacological properties (England & de Groot, 2009). Evidence indicates that the sensory neuronal Nav1.7, Nav1.8 and Nav1.9 isoforms are important in inflammatory and neuropathic pain (Lai et al. 2002; Priest et al. 2005; Cox et al. 2006; Dib-Hajj et al. 2008b). Recently, mutations in Nav1.7 that give rise to three separate pain disorders have been identified. Congenital insensitivity to pain (CIP) is a result of loss-of-function mutations characterized by a complete inability to sense painful stimuli (Cox et al. 2006). In contrast, inherited erythromelalgia (IEM) and paroxysmal extreme pain disorder (PEPD) are distinct severe pain syndromes associated with gain-of-function mutations. IEM is characterized by episodes of burning pain, erythema and mild swelling in the extremities (Waxman & Dib-Hajj, 2005), whereas PEPD is characterized by severe rectal, ocular and mandibular pain (Fertleman et al. 2007). Sensory neurons expressing either PEPD or IEM mutant channels are hyperexcitable (Rush et al. 2006; Dib-Hajj et al. 2008a). However, the two classes of mutations induce hyperexcitability via distinct mechanisms. IEM mutations produce a hyperpolarizing shift in the voltage dependence of activation and slow the rate of deactivation, resulting in a lowered action-potential threshold (Cummins et al. 2004; Dib-Hajj et al. 2005; Choi et al. 2006). In contrast, PEPD mutations destabilize fast inactivation via a depolarizing shift in steady-state fast inactivation, slowed rate of open-channel fast inactivation and persistent currents (Fertleman et al. 2006; Dib-Hajj et al. 2008a; Jarecki et al. 2008).
After opening, Nav channels normally undergo inactivation within milliseconds via a ‘hinged lid’ mechanism dependent on an inactivation gate composed of four amino acids (IFMT) connecting domains 3 and 4 (Vassilev et al. 1988; West et al. 1992). In this model, the inactivation gate occludes the pore of the channel by binding to nearby ‘docking sites’ (Fig. 1A). Channels are normally refractory following inactivation until the membrane potential has been sufficiently hyperpolarized. Thus, altering the stability of inactivation can have profound effects on neuronal excitability. Under certain conditions channels can re-open during repolarization to intermediate potentials allowing a surge of inward current (resurgent current). Resurgent sodium currents were first identified in cerebellar Purkinje neurons (Raman & Bean, 1997) and more recently in dorsal root ganglion (DRG) neurons (Cummins et al. 2005). In the cerebellum, these currents are proposed to flow following relief of ultra-fast open-channel block by an endogenous intracellular blocking particle, postulated to be the C-terminal portion of the auxiliary Navβ4 subunit (Grieco et al. 2005; Bant & Raman, 2010), and may contribute to high frequency firing (Raman & Bean, 1997; Khaliq et al. 2003). Nav1.6 is the predominant carrier of resurgent current in cerebellar (Raman et al. 1997) and DRG (Cummins et al. 2005) neurons, although a recent study from our lab suggests that other Nav isoforms can carry resurgent currents under conditions which slow the rate of fast inactivation. Indeed, a PEPD mutation, Nav1.7-I1461T, enhanced resurgent currents (Jarecki et al. 2010). Therefore, we questioned whether (1) additional PEPD mutations that destabilize inactivation can enhance resurgent currents and (2) if the location(s) of the PEPD mutations affects resurgent current properties. To verify the hypothesis that inherited mutations that slow inactivation increase resurgent currents, we also tested whether IEM mutations endow Nav1.7 channels with the ability to generate resurgent currents.
Figure 1. Mutations within Nav1.7 have differential effects on current properties.
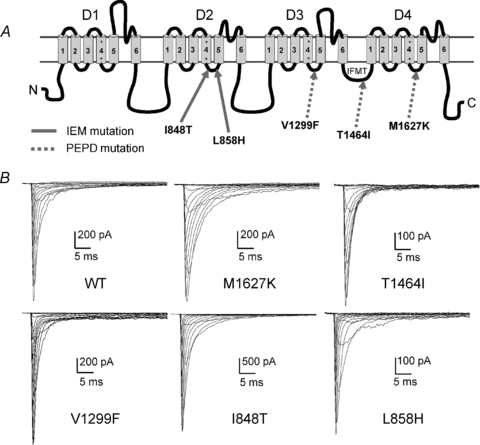
A, linear representation of the Nav α-subunit structure. Mutations implicated in IEM are indicated with continuous arrows, and mutations implicated in PEPD are indicated with dashed arrows. Putative ‘docking sites’ for binding by the inactivation gate (IFMT) are located within the cytoplasmic linkers connecting S4 and S5 of D3 (Smith & Goldin, 1997) and D4 (Lerche et al. 1997) and within the mouth of the pore within D4/S6 (McPhee et al. 1995). B, current traces recorded from representative HEK293 cells expressing either Nav1.7 WT or mutant channels. Cells were held at −80 mV, and currents were elicited with 50 ms test pulses to potentials ranging from −80 to +60 mV.
In this study, we used stably transfected HEK293 cells, which have been demonstrated to produce resurgent currents with Nav1.1 (Aman et al. 2009) and Nav1.5 (Wang et al. 2006) if the Navβ4 peptide is present in the recording pipette. This model system allows for exposure to a known concentration of the Navβ4 peptide thus isolating only differences in the gating properties conferred by the channel mutations. Using this expression system, we show for the first time that inclusion of the Navβ4 peptide in the recording pipette is sufficient for generating resurgent currents from Nav1.7 channels. All three PEPD mutations differentially enhanced resurgent currents, whereas the IEM mutations did not enhance resurgent currents compared to wild-type. Based on our data we propose that impaired inactivation is a major determinant of resurgent current generation in Nav1.7 channels, but that the location of the mutations may affect this enhancement.
Methods
Ethical information
HEK293 cells were obtained from ATCC, Manassas, VA, USA. Use of the HEK293 cells was approved by the Institutional Biosafety Committee and conformed to the ethical guidelines for the National Institutes of Health for the use of human-derived cell lines.
Preparation of stably transfected cell lines
Mutations were inserted into the plasmid encoding Nav1.7 (Klugbauer et al. 1995) using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). HEK293 cells were grown under standard tissue culture conditions (5% CO2; 37°C) in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Stable cell lines expressing human Nav1.7 (Nav1.7 wild-type (WT), T1464I, M1627K, V1299F, L858H and I848T) channels were generated in HEK293 cells using the calcium phosphate precipitation transfection technique and antibiotic selection. The calcium phosphate–DNA mixture was added to the cell culture medium and left for 15–20 h, after which time the cells were washed with fresh medium. After 48 h, antibiotic (G418, Geneticin; Life Technologies, Gaithersburg, MD, USA) was added to select for neomycin-resistant cells and establish stable cell lines. After approximately 3 weeks in G418, colonies were picked, split and subsequently tested for channel expression using whole-cell patch-clamp recording techniques. Occasionally, HEK293 cells were grown at 28°C overnight to increase channel expression.
Whole-cell patch clamp recordings
Whole-cell patch clamp recordings were conducted at room temperature (∼22°C) after obtaining a gigaohm seal using a EPC-10 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Data were acquired on a Windows-based Pentium IV computer using the Pulse program (v. 8.80, HEKA Electronik). Fire-polished electrodes (1.0–1.6 MΩ) were fabricated from 1.7 mm capillary glass (VWR International, West Chester, PA, USA) using a Sutter P-97 puller (Sutter Instrument Co., Novato, CA, USA). A caesium aspartate dominant intracellular solution consisted of (in mm): 20 CsCl, 100 caesium aspartate, 10 NaCl, 4 Hepes, 4 EGTA, 0.4 CaCl2, 2 Mg-ATP, 0.3 Li-GTP, pH 7.3 (adjusted with CsOH). The standard bathing solution consisted of (in mm): 140 NaCl, 1 MgCl2, 3 KCl, 1 CaCl2, 10 Hepes, 10 glucose, pH 7.3 (adjusted with NaOH). Cells on 35 mm plastic culture dishes were bathed in 2 ml of bathing solution and transferred to the recording platform. Voltage errors were minimized (<5 mV) using 70–80% series resistance compensation during voltage-clamp recordings. Passive leak currents were linearly cancelled by digital P/–5 subtraction. Cells were held at a membrane potential of −80 mV, and 100 ms conditioning pre-pulses to −100 mV preceded the start of current–voltage (I–V) and resurgent current protocols to ensure increased availability of channels. Membrane currents were filtered at 5 kHz and sampled at 10–20 kHz. Data were not recorded before 5 min after whole-cell access to allow adequate time for the intracellular recording solution to equilibrate into the cell. Data recordings did not last more than 15 min and cells were not held in the standard bathing solution for more than 90 min.
Resurgent current measurement
Resurgent currents are not observed in HEK293 cells expressing Nav1.7 alone or co-transfected with the full-length Navβ4 subunit (see online supplemental material, Supplemental Fig. 1). However, resurgent currents are reliably observed with inclusion of the C-terminal portion of the Navβ4 subunit (Navβ4 peptide). Therefore, to generate Nav1.7-mediated resurgent currents, the Navβ4 peptide (KKLITFILKKTREK-OH (Biopeptide Co., San Diego, CA, USA); 10 mm stock in ddH2O; 100 μm final concentration) was included in the intracellular solution.
For all cells identified with resurgent current in this study, maximal peak resurgent currents were produced within a window of repolarization potentials from −5 to −40 mV and were first observed around +10 mV. These currents display unique gating kinetics with a noticeably slow onset and decay phase, unlike classic Nav tail currents, which are observed instantaneously during hyperpolarizing steps and decay rapidly. Additionally, resurgent currents display a distinctly non-monotonic I–V relationship whereas simple tail currents display a linear I–V relationship. Currents which did not meet both of these criteria were not classified as resurgent currents and therefore were excluded from the analysis of resurgent currents. Resurgent current amplitudes were measured after 1.5 ms into the repolarizing test pulse in order to avoid contamination from tail currents and were measured relative to the leak subtracted baseline. Resurgent current traces represent an average of five sweeps at each repolarization potential. The relative resurgent currents amplitudes were calculated by dividing the peak resurgent currents by the average peak transient current and represented as a percentage of the peak transient current. Peak transient currents represent the average current measured at +20 mV from I–V recordings obtained immediately before and after the resurgent current protocol. The test potential to +20 mV was selected because the I–V relationship is linear for all of the Nav1.7 constructs at this voltage and thus less subject to voltage-clamp errors. The average resurgent current amplitude for each Nav1.7 construct was calculated using only data from cells in which resurgent currents were detected.
Data analysis
Data were analysed using the Pulsefit (v. 8.80, HEKA Elektronik), Origin (v. 8.0, OriginLab Corp., Northhampton, MA, USA), and Microsoft Excel software programs. Currents were analysed in PulseFit and filtered at 1000 Hz to reduce noise. Decay time constants for open-channel fast inactivation were measured from I–V traces at +20 mV and fitted to a single-exponential. The midpoints of activation and inactivation were determined by fitting the data with a Boltzmann function. For the data presented in Fig. 4, a good correlation is defined as R2 > 0.5. All data are shown as means ±s.e.m. Statistical significance was assessed with Student's unpaired t test or one-way ANOVA with Tukey's post hoc test where mentioned (* or †, P < 0.05; # or ‡, P < 0.01).
Figure 4. Correlation between resurgent current amplitude and the decay time constant for Nav1.7 WT and mutant channels.
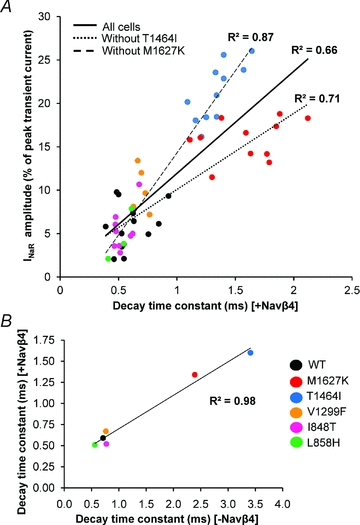
A, each cell which generated resurgent current (n= 52) across Nav1.7 WT and mutant channels is plotted here with its respective decay time constant in the presence of the Navβ4 peptide. The continuous line represents the linear trendline including all cells. The dotted line represents the linear trendline incorporating all cells except T1464I (n= 41) and the dashed line represents the linear trendline incorporating all cells except M1627K (n= 41) to demonstrate that the improved correlation when the subgroups are excluded. B, average decay time constants for each channel construct in the presence (y-axis) and absence (x-axis) of the Navβ4 peptide.
Results
PEPD and IEM mutations differentially affect voltage-dependent gating properties of Nav1.7
The electrophysiological properties of Nav1.7 WT, PEPD (V1299F, T1464I and M1627K) and IEM (I848T and L858H) mutant channels (Fig. 1A) were investigated using whole-cell voltage clamp recordings in HEK293 cells. Figure 1B shows representative whole-cell sodium currents for each construct. The voltage dependence of activation and steady-state inactivation were examined to determine if the Navβ4 peptide differentially affected PEPD and IEM channel transitions between conducting and non-conducting states (Supplemental Fig. 2, Table 1). In the absence of the Navβ4 peptide, there was no significant shift in the midpoint of activation in any of the PEPD mutant constructs compared to WT. We next compared the midpoint of activation for each of the individual PEPD constructs with and without the Navβ4 peptide (100 μm) and observed no significant difference (Table 1). In contrast to the PEPD mutants, both IEM mutant channels display significantly hyperpolarized shifts in the midpoint of activation compared to WT in the absence of Navβ4 peptide, consistent with our previous reports for I848T and L858H expressed in HEK293 cells (Cummins et al. 2004). Inclusion of the Navβ4 peptide in the pipette did not alter the voltage dependence of I848T activation, but induced a significantly larger hyperpolarizing shift for L858H (Table 1). The Navβ4 peptide also induced a slight, but significant, decrease in the activation slope factor for T1464I, I848T and L858H, although it is unclear as to the physiological significance of this change. Inclusion of the Navβ4 peptide in the pipette did not significantly alter the peak current amplitude or the current density for WT, PEPD or IEM Nav1.7 cells (Supplemental Table 1).
Table 1.
Biophysical properties of WT, PEPD and IEM Nav1.7 channels
| No peptide | |||||
|---|---|---|---|---|---|
| Channel | V1/2 activation (mV)a | Slope (mV/e-fold) | V1/2 inactivation (mV)b | Slope (mV/e-fold) | n |
| Nav1.7 | −11.6 ± 1.1 | 5.6 ± 0.1 | −54.0 ± 1.2 | 9.7 ± 0.4 | 12 |
| Nav1.7-M1627K | −10.0 ± 1.5 | 5.7 ± 0.1 | −33.8 ± 1.4# | 6.2 ± 0.2# | 9 |
| Nav1.7-T1464I | −10.7 ± 1.2 | 6.7 ± 0.2# | −47.3 ± 0.9# | 5.6 ± 0.2# | 8 |
| Nav1.7-V1299F | −9.9 ± 0.8 | 6.4 ± 0.1# | −40.9 ± 0.7# | 5.6 ± 0.2# | 9 |
| Nav1.7-I848T | −21.1 ± 0.9# | 6.4 ± 0.3* | −61.0 ± 1.7# | 8.0 ± 0.3# | 8 |
| Nav1.7-L858H | −15.6 ± 0.8# | 7.6 ± 0.1# | −55.5 ± 1.5 | 8.6 ± 0.4 | 8 |
| Navβ4 C-terminal peptide (100 μm) | |||||
| Channel | V1/2 activation (mV)a | Slope (mV/e-fold) | V1/2 inactivation (mV)b | Slope (mV/e-fold) | n |
| Nav1.7 | −14.4 ± 1.4 | 5.6 ± 0.2 | −57.5 ± 1.9 | 11.0 ± 0.6 | 14 |
| Nav1.7-M1627K | −10.1 ± 0.7* | 6.0 ± 0.2 | −36.6 ± 1.1# | 6.7 ± 0.3# | 11 |
| Nav1.7-T1464I | −13.3 ± 0.9 | 6.2 ± 0.1*† | −47.5 ± 0.9# | 5.7 ± 0.4# | 11 |
| Nav1.7-V1299F | −11.4 ± 1.1 | 6.3 ± 0.3* | −42.2 ± 1.0# | 6.1 ± 0.3# | 11 |
| Nav1.7-I848T | −23.9 ± 1.3# | 5.5 ± 0.3† | −60.5 ± 1.5 | 7.9 ± 0.2# | 10 |
| Nav1.7-L858H | −19.5 ± 0.9#‡ | 7.2 ± 0.2#† | −56.7 ± 1.1 | 9.4 ± 0.2* | 8 |
The voltage dependence of activation was examined using a series of 50 ms depolarizing test pulses from −80 mV to +60 mV. The midpoint of activation was estimated by fitting the data with a Boltzmann function.
The voltage dependence of steady-state fast inactivation was examined using a series of 200 ms conditioning pre-pulses from −120 mV to +30 mV, followed by a 20 ms test pulse to +15 mV to assess channel availability. The midpoint of activation was estimated by fitting the data with a Boltzmann function.
P < 0.05,
P < 0.01 from Nav1.7.
P < 0.01 from no peptide by Student's unpaired t test.
The three PEPD mutants all displayed significant depolarizing shifts in the midpoint of steady-state inactivation compared to WT in the absence of the Navβ4 peptide. These results are consistent with previous reports for T1464I (Dib-Hajj et al. 2008a), M1627K (Fertleman et al. 2006) and V1299F (Jarecki et al. 2008) expressed in HEK293 cells. However, when comparing each individual channel construct, there was no change in the midpoint of inactivation or the slope factor with the addition of the Navβ4 peptide (Table 1). Overall, the Navβ4 peptide did not dramatically alter the gating properties of wild-type or mutant constructs.
The Navβ4 peptide induces ultra-fast open-channel block
PEPD mutations destabilize fast inactivation in part by slowing the transition from the open to inactivated state, as measured as an increase in the decay time constant (Dib-Hajj et al. 2008a; Jarecki et al. 2008). We opted to focus on the decay phase of the current elicited from a depolarizing test pulse to +20 mV because at this potential the current–voltage relationship is linear for all constructs, and channel open probability is maximized (Supplemental Fig. 2). Furthermore, at more depolarized potentials the currents are smaller and, as such, the signal-to-noise ratio would be decreased. In the absence of the Navβ4 peptide, PEPD mutations in both the IFMT motif (T1464I) and the D4/S4–S5 intracellular loop (M1627K) exhibit significantly slower decay time constants compared to WT (Fig. 2), although a mutation in the neighbouring D3/S4–S5 intracellular loop (V1299F) did not significantly alter the decay time constant at +20 mV. Consistent with previous reports, the decay time constant was not different for I848T; however the other IEM mutant, L858H, exhibited a significantly faster rate of inactivation compared to WT. We additionally measured the decay time constant at potentials between −20 and +40 mV (Supplemental Fig. 3A) and observed that T1464I and M1627K are slower than WT at nearly every potential tested. Interestingly, V1299F is slower than WT only at +30 and +40 mV. Nevertheless, the differences in decay time constant in relation to WT for both T1464I and M1627K are substantially larger than that for V1299F.
Figure 2. Effects of Nav1.7 mutations and the Navβ4 peptide on open channel fast inactivation.
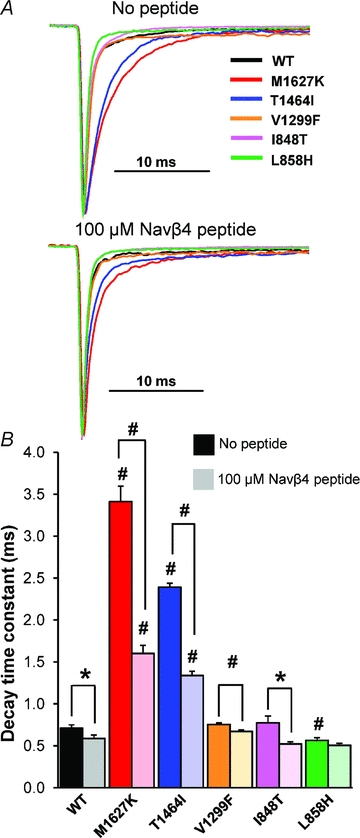
A, normalized current traces elicited by a step depolarization to +20 mV from representative HEK293 cells expressing either Nav1.7 WT or mutant channels in the absence (top) and presence (bottom) of the Navβ4 peptide in the recording pipette. B, bar graph displaying the average decay time constants in the absence (dark shaded bars) and presence (light shaded bars) of Navβ4 in the recording pipette (n= 8–14, *P < 0.05, #P < 0.01 by Student's unpaired t test).
Generation of resurgent current is thought to be dependent on ultra-fast open-channel block mediated by a putative blocking particle, Navβ4, binding before fast inactivation (occlusion of the pore by the IFMT gate) occurs. Ultra-fast open-channel block is evident by faster decay kinetics in cells that generate resurgent current compared to cells that do not produce these currents (Grieco et al. 2002, 2005; Wang et al. 2006). Consistent with previous observations with other Nav isoforms, we observed significantly faster decay time constants with Navβ4 peptide in the recording pipette (Fig. 2 and Supplemental Figs 3B and 4). The change in decay kinetics was most evident in the M1627K and T1464I mutant channels, which also displayed the slowest time constants in the absence of the peptide. As seen in Supplemental Fig. 3C, for T1464I and M1627K, the difference in decay time constants with and without the peptide is more apparent with stronger depolarizations. The IEM-L858H mutant did not exhibit significantly different rates of decay with and without the peptide. The IEM-I848T mutant exhibited faster decay kinetics with the peptide at nearly every potential tested. Together, the data presented here demonstrate for the first time that inclusion of the Navβ4 peptide in the recording pipette accelerates the transition of the Nav1.7 channel from a conducting to non-conducting state.
PEPD mutations differentially enhance resurgent currents in HEK293 cells with inclusion of the Navβ4 peptide in the recording pipette
Our lab previously reported that missense mutations which slow the transition from the open to the inactivated state, such as the PEPD-I1461T mutation, enhance resurgent currents (Jarecki et al. 2010). Therefore, we hypothesized those additional PEPD mutations which increase the decay time constant of open-channel fast inactivation would enhance resurgent currents. Conversely, we postulated that IEM mutations which do not slow the rate of inactivation would not enhance resurgent currents. For reasons that are unclear, heterologous expression systems such as HEK293 cells are not capable of generating resurgent currents with co-expression of the full-length Navβ4 subunit with the Nav1.1 (Aman et al. 2009), Nav1.6 (Chen et al. 2008) or Nav1.7 isoforms (Supplemental Fig. 1). However, the Navβ4 C-terminal peptide can induce resurgent currents in Nav1.1 and Nav1.5 channels expressed in HEK293 cells when it is included in the recording pipette (Wang et al. 2006; Aman et al. 2009). Therefore, to determine whether the mutant and WT Nav1.7 constructs are capable of generating enhanced resurgent currents, we conducted experiments utilizing a resurgent current protocol (Fig. 3D) in the absence and presence of the Navβ4 peptide (100 μm) in the recording pipette.
Figure 3. Navβ4 peptide produces resurgent currents in Nav1.7 channels expressed in HEK293 cells.
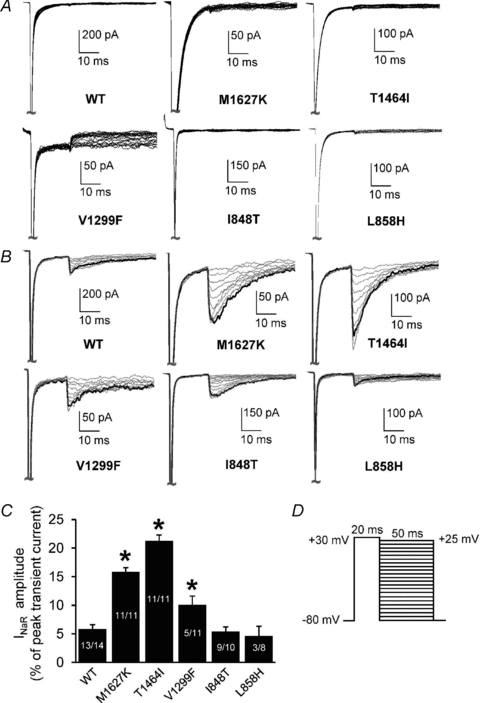
A and B, resurgent current traces recorded from representative HEK293 cells expressing either Nav1.7 WT or mutant channels in the absence (A) and presence (B) of Navβ4 in the recording pipette. Note that the current amplitudes are scaled to reflect the relative size of resurgent currents in relation to WT and to better compare the currents between each construct in the absence and presence of the peptide. C, resurgent current amplitude, as measured as a percentage of the average peak transient current elicited at +20 mV obtained immediately prior to and following the resurgent current protocol as shown in D. For cells recorded in the absence of Navβ4: WT, n= 12; M1627K, n= 9; T1464I, n= 8; V1299F, n= 9; I848T, n= 8; L858H, n= 8. The n values for cells recorded in the presence of Navβ4 are shown on the bars in C. *P < 0.05 by one-way ANOVA and Tukey's post hoc test compared to WT.
Resurgent currents were not observed in any of the constructs in the absence of the Navβ4 peptide (Fig. 3A). Interestingly, varying degrees of resurgent currents were detected in all channel constructs with Navβ4 peptide present in the recording pipette (Fig. 3B and C). Resurgent currents were detected in 93% of WT cells and in every T1464I and M1627K expressing cell examined but in only 45% of V1299F expressing cells. The PEPD-T1464I mutant exhibited the largest enhancement in resurgent current amplitude (∼21% of peak transient current) compared to WT (∼6%). The other PEPD mutants, M1627K (∼16%) and V1299F (∼10%), also enhanced resurgent current amplitude relative to WT. Conversely, neither IEM mutation enhanced resurgent currents compared to WT channels. The IEM-I848T mutant exhibited resurgent currents in 90% of cells tested and the resurgent current amplitude was ∼5% of peak transient current. The IEM-L858H mutant only produced detectable resurgent current in 38% of cells tested and the resurgent current amplitude was also ∼5% of peak transient current. These results demonstrate that of the mutations investigated here, only PEPD mutants increase resurgent current amplitude relative to WT.
The decay time constant is correlated with resurgent current amplitude
To determine whether the decay time constant (in the presence of Navβ4 peptide at +20 mV) correlates with the resurgent current amplitude, all cells for each construct that generated resurgent current were displayed on a scatter plot and fitted with a linear trendline. As seen in Fig. 4A, the decay time constant correlates reasonably well with resurgent current amplitude (R2= 0.66). However, when either the T1464I or M1627K groups are excluded from the plot, and the plot is re-fitted, the correlation is substantially improved (T1464I excluded, R2= 0.71; M1627K excluded, R2= 0.87). This difference is readily apparent in that the M1627K channels exhibit significantly smaller resurgent currents (∼16%) compared to T1464I (∼21%), yet display a slower decay time constant.
It should be noted that in the presence of the Navβ4 peptide, the decay time constant is not a direct measure of the rate of normal fast inactivation mediated by the inactivation gate, but rather the rate of ultra-fast open-channel block mediated by the Navβ4 peptide. As such, we cannot directly compare the rate of normal fact inactivation to resurgent current amplitude on an individual cell basis because we are unable to record the decay time constant in an individual cell in the presence and absence of the Navβ4 peptide. However, as shown in Fig. 4B, the average decay time constants (from Fig. 2) for each construct in the presence and absence of the Navβ4 peptide are strongly correlated (R2= 0.98), suggesting that the rate of ultra-fast open-channel block in the presence of the Navβ4 peptide is an excellent predictor of the relative rate of normal fast inactivation.
T1464I maintains a more stable open-channel block compared to M1627K and WT Nav1.7
Resurgent currents are voltage and time dependent, with large currents favoured by brief, strong depolarizations and a decrease in amplitude associated with longer duration pulses (Raman & Bean, 2001; Wang et al. 2006). Thus, it is believed that over time, the inactivation gate will outcompete the blocking particle, favouring normal fast inactivation during prolonged depolarizations. The data shown in Fig. 4A suggest that the relationship between the rate of current decay and resurgent current amplitude may be affected by the location of the mutation and the mechanism by which inactivation is impaired. We hypothesized that a mutation (T1464I) within the inactivation gate would differentially affect the ability of the IFMT loop to outcompete the Navβ4 peptide compared to a mutation (M1627K) within a putative ‘docking site’. We predicted the location-specific effects to be manifested by differences in resurgent current amplitude across longer depolarizing pulse durations between the two groups. As seen in Fig. 5, the WT, T1464I and M1627K channels all display reduced resurgent current amplitudes with longer duration pulses. However, the relative magnitude of the decrease in resurgent current amplitude over time was significantly less pronounced for T1464I compared to WT and M1627K. These results suggest that the T1464I mutation within the inactivation gate hinders the ability of the channel to ultimately transition to a stabilized inactivated state to a greater extent than the mutation in a putative ‘docking site’ (M1627K). As a result, there is likely to be a higher percentage of channels that undergo open-channel block by the peptide for the T1464I mutant, as indicated by the large enhancement in resurgent current amplitude noted previously (Fig. 3).
Figure 5. Dependence of resurgent current amplitude on duration of depolarizing voltage step between Nav1.7 WT and PEPD mutant channels.
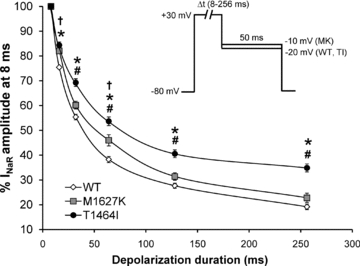
Cells expressing WT, T1464I and M1627K channels were assayed for their ability to generate resurgent current using a depolarizing conditioning pulse to +30 mV at different pulse durations (8–256 ms) and then repolarized to a potential near which the peak resurgent current amplitude was observed for each channel construct in order to maximize the signal-to-noise ratio (see inset). For each individual recording, resurgent current amplitudes were normalized to the current amplitude at 8 ms and presented as a percentage of that amplitude and averaged for all cells in each group (n= 8–9, *P < 0.05 TI different from WT; #P < 0.05 TI different from MK, †P < 0.05 MK different from WT by one-way ANOVA and Tukey's post hoc test).
Discussion
PEPD and IEM mutations differentially affect the voltage-dependent gating properties of Nav1.7. Although both sets of mutations can increase the excitability of sensory neurons, they are associated with distinct syndromes characterized by pain in different regions of the body. We asked if resurgent currents, which contribute to increased action potential firing in neurons, might be differentially enhanced by PEPD and IEM mutations. For the first time, we demonstrate that Nav1.7 channels stably expressed in HEK293 cells can produce resurgent currents with inclusion of the Navβ4 peptide in the recording pipette. We did not observe an enhancement in resurgent current amplitude with either of the two IEM mutations compared to WT Nav1.7 channels. In contrast, we show that all three PEPD mutations exhibited significantly larger resurgent currents compared to WT channels, although to varying degrees. Furthermore, our data indicate that the amplitude of resurgent currents depends on not only the extent to which inactivation is impaired, but also the mechanism by which inactivation is impaired. Based on these data, we hypothesize that resurgent currents could contribute to modulating the neuronal hyperexcitability and pain associated with PEPD.
The IEM mutations did not enhance resurgent current generation. Cells expressing the L858H mutation displayed resurgent currents less frequently (3 of 8 cells) compared to WT (13 of 14 cells), which may be a consequence of the faster current decay kinetics exhibited by L858H. Although cells expressing the I848T mutations displayed resurgent currents in nearly every cell tested (9 of 10 cells), resurgent current amplitude was not different from WT. The I848T and L8585H mutant channels do not exhibit altered voltage dependence of channel inactivation, but do exhibit a hyperpolarized shift in the voltage dependence of activation (Table 1) and considerably slower deactivation (transition from open to closed states) (Cummins et al. 2004) of Nav1.7 sodium currents. Together, these results suggest that changes in channel activation and deactivation are not sufficient to play a significant role in modulating resurgent current generation.
Two of the PEPD mutations (T1464I and M1627K) substantially and reliably enhanced resurgent currents compared to WT. Both of these mutations significantly slow the rate of fast inactivation. Fast inactivation of sodium channels occurs within milliseconds after channel opening and involves a structural rearrangement of the channel resulting in binding of the inactivation gate to a nearby region (docking site), occluding the channel pore and preventing further sodium influx (Catterall, 2000). Mutation of residues within the inactivation gate or putative docking sites, such as with PEPD, is likely to result in impaired interactions between these two regions of the channel, thus destabilizing inactivation (Fertleman et al. 2006; Dib-Hajj et al. 2008a; Jarecki et al. 2008). The current model for resurgent current generation suggests that a blocking particle competes with the inactivation gate for binding to or near the channel pore. If the blocking particle binds before normal inactivation occurs, the channel undergoes open-channel block thereby preventing the inactivation gate from binding (Grieco et al. 2005). Our data on the rate of current decay in the absence and presence of the Navβ4 peptide clearly indicate that with Nav1.7, the Navβ4 peptide is capable of occluding the pore more quickly than the inactivation gate resulting in ultra-fast open-channel block. With the T1464I and M1627K mutants, the decay time constant at more positive potentials in the presence of the peptide is substantially accelerated compared to in the absence of the peptide (Supplemental Fig. 4), suggesting that open-channel block is voltage dependent. Alternatively, inclusion of the Navβ4 peptide may facilitate binding of the inactivation gate; however this scenario is unlikely considering that the inactivation gate cannot unbind from the pore until the cell has been sufficiently hyperpolarized for several milliseconds. Indeed, in the absence of the Navβ4 peptide, no current is detected following repolarization of the membrane potential in the WT or mutant Nav1.7 channels (Fig. 3A), and thus unbinding of the inactivation gate does not occur at intermediate potentials at which resurgent currents are detected. Furthermore, Wang et al. (2006) demonstrated that Navβ4 can block inactivation-deficient mutant Nav1.5 channels, demonstrating that the Navβ4 peptide alone can block sodium channels.
Open-channel block is voltage and time dependent, with block favoured by brief, strong depolarizations. Over time, it is believed that the inactivation gate outcompetes the blocking particle, with long depolarizations favouring normal inactivation and thus smaller resurgent currents (Raman & Bean, 2001). We demonstrate that over time, the resurgent currents seen in WT and M1627K decrease in amplitude to a similar degree (Fig. 5), suggesting that any structural or functional change induced by the M1627K mutation does not significantly impair the ability of the inactivation gate to outcompete the blocking particle over time. Thus, the slower decay time constant seen in M1627K may be the determining factor for the large difference in resurgent current amplitude compared to WT. However, for T1464I, resurgent current amplitude decreased less over time compared to WT and M1627K, suggesting that the T1464I mutation reduces the ability of the inactivation gate to outcompete Navβ4 for binding to the channel and occluding the pore. We hypothesize that because the intracellular loop to which the M1627K mutation is localized is just one of several putative docking sites for the inactivation gate, normal inactivation may proceed, albeit slowly, through binding to the remaining unaltered docking site(s). In contrast, the T1464I mutation lies within the inactivation gate, and thus stabilization of inactivation (and displacement of Navβ4 over time) proceeding through binding of the inactivation gate to the docking sites is likely to be impaired. As such, enhanced stability of Navβ4 binding to the channel, and hence more channels undergoing open-channel block, may provide an explanation for the larger resurgent currents seen with T1464I.
The PEPD-V1299F mutation also enhanced resurgent currents compared to WT channels, but to a much lesser extent than the other PEPD mutations. Interestingly, in our present study, the V1299F mutation exhibited slightly slower decay kinetics compared to WT only at +30 and +40 mV, whereas the M1627K and T1464I channels exhibited substantially slower kinetics at nearly every potential tested. These results may partially explain why resurgent currents were observed less frequently (5 of 11 cells) for V1299F and were significantly smaller compared to the resurgent currents observed with the M1627K and T1464I mutations. The observation that V1299F enhanced resurgent current amplitude while only slightly altering the rate of fast inactivation indicates that other factors may impact resurgent current generation. In addition to the depolarizing shift in the voltage dependence of steady-state fast inactivation, V1299F displayed enhanced persistent currents (5.3 ± 0.3% normalized current remaining at 45 ms) compared to WT (1.0 ± 0.2%), indicating that changes in inactivation that result in increased persistent currents may also contribute to resurgent current generation, albeit to a lesser extent than alterations that substantially slow the rate of inactivation. Consequently, destabilized inactivation in general (not only changes in the rate of fast inactivation) may be important in the mechanism of resurgent current generation. These data raise the possibility that the extent to which specific PEPD mutations impair inactivation is likely to be subject to modulation and therefore we predict that the relative amplitude of resurgent currents generated by PEPD mutant channels is also subject to modulation. We speculate that modulation of resurgent currents in nociceptive afferents innervating different regions of the body might be one factor contributing to the regional distribution of abnormal pain sensations associated with PEPD.
Resurgent currents are proposed to facilitate high frequency firing in Purkinje neurons by providing a depolarizing input at moderately negative potentials to drive cells to firing threshold through rapid recovery from open-channel block (Raman & Bean, 2001; Khaliq et al. 2003). We previously demonstrated that a PEPD mutation localized to the inactivation gate (Nav1.7-I1461T) that produces resurgent currents in transfected DRG neurons increases action-potential repetitive firing in simulated neurons (Jarecki et al. 2010). M1627K channels expressed in DRG neurons render these neurons hyperexcitable in part by lowering the threshold for action-potential firing (Dib-Hajj et al. 2008a), and we hypothesize that enhanced resurgent currents play a role in mediating this hyperexcitability. It is currently unknown whether the V1299F, T1464I and M1627K mutations confer differential degrees of hyperexcitability as predicted by the differential enhancement in resurgent currents. Future studies are necessary in order to determine if there is a direct correlation between the extent of neuronal hyperexcitability and the resurgent current amplitude within individual neurons expressing these mutant channels.
Although inclusion of the C-terminal Navβ4 peptide in the intracellular solution is sufficient for generating resurgent currents in Nav1.7 channels, co-expression of the full-length Navβ4 subunit with Nav1.7 in HEK293 cells does not yield resurgent currents (Supplemental Fig. 1). The reasons for this discrepancy are beyond the scope of this study, although several hypotheses have been suggested (Aman et al. 2009). Resurgent currents seen in HEK293 cells appear very similar to resurgent currents seen in Purkinje and DRG neurons, yet several caveats arise with investigating resurgent current mechanisms using a truncated version of the Navβ4 subunit in a heterologous expression system. First, the peptide may not be subjected to the same modulation as the full-length protein. As such, additional modulation may be important in determining resurgent current characteristics between the PEPD mutants. Second, although the Navβ4 subunit seems to be crucial to generation of resurgent currents in cerebellar granule cells, other blocking particles may be important in different neuronal populations and, if so, these may interact differently with distinct PEPD mutations. Furthermore, it remains a possibility that in the native system, such as with DRG neurons, the C-terminal portion of the Navβ4 tail may be cleaved before interacting with the channel.
Overall, our data demonstrate that localization of missense mutations within the α-subunit of the channel confers differential effects on voltage-dependent gating properties and resurgent current characteristics. Channels expressing mutations that destabilize fast inactivation, such as those associated with PEPD, have an increased likelihood of undergoing ultra-fast open-channel block resulting in enhanced resurgent currents. As such, selective manifestation of resurgent currents in Nav1.7 channels harbouring PEPD mutations may contribute to the differential pathophysiologies displayed with the different PEPD mutations and more importantly, between PEPD and IEM. Furthermore, drugs that selectively target resurgent currents could result in improved selectivity in the treatment of PEPD or other neuropathies displaying enhanced resurgent currents while minimizing unwanted side effects.
Acknowledgments
Thanks to James O. Jackson II for assistance in generating the stable Nav1.7 HEK293 cell lines. J.W.T. was supported by the 2010 PhRMA Foundation Post-Doctoral Fellowship in Pharmacology/Toxicology Award. This work was also supported by NIH research grant NS053422 (T.R.C.).
Glossary
Abbreviations
- CIP
congenital insensitivity to pain
- DRG
dorsal root ganglion
- IEM
inherited erythromelalgia
- IFMT
isoleucine, phenylalanine, methionine, threonine
- INaR
resurgent current
- Nav
voltage-gated sodium channel
- PEPD
paroxysmal extreme pain disorder
- WT
wild-type
Author contributions
Conception and design of experiments: J.W.T., B.W.J., A.D.P., T.R.C. Collection of data: J.W.T. (performed in the lab of Dr Cummins at the Stark Institute). Analysis and interpretation of data: J.W.T., A.D.P., T.R.C. Drafting of article: J.W.T., T.R.C., B.W.J., A.D.P.
Supplementary material
Supplemental Table 1.
Supplemental Figure 1.
Supplemental Figure 2.
Supplemental Figure 3.
Supplemental Figure 4.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM. Regulation of persistent Na current by interactions between β subunits of voltage-gated Na channels. J Neurosci. 2009;29:2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bant JS, Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel β4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A. 2010;107:12357–12362. doi: 10.1073/pnas.1005633107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T, Catterall WA. Functional properties and differential neuromodulation of Nav1.6 channels. Mol Cell Neurosci. 2008;38:607–615. doi: 10.1016/j.mcn.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Dib-Hajj SD, Waxman SG. Inherited erythermalgia: limb pain from an S4 charge-neutral Na channelopathy. Neurology. 2006;67:1563–1567. doi: 10.1212/01.wnl.0000231514.33603.1e. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005;579:2166–2170. doi: 10.1016/j.febslet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. 2004;24:8232–8236. doi: 10.1523/JNEUROSCI.2695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Estacion M, Jarecki BW, Tyrrell L, Fischer TZ, Lawden M, Cummins TR, Waxman SG. Paroxysmal extreme pain disorder M1627K mutation in human Nav1.7 renders DRG neurons hyperexcitable. Mol Pain. 2008a;4:37. doi: 10.1186/1744-8069-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, Marshall L, Waxman SG. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128:1847–1854. doi: 10.1093/brain/awh514. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Waxman SG. Genetics and molecular pathophysiology of Nav1.7-related pain syndromes. Adv Genet. 2008b;63:85–110. doi: 10.1016/S0065-2660(08)01004-3. [DOI] [PubMed] [Google Scholar]
- England S, de Groot MJ. Subtype-selective targeting of voltage-gated sodium channels. Br J Pharmacol. 2009;158:1413–1425. doi: 10.1111/j.1476-5381.2009.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG. Sodium channel α-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res. 1997;45:71–82. doi: 10.1016/s0169-328x(96)00241-0. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg-Olofsson O, Elmslie FV, Griesemer DA, Goutieres F, Kirkpatrick M, Malmros IN, Pollitzer M, Rossiter M, Roulet-Perez E, Schubert R, Smith VV, Testard H, Wong V, Stephenson JB. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome) Neurology. 2007;69:586–595. doi: 10.1212/01.wnl.0000268065.16865.5f. [DOI] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Grieco TM, Afshari FS, Raman IM. A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar purkinje neurons. J Neurosci. 2002;22:3100–3107. doi: 10.1523/JNEUROSCI.22-08-03100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki BW, Piekarz AD, Jackson JO, 2nd, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest. 2010;120:369–378. doi: 10.1172/JCI40801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki BW, Sheets PL, Jackson JO, 2nd, Cummins TR. Paroxysmal extreme pain disorder mutations within the D3/S4–S5 linker of Nav1.7 cause moderate destabilization of fast inactivation. J Physiol. 2008;586:4137–4153. doi: 10.1113/jphysiol.2008.154906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95:143–152. doi: 10.1016/s0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, Lehmann-Horn F. Role in fast inactivation of the IV/S4–S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. J Physiol. 1997;505:345–352. doi: 10.1111/j.1469-7793.1997.345bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for transmembrane segment IVS6 of the sodium channel α subunit in fast inactivation. J Biol Chem. 1995;270:12025–12034. doi: 10.1074/jbc.270.20.12025. [DOI] [PubMed] [Google Scholar]
- Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, Ritter AM, Liberator P, Iyer LM, Kash SF, Kohler MG, Kaczorowski GJ, MacIntyre DE, Martin WJ. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A. 2005;102:9382–9387. doi: 10.1073/pnas.0501549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103:8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4–S5. Biophys J. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Wang GK, Edrich T, Wang SY. Time-dependent block and resurgent tail currents induced by mouse β4(154–167) peptide in cardiac Na+ channels. J Gen Physiol. 2006;127:277–289. doi: 10.1085/jgp.200509399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Dib-Hajj S. Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol Med. 2005;11:555–562. doi: 10.1016/j.molmed.2005.10.004. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.