

Abstract
Interferons (IFNs) inhibit cell growth in a Stat1-dependent fashion that involves regulation of c–myc expression. IFN–γ suppresses c–myc in wild-type mouse embryo fibroblasts, but not in Stat1-null cells, where IFNs induce c–myc mRNA rapidly and transiently, thus revealing a novel signaling pathway. Both tyrosine and serine phosphorylation of Stat1 are required for suppression. Induced expression of c–myc is likely to contribute to the proliferation of Stat1-null cells in response to IFNs. IFNs also suppress platelet-derived growth factor (PDGF)-induced c–myc expression in wild-type but not in Stat1-null cells. A gamma-activated sequence element in the promoter is necessary but not sufficient to suppress c–myc expression in wild-type cells. In PKR-null cells, the phosphorylation of Stat1 on Ser727 and transactivation are both defective, and c–myc mRNA is induced, not suppressed, in response to IFN–γ. A role for Raf–1 in the Stat1-independent pathway is revealed by studies with geldanamycin, an HSP90-specific inhibitor, and by expression of a mutant of p50cdc37 that is unable to recruit HSP90 to the Raf–1 complex. Both agents abrogated the IFN–γ-dependent induction of c–myc expression in Stat1-null cells.
Keywords: c–jun/cell proliferation/geldanamycin/p50cdc37/PKR
Introduction
Interferons (IFNs) are pleiotropic cytokines that mediate anti-viral responses, inhibit proliferation and participate in immune surveillance and tumor suppression (Farrar and Schreiber, 1993; Stark et al., 1998). Transcriptional regulation in response to IFNs is mediated by the Jak–Stat pathway (for recent reviews, see Leaman et al., 1996; Darnell, 1997; Stark et al., 1998). Negative as well as positive regulation of gene expression in response to IFN–γ has been reported (Der et al., 1998; Sharma and Iozzo, 1998). IFN–α and -β activate Stat1 and Stat2, which, with p48, form the transcription factor ISGF3, which binds to IFN–stimulated response elements. Activated Stat1 dimers translocate into the nucleus and bind to gamma-activated sequence (GAS) elements. All IFNs cause phosphorylation of Stat1 on Tyr701 and Ser727 (Wen et al., 1995) and both phosphorylations are required for maximal transactivation (Wen et al., 1995; Wen and Darnell, 1997). Stat1 dimers function through interaction with transcriptional co-activators such as CBP/p300, Nmi and MCM–5, as well as with other transcription factors, including p48 and SP1 (Look et al., 1995; Horvath et al., 1996; Zhang et al., 1996, 1998; Zhu et al., 1999). In addition to the IFNs, many growth factors and cytokines also activate Stat1 (Schindler and Darnell, 1995).
Targeted disruption in mice has confirmed that Stat1 is obligatory for signaling in response to all IFNs and has revealed that Stat1 is also involved in immune surveillance and tumor suppression (Durbin et al., 1996; Meraz et al., 1996; Kaplan et al., 1998). Some tumor cells and tumor-derived cell lines express little or no Stat1 mRNA or protein (L.H.Wong et al., 1997; Abril et al., 1998; Sun et al., 1998) or fail to activate Stat1 following treatment with IFNs (Lucas et al., 1998). IFNs inhibit the growth of many cell types (Balkwill and Taylor-Papadimitriou, 1978; Lin et al., 1986; Kimchi, 1992), and Stat1 that is fully active transcriptionally is required for this effect (Bromberg et al., 1996). The inhibition of cell growth correlates with the regulation of several cell cycle regulatory genes by IFNs. mRNAs encoding cyclin D and cdc25A decrease in response to IFN–α and -β, and expression of the cyclin-dependent kinase (CDK) inhibitor p21waf1 is up-regulated by IFN–γ in epidermal carcinoma and glioblastoma cell lines (Chin et al., 1996; Tiefenbrun et al., 1996; Kominsky et al., 1998). In contrast, the inhibition by IFN–γ of the growth of the colon carcinoma cell line HCT116 is independent of p21 (Sharma and Iozzo, 1998). IFN–γ can stimulate rather than suppress the growth of certain cells (Caux et al., 1992; Shiohara et al., 1993), but the basis of this paradoxical activity is unclear. Stat1, and indeed other Stats, can also mediate negative regulation of gene expression in response to effectors other than the IFNs. For example, EGF-induced proliferation correlates with the transient activation of Stat1, whereas EGF-mediated growth suppression correlates with its sustained activation (Bromberg et al., 1998).
c–myc, a transcription factor that helps to regulate proliferation, is induced rapidly and transiently by many growth factors and cytokines (Spencer and Groudine, 1991; Bouchard et al., 1998; Dang, 1999). The expression of c–myc is aberrant in a variety of human tumors (Marcu et al., 1992). Its ectopic expression overrides both the G1 and S check points, promoting genomic instability and tumorigenesis (Chernova et al., 1998; Felsher and Bishop, 1999). c–myc regulates the G1–S transition by activating cyclin–CDK complexes and, together with its dimerization partner max, transactivates genes required for entry into S–phase (Blackwood and Eisenman, 1991; Grandori and Eisenman, 1997; Obaya et al., 1999). Treatment with IFN–α and -β abolishes the formation of transcription factor complexes on the E2F site of the c–myc promoter and suppresses c–myc expression in both Daudi and M1 cells (Resnitzsky and Kimchi, 1991; Melamed et al., 1993).
The constitutive expression of ectopic c–myc overcomes IFN–γ-mediated arrest of macrophages and vascular smooth muscle cells, indicating that c–myc is likely to be involved in the inhibition of proliferation mediated by IFN–γ (Bennett et al., 1994; Vairo et al., 1995). We now find that IFN–γ inhibits the expression of c–myc in wild-type cells, an effect that is mediated by consensus GAS elements in the c–myc promoter to which Stat1 homodimers bind. Furthermore, in Stat1-null cells, both c–myc and c–jun are induced transiently and rapidly by IFNs, revealing a novel signaling pathway. In IFN–γ-treated PKR-null mouse cells, serine phosphorylation of Stat1 is defective, transactivation is impaired and c–myc mRNA is induced, not suppressed. Furthermore, inhibitors of Raf–1 activation abrogate the IFN–dependent induction of c–myc in Stat1-null cells, indicating that Raf–1 is important in Stat1–independent signaling.
Results
Regulation of c–myc gene expression by IFN–γ in wild-type and Stat1-null mouse embryo fibroblasts (MEFs)
To determine if c–myc is a target of IFN–γ-mediated signaling, we examined c–myc mRNA levels in wild-type and Stat1-null MEFs. In wild-type cells that were serum-starved for 36 h, IFN–γ treatment decreased c–myc mRNA expression by 4–fold in 3 h (Figure 1C). In contrast, c–myc mRNA was induced 6–fold by IFN–γ in Stat1-null cells, rapidly and transiently (Figures 1A, B and 2B). Treatment with IFN–β also suppressed the induction of c–myc by platelet-derived growth factor (PDGF) in wild-type cells and induced c–myc expression in Stat1-null cells (Figure 2C). These results are in accord with the suppression by IFN–γ of cell growth in wild-type cells and with the loss of growth inhibition in Stat1-null cells (Bromberg et al., 1996). Since other immediate-early genes are also induced transiently and rapidly in response to growth factors such as PDGF (Greenberg and Ziff, 1984), we investigated the induction by IFN–γ of genes in the fos and jun families in Stat1-null cells. c–jun was induced rapidly by IFN–γ in Stat1-null but not wild-type MEFs, whereas c–fos and jun-B were not induced in either Stat1-null or wild-type cells (Figure 1D).
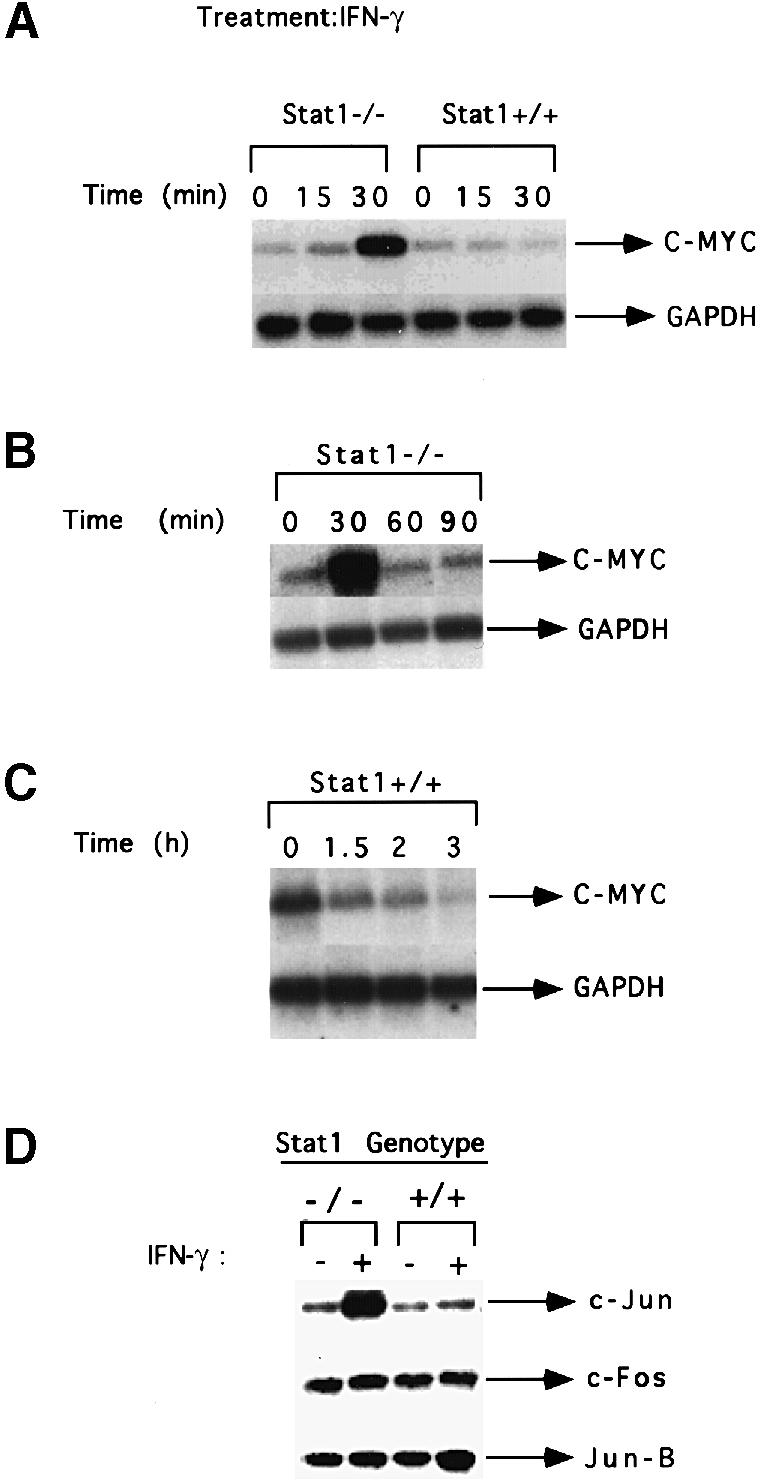
Fig. 1. c–myc mRNA expression in response to IFN–γ in Stat1-null and wild-type MEFs. (A) Subconfluent, serum-starved MEFs were either untreated or treated with 1000 IU/ml of murine IFN–γ for 15 or 30 min. c–myc and GAPDH mRNA levels were analyzed by Northern blotting. (B) Stat1-null cells were treated with murine IFN–γ (1000 IU/ml). c–myc and GAPDH mRNA levels were determined as above. (C) Wild-type cells were treated with murine IFN–γ (1000 IU/ml). c–myc and GAPDH mRNA levels were determined as above. (D) Subconfluent, serum-starved fibroblasts were either untreated or treated with 1000 IU/ml of murine IFN–γ for 30 min. Northern blot analyses were conducted with the probes indicated.
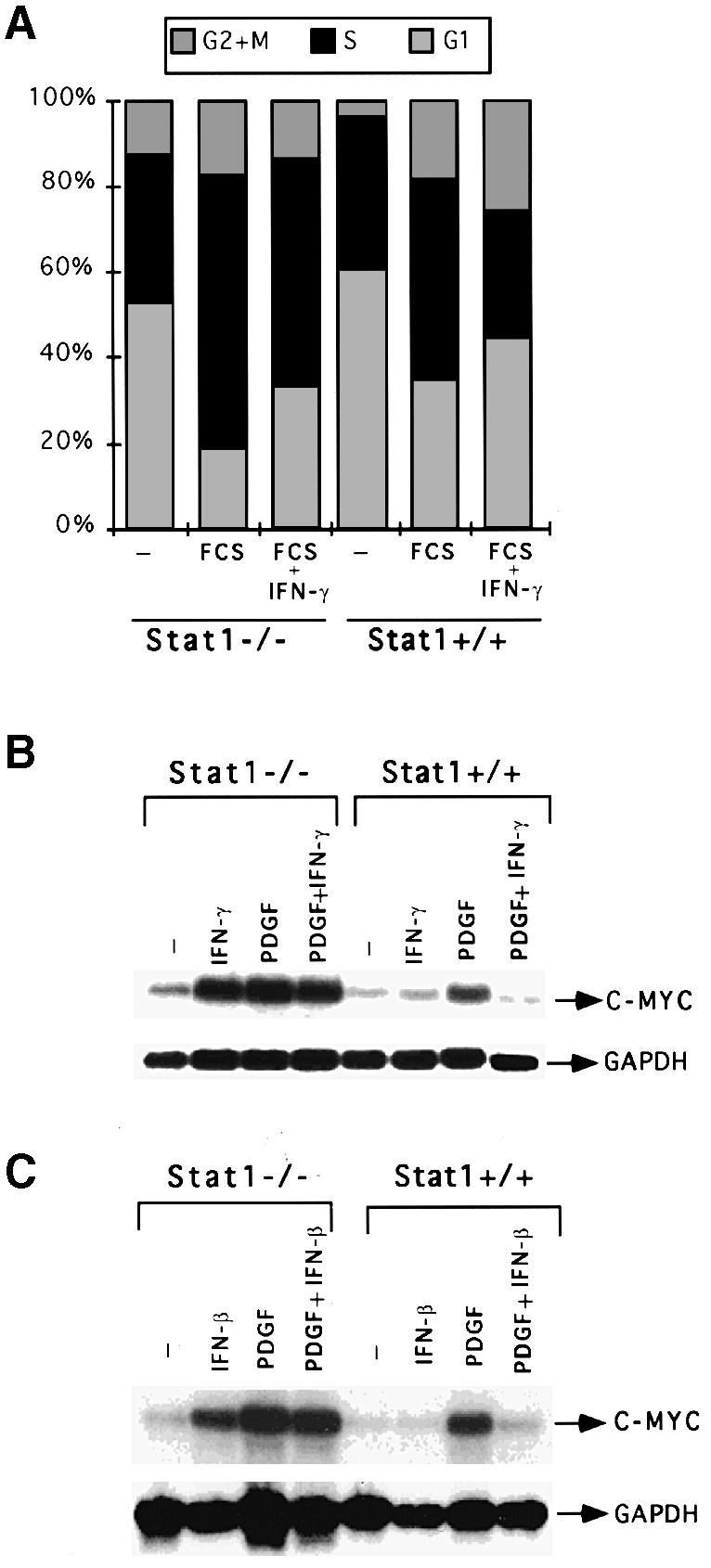
Fig. 2. Effects of growth factor and IFN treatment on c–myc regulation. (A) MEFs were grown to 20% confluence in DMEM with 10% FCS. The cells were serum-starved in DMEM with 0.1% FCS for 48 h. Cells were either untreated (–) or treated with 10% FCS, alone (FCS) or with 1000 IU/ml of murine IFN–γ (FCS+IFN–γ). Twenty-four hours later, the cells were stained with propidium iodide and the DNA content was analyzed by flow cytometry. The percentage of cells in the G1, S and G2+M parts of the cell cycle are indicated in each histogram. (B) Stat1-null or wild-type MEFs were either untreated or treated for 30 min with 1000 IU/ml of IFN–γ alone, 200 ng/ml of PDGF alone, or PDGF plus IFN–γ. Northern transfers were hybridized with c–myc or GAPDH probes. (C) Stat1-null or wild-type MEFs were either untreated or treated for 30 min with 1000 IU/ml of IFN–β alone, 200 ng/ml of PDGF alone, or PDGF plus IFN–β. Northern transfers were hybridized with c–myc or GAPDH probes.
Regulation of PDGF-dependent induction of c–myc by IFNs in wild-type and Stat1-null MEFs
PDGF, a major mitogen in serum, induces c–myc rapidly and transiently (Greenberg and Ziff, 1984). Treatment with IFN–α and -β has been found to abrogate the induction by PDGF of c–myc and entry into S–phase in mouse fibroblasts (Einat et al., 1985). We investigated the effect of IFN–γ on cell cycle progression in wild-type and Stat1-null MEFs. Cells at 20% confluence were serum-starved in 0.1% serum for 48 h, subsequently returned to medium with 10% serum, with or without IFN–γ, and examined after 24 h (their approximate doubling time) for cell cycle distribution. Only 29% of the wild-type cells were in S–phase with serum and IFN–γ, whereas 52% of the Stat1-null cells were in S–phase under the same conditions (Figure 2A). These results indicate that the effect of IFN–γ in limiting cell cycle progression depends on Stat1. Next, we examined the effect of IFN–γ or IFN–β on the PDGF-dependent induction of c–myc in wild-type and Stat1-null MEFs. PDGF induced c–myc expression in both types of cells. However, the induction was ∼2–fold higher in Stat1-null cells than in wild-type cells. Simultaneous treatment with IFN–γ or IFN–β abrogated the induction by PDGF of c–myc in wild-type but not in Stat1-null cells (Figure 2B and C). These results indicate that suppression of the PDGF-dependent induction of c–myc by IFNs depends on Stat1. Although PDGF or IFNs induced c–myc expression independently in Stat1-null cells, simultaneous treatment was not additive or synergistic, indicating that these ligands may utilize similar signaling pathways (Figure 2B and C).
IFN–γ regulates the expression of c–myc but not the CDK inhibitor p21 in human fibrosarcoma cells
Human fibrosarcoma cells expressing Stat1 (2fTGH), lacking Stat1 (U3A) or reconstituted with Stat1 (U3A variants) have been used extensively to study the roles of Stat1 in a variety of biological responses (Horvath et al., 1996; Kumar et al., 1997b). We investigated the IFN–γ-dependent regulation of c–myc expression in these cell lines. As in MEFs, the expression of c–myc was suppressed by IFN–γ in 2fTGH and induced in U3A cells (Figure 3A). Since the cyclin–CDK inhibitor p21waf1 has been suggested to mediate growth arrest in response to IFN–γ in epidermal carcinoma and glioblastoma cells (Chin et al., 1996; Kominsky et al., 1998), we investigated the response of the p21 gene to IFN–γ in 2fTGH and U3A cells. Northern and Western blot analyses revealed that p21 expression was not significantly increased by IFN–γ treatment in either cell line (Figure 3).
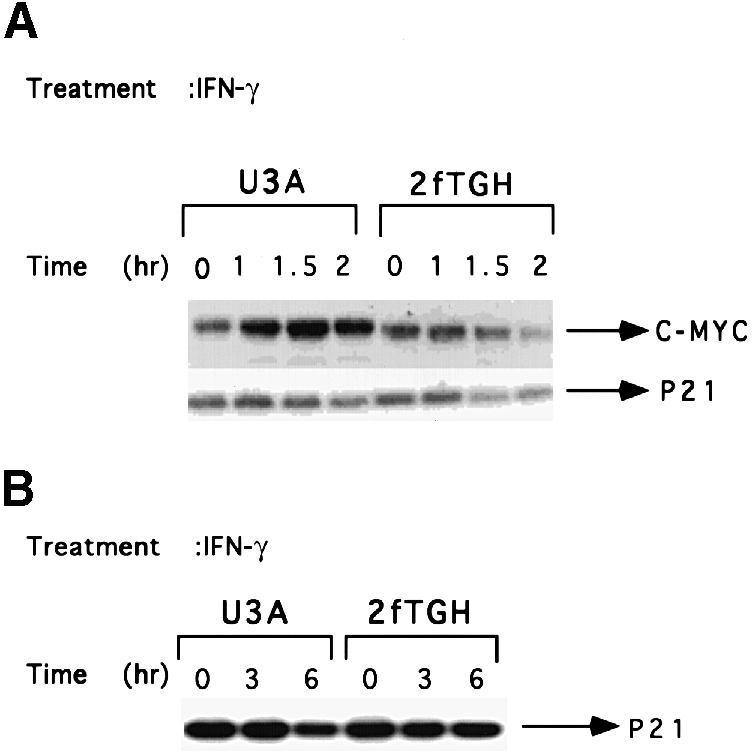
Fig. 3. Regulation of c–myc but not p21waf1 expression by IFN–γ in 2fTGH and U3A cells. (A) Subconfluent, serum-starved cells were treated with 1500 IU/ml of human IFN–γ for 60 to 120 min. Northern transfers were hybridized with the probes indicated. (B) Cells were either untreated or treated with 1500 IU/ml of human IFN–γ for 3 or 6 h. p21waf1 levels were determined by Western blot analysis.
Both tyrosine and serine phosphorylation of Stat1 are required to suppress c–myc expression
We examined the effects of IFN–γ in U3A cells reconstituted with Stat1 variants lacking the tyrosine phosphorylation site 701 or the serine phosphorylation site 727, which are in the transactivation domain of Stat1. c–myc expression was induced, not suppressed, by IFN–γ in these cell lines, indicating that these two amino acid residues, required for Stat1-dependent transactivation, are also required to suppress c–myc expression (Figure 4). In control U3A cells reconstituted with wild-type Stat1, the expression of c–myc was suppressed similarly to parental 2fTGH cells (Figure 4). In U4A or γ2A cells, lacking Jak1 and Jak2, respectively, neither induction nor suppression of c–myc was observed in response to IFN–γ, indicating that both of these kinases are required for both Stat1-dependent and Stat1-independent regulation of c–myc expression (data not shown).
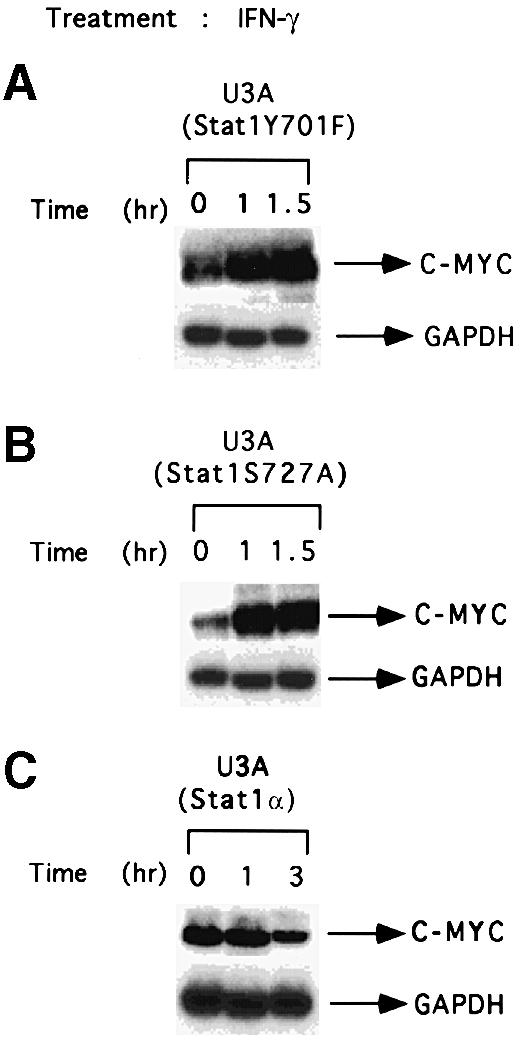
Fig. 4. Stat1 domains required to suppress expression of c–myc mRNA. U3A cells reconstituted with a Stat1 tyrosine (U3A Stat1 Y701F) or serine (U3A Stat1 S727A) phosphorylation site mutant or with wild-type Stat1α (Kumar et al., 1997b; see for expression levels) were treated with 1500 IU/ml of human IFN–γ. c–myc and GAPDH RNA levels were determined by Northern blot analysis.
A consensus GAS element in the c–myc promoter is necessary but not sufficient for negative regulation of expression in response to IFN–γ
A series of promoter deletions linked to a luciferase reporter was used to identify the elements responsible for suppression of c–myc expression in 2fTGH cells. Sequences between –1138 and –1100 were required to mediate a 5–fold reduction in luciferase expression in response to IFN–γ (Figure 5A). Sequence analysis revealed a consensus GAS element at –1107 to –1099, with strong homology to other GAS elements (Table I). A concatamer containing seven copies of the c–myc GAS element trans-activated the expression of the same reporter by 70–fold, showing that this GAS element lacks intrinsic repressor activity (Figure 5B). Therefore, this element is necessary but not sufficient for c–myc suppression. It is likely that suppression involves the interaction of Stat1 bound to the GAS element with a co-repressor bound elsewhere in the c–myc promoter. A 1.7 kb c–myc promoter fragment linked to the cell surface marker cd2 was stably transfected into NIH 3T3 fibroblasts. Treatment with PDGF induced cd2 expression, whereas IFN–γ abrogated this induction of expression by PDGF (Figure 5C), in accord with the results of Northern analysis in MEFs (Figure 2).

Fig. 5. Identification of a GAS element in the c–myc promoter that is necessary but not sufficient for suppression. (A) The full-length 1.7 kb c–myc promoter or two deletion constructs were linked to luciferase and transfected transiently into 2fTGH cells. Luciferase activity was determined with or without IFN–γ treatment (1500 IU/ml for 6 h). The data shown represent duplicate experiments in three independent trials (standard deviations). (B) Constructs containing eight copies of the consensus GAS (TTCTCGGAA) or seven copies of the c–myc GAS (TTCTGGGAA) linked to luciferase were transfected transiently into 2fTGH cells. Luciferase activity was determined with or without IFN–γ treatment (1500 IU/ml for 6 h). Data are presented from three independent experiments, with standard deviations. (C) NIH 3T3 cells stably transfected with the 1.7 kb c–myc promoter linked to the cell-surface protein cd2 were serum-starved and treated with PDGF alone (200 ng/ml), PDGF plus IFN–γ (1000 IU/ml) or were untreated. cd2 expression was determined by FACScan analysis.
Table I. Comparison of functional GAS elements.
| Gene | Species | GAS sequence |
|---|---|---|
| c–myca | mouse | T T C T G G G A A |
| ICAM-1 | human | T T C C C G G A A |
| IRF–1 | mouse | T T C C C C G A A |
| ICSBP | human | T T C T C G G A A |
| FcγR1 | human | T T C C C A G A A |
| IFP 53 | human | T T C T C A G A A |
aThe c–myc sequence is from Roussel et al. (1994) and the others are from Schindler and Darnell (1995).
Electrophoretic mobility shift assays (EMSAs) indicate that Stat1 bound as a homodimer to the c–myc GAS element in extracts of IFN–γ-treated 2fTGH but not U3A cells (Figure 6A). Binding was abolished by pre-incubation with either anti-Stat1 or unlabeled competitor oligonucleotide, indicating that the interaction is specific (Figure 6B). EMSAs with extracts of NIH 3T3 cells revealed that IFN–γ stimulated the binding of Stat1 homodimers to the c–myc GAS element, whereas PDGF did not generate any complex (Figure 6C). In response to PDGF, Stat1 and Stat3 homodimers and Stat1–Stat3 heterodimers are formed on a high-affinity GAS element such as the SIE, and the complexes with Stat3 predominate (Vignais et al., 1996). Apparently the relatively small amount of Stat1 dimer formed in response to PDGF was not sufficient for us to observe binding to the c–myc GAS element under the conditions employed.
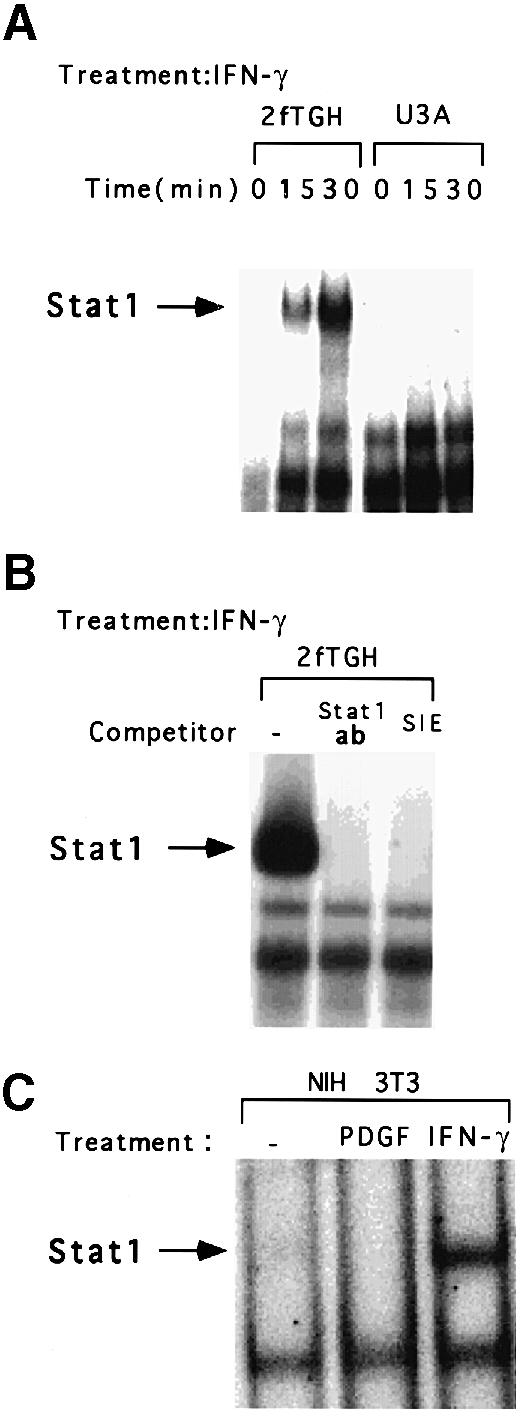
Fig. 6. Binding of Stat1 to the c–myc GAS. (A) EMSAs were performed with whole-cell extracts prepared from U3A or 2fTGH cells, not treated or treated for 15 or 30 min with 1000 IU/ml of IFN–γ. (B) EMSAs were performed with whole-cell extracts from 2fTGH cells treated with 1000 IU/ml of IFN–γ. The extracts were pre-incubated with anti-Stat1 or with a 100–fold molar excess of the unlabeled SIE (m67) GAS. (C) EMSAs were performed with whole-cell extracts from NIH 3T3 cells, treated for 30 min with 200 ng/ml of PDGF, 1000 IU/ml of murine IFN–γ, or not treated.
Role of PKR in Stat1-dependent suppression and transactivation in response to IFN–γ
The IFN–inducible, double-stranded, RNA-activated protein kinase PKR plays an important role in anti-viral and anti-proliferative responses to the IFNs (Williams, 1995; Clemens and Elia, 1997). A dominant-negative variant of PKR can abrogate the IFN–α-mediated inhibition of c–myc expression and the inhibition of cellular proliferation in M1 myeloid leukemia cells (Raveh et al., 1996). PKR-null cells are defective in activating the transcription factors IRF–1 and NF-κB in response to double-stranded RNA and in signaling in response to IFN–γ (Yang et al., 1995; Kumar et al., 1997a). Furthermore, the activation of the GBP and IRF–1 promoters in response to IFN–γ is defective in PKR-null cells (Kumar et al., 1997a). Since these promoters are complex, requiring NF-κB and IRF–1 in addition to Stat1, we analyzed transcription driven by an oligomeric GAS element alone to determine the response to IFN–γ in PKR-null and wild-type cells. Stat1-dependent transactivation was 4–fold lower in PKR-null cells than in wild-type cells (Figure 7A). EMSAs revealed that the binding of Stat1 to DNA was not defective in PKR-null cells, either in the absence (Figure 7B) or in the presence of serum (Kumar et al., 1997a). However, the IFN–γ-stimulated phosphorylation of Stat1 on Ser727 was defective in serum-starved PKR-null cells (Figure 7C). Also consistent with a defect in Stat1 activation in PKR-null cells, treatment with IFN–γ led to the induction of c–myc mRNA, rather than to the suppression seen in wild-type cells, with kinetics similar to those observed in Stat1-null cells (Figure 7D). These results reveal that both Stat1 and PKR are required for IFN–γ to suppress c–myc expression.
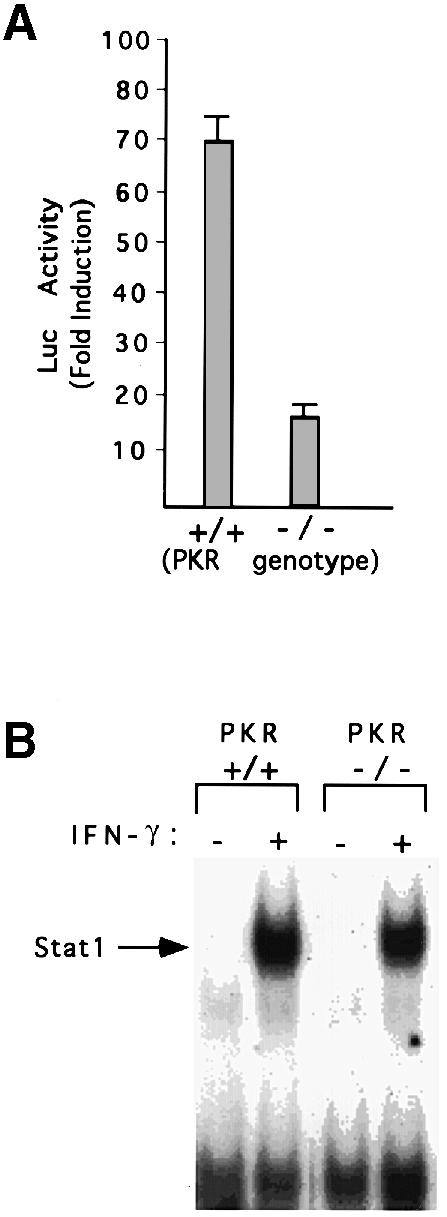

Fig. 7. Defective Stat1 activation and serine phosphorylation in PKR-null cells. (A) 8× con GAS linked to luciferase was transiently transfected into wild-type or PKR-null MEFs. Luciferase activity was determined with or without murine IFN–γ (1000 IU/ml) for 6 h. Results are presented with standard deviations from three independent experiments. (B) Whole-cell extracts were prepared from serum-starved wild-type or PKR-null cells, with or without treatment with 1000 IU/ml of murine IFN–γ. Stat1 binding to the SIE (m67) GAS was determined by EMSA. (C) Extracts of serum-starved cells, either untreated or treated with IFN–γ (1000 IU/ml for 20 min) were immunoprecipitated with anti-Stat1. The transfer was probed first with an antibody specific for a Stat1 peptide that includes phosphorylated Ser727 and then reprobed with anti-Stat1. (D) RNA from MEFs untreated or treated with murine IFN–γ (1000 IU/ml) was analyzed by the Northern procedure.
IFN–dependent induction of c–myc expression in Stat1-null cells is inhibited by geldanamycin or dominant-negative p50cdc37
Raf1 activation is critical for the mitogen-induced signaling pathways that mediate the induction of c–myc expression (Kerkoff et al., 1998; Aziz et al., 1999). p50cdc37 and HSP90 are critical partners in the activation of Raf–1 in mammalian cells (Grammatikakis et al., 1999). Pre-treatment with geldanamycin, an HSP90-specific inhibitor, or overexpression of dominant-negative p50cdc37 prevents the association of Raf–1 with p50cdc37 and HSP90, and the activation of Raf–1 and MAPK in response to growth factors (Grammatikakis et al., 1999). Pre-treatment with geldanamycin prevented the induction of c–myc expression by IFN–γ, IFN–β or PDGF in Stat1-null cells (Figure 8A). Similarly, expression of dominant-negative p50cdc37 also prevented the induction of c–myc expression by IFN–γ in Stat1-null cells (Figure 8B). These results suggest that Raf–1 activation is critical for the induction of c–myc expression by IFNs in Stat1-null cells.
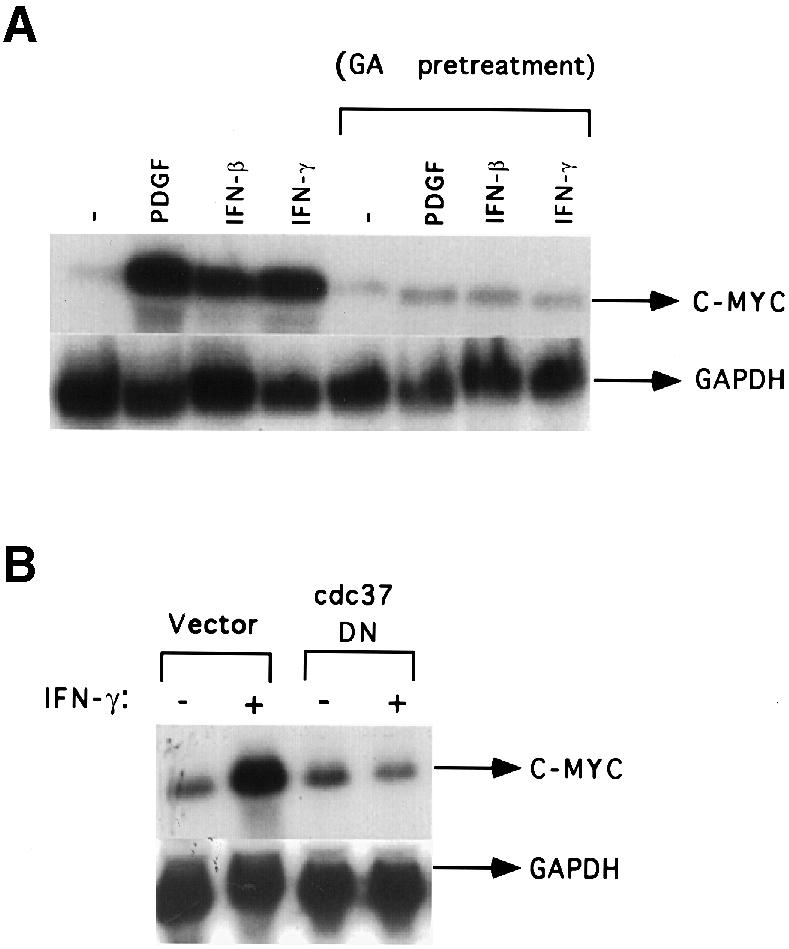
Fig. 8. Pre-treatment with geldanamycin or dominant-negative p50cdc37 abrogates the induction of c–myc by IFN–γ. (A) Stat1-null MEFs were serum-starved and either untreated or pre-treated with geldanamycin (GA, 2 μg/ml) for 5 h. The cells were unstimulated or stimulated with IFN–γ, IFN–β or PDGF for 30 min. c–myc and GAPDH mRNA levels were determined by Northern blot analysis. (B) Stat1-null MEFs were transiently transfected with vector alone or vector encoding dominant-negative p50cdc37. The transfected cells were serum-starved for 24 h and either unstimulated or stimulated with IFN–γ for 30 min. c–myc and GAPDH RNA levels were determined as above.
Discussion
c–myc and c–jun, required for cell cycle progression (Grandori and Eisenman, 1997; Obaya et al., 1999; Wisdom et al., 1999), are important targets of Stat1-independent responses to IFN–γ. c–jun is required for progression through G1 and for trans-activation of the cyclin D1 gene in fibroblasts (Wisdom et al., 1999), thus helping to provide a link between the response to growth factors and cell cycle regulation. Deregulated expression of c–myc and c–jun is likely to be important in the abnormal proliferation of Stat1-null cells in response to IFN–γ, which can serve as a growth factor for some cells (Caux et al., 1992; Shiohara et al., 1993). IFN–γ suppressed the expression of c–myc in wild-type cells and is likely to be important in regulating the switch between growth arrest and proliferation. IFN–γ or -β abrogated the induction by PDGF of c–myc expression in wild-type cells but not in Stat1-null cells, suggesting that Stat1 is required for this response. IFN–α also abrogated the induction of c–myc by PDGF in NIH 3T3 fibroblasts and in Kaposi's sarcoma cells (Einat et al., 1985; Koster et al., 1996), although the direct involvement of Stat1 in these responses was not demonstrated. Our results also indicate that c–myc, but not the CDK inhibitor p21waf1, is a target of regulation by IFN–γ in human fibrosarcoma cells. p21 has been implicated as a mediator of IFN–γ-dependent growth arrest in epidermal carcinoma and glioblastoma but not colon carcinoma (HCT116) cell lines (Chin et al., 1996; Kominsky et al., 1998; Sharma and Iozzo, 1998). p21 is induced by IFN–γ in tumor cell lines harboring mutated p53 but not in cell lines expressing wild-type p53 such as HT1080 (from which 2fTGH and U3A cells are derived), where its basal expression is high, indicating that the regulation of p21 expression by p53 is dominant over IFN–γ-mediated regulation of this gene. Studies in U3A cell variants indicate that tyrosine and serine phosphorylation sites in the C-terminal transactivation domain of Stat1 are required to suppress c–myc expression. These results are consistent with previous data suggesting that transcriptionally competent Stat1 is required for the anti-proliferative effect of IFNs (Bromberg et al., 1996).
Transient transfection of a c–myc promoter fragment linked to luciferase revealed that a consensus GAS element –1107 to –1099 (relative to the P1 promoter) is required for c–myc suppression. This element differs from the previously identified GAS element, which binds to Stat3 preferentially, overlaps the E2F element and functions in the IL–6- and gp130-mediated transactivation of c–myc (Kiuchi et al., 1999). Stat1 binds to the upstream GAS as a homodimer in extracts of IFN–γ-treated wild-type cells. The upstream element is necessary but not sufficient for suppression of c–myc since it lacks intrinsic repressor activity. Therefore, Stat1 is likely to interact with a co-repressor bound to another site in the c–myc promoter to inhibit expression. A likely candidate is Blimp–1, a member of the Groucho family of co-repressors that binds to the PRF site of the c–myc promoter and mediates repression (Lin et al., 1997; Ren et al., 1999). Since Blimp–1 is expressed exclusively in B-lymphocytes, other Groucho family members may participate in repressing c–myc expression in other cell types. Another candidate is MBP-1, which represses c–myc expression when bound to the E2F site (Ray and Miller, 1991). IFN–γ inhibits the transcription of several genes, including those encoding perlecan, bullous pemphigoid antigen 1 and cyclin A (Tamai et al., 1995; Sharma and Iozzo, 1998; Sibinga et al., 1999). Transcriptional repression of the perlecan gene by IFN–γ requires functional Stat1 and a promoter region containing multiple GAS elements. However, the binding of Stat1 to these GAS elements has not been reported and thus the mechanism of repression is not known (Sharma and Iozzo, 1998).
In NIH 3T3 cells stably expressing cd2 under the control of the 1.7 kb c–myc promoter, treatment with PDGF induced cd2 expression, and simultaneous treatment with IFN–γ abrogated this induction. The induction of gene expression by PDGF depends on several signal transduction pathways, among which is the ras/MAPK pathway, and involves the E2F site of the c–myc promoter (Sacca and Cochran, 1990; Claesson-Welsh, 1994). ras/MAPK-activated Ets factors have been proposed to mediate c–myc expression in response to growth factor stimulation (Roussel et al., 1994; Aziz et al., 1999; Cheng et al., 1999). The abrogation by IFN–γ of c–myc induction in response to PDGF might involve a competition between promoter-bound Ets factors and Stat1 dimers for co-activators such as CBP/p300 (Horvai et al., 1997). PDGF activates the formation of complexes involving Stat1, Stats1 and 3 and Stat3 on the SIE element of the c–fos gene (Vignais et al., 1996). However, PDGF did not induce the formation of Stat complexes on the c–myc GAS element, indicating that the abrogation by IFN–γ of c–myc induction in response to PDGF does not involve a competition between different Stat dimers for the GAS site.
PKR is involved in regulating anti-viral, anti-proliferative and tumor suppressor functions (Clemens and Elia, 1997), and PKR-null cells are defective in activating IRF–1 and NF-κB in response to double-stranded RNA (Kumar et al., 1997a). The defective activation of the GBP and IRF–1 promoters by IFN–γ in PKR-null cells can be rescued by expressing wild-type but not mutant PKR, indicating that PKR is also required in IFN–γ-dependent signaling (Kumar et al., 1997a). In extracts of IFN–γ treated cells, a decrease in the mobility of PKR in SDS–PAGE gels was observed, consistent with its phosphorylation (Kumar et al., 1997a). A dominant-negative derivative of PKR abrogated both the IFN–α-mediated downregulation of c–myc expression and the inhibition of cell growth (Raveh et al., 1996). Our results indicate that PKR-null cells are defective in phosphorylating Stat1 on Ser727, and that Stat1-dependent transactivation is 4–fold lower in PKR-null than in wild-type cells, an effect comparable to the reduction observed for the S727A mutant of Stat1 (Zhang et al., 1998). Furthermore, in PKR-null cells, c–myc mRNA is induced transiently and rapidly in response to IFN–γ, just as it is in Stat1-null cells. Therefore, both Stat1 and PKR are required to suppress the expression of c–myc in wild-type cells. PKR associates with Stat1 both in vitro and in vivo, although it does not phosphorylate Stat1 directly (A.H.Wong et al., 1997). Therefore, PKR may be part of a kinase cascade involved in phosphorylating Stat1 on Ser727 in response to IFN–γ. The phosphorylation of Stat1 on Ser727 is required for maximal transactivation and recruitment of the transcription co-factor MCM–5 (Wen et al., 1995; Zhang et al., 1998), also a component of the DNA replication licensing factor. The recruitment of MCM–5 from origins of DNA replication to the transcriptional machinery, mediated by Stat1 in response to IFN–γ, has been suggested as a mechanism for suppression of proliferation (Zhang et al., 1998). Nmi, originally identified because it interacts with N-myc, was later shown to be inducible by IFN–α and also to enhance Stat1-dependent transactivation (Bao and Zervos, 1996; Lebrun et al., 1998; Zhu et al., 1999). Whether PKR is involved in regulating Nmi or MCM–5 is not known at present.
The kinases directly responsible for phosphorylating Stat1 on Ser727 are not known. Stat1 serine phosphorylation is enhanced by treatment with IFN–γ or LPS, or by serum stimulation (Wen et al., 1995; Kovarik et al., 1998; Takaoka et al., 1999). The Ser727 phosphorylation site lies within an MAPK consensus sequence and the MAPK Erk2 has been proposed to phosphorylate this residue (David et al., 1995). The IFN–γ-induced activation of Erk2, serine phosphorylation of Stat1 and Stat1-dependent transactivation are strongly inhibited by overexpression of a dominant-negative form of the protein tyrosine kinase Pyk2 in a Jak2-dependent manner (Takaoka et al., 1999). Evidence for a Stat1 serine kinase that depends on Jak2 and is distinct from MAPK has also been presented (Zhu et al., 1997). These results indicate that the phosphorylation of Stat1 on serine is likely to be regulated by kinase cascades rather than by a single kinase.
Genetic analyses in yeast and Drosophila have shown that p50cdc37 functions both in the cell cycle and in the ras/raf/MAPK pathway in close cooperation with its partner HSP90 (Reed, 1980; Cutforth and Rubin, 1994). Recent studies have shown that p50cdc37 is the primary determinant of HSP90 recruitment to Raf–1 and of the activation of Raf–1 by serum and growth factors in mammalian cells (Grammatikakis et al., 1999). Co-expression of p50cdc37 strongly potentiated the v-src-mediated activation of Raf–1 and, conversely, dominant-negative p50cdc37, unable to recruit HSP90 into the Raf–1 complex, abrogated the activation of Raf–1 (Grammatikakis et al., 1999). Inhibition of Raf–1 activation by pre-treatment with geldanamycin or expression of dominant-negative p50cdc37 abrogated the induction of c–myc by IFN–γ in Stat1-null cells, suggesting that Raf–1 activation is critical for this pathway. Jak1 and Jak2 are also implicated in the regulation of c–myc expression in response to IFN–γ. Conditional dimerization of Jak1 or Jak2, leading to their activation, can stimulate the c–myc promoter (Mizuguchi and Hatakeyama, 1998; Mohi et al., 1998). It is likely that, in the absence of Stat1, IFN–γ-activated Jak1 and Jak2 phosphorylate as yet unknown signaling molecules that can activate c–myc expression through Raf–1. Both IFN–γ and IFN–β mediate the activation of c–myc in the absence of Stat1 and it remains to be determined whether the signals emanating from the two different receptors are the same or different.
A global expression study has shown that growth factor-dependent stimulation of a mutant PDGF receptor with a restored ras–GAP binding site promotes the induction of IFN–γ-responsive genes rather than of immediate-early genes (Fambrough et al., 1999), revealing a link between PDGF- and IFN–γ-dependent pathways. We have identified several additional genes repressed in wild-type cells or induced in U3A cells in response to IFN–γ (Der et al., 1998; C.V.Ramana and G.R.Stark, unpublished data). Characterization of the promoters of these genes and further analysis of the c–myc and c–jun promoters should also help to identify the components of this novel pathway.
Materials and methods
Reagents and cell culture
Recombinant human and mouse IFN–γ were purchased from Boehringer Mannheim. PDGF–BB and murine IFN–β were purchased from Gibco-BRL and PBL, respectively. The human fibrosarcoma cell lines 2fTGH, U3A, U4A, γ2A and derivatives of U3A reconstituted with Stat1 variants were described by Kumar et al. (1997b). The HT1080 parents of all these cells have wild-type p53, which drives high expression of p21 (our unpublished data). MEFs from littermates of Stat1-null, PKR-null and the corresponding wild-type mice were obtained as described by Meraz et al. (1996) and Kumar et al. (1997a). NIH 3T3 mouse fibroblasts were obtained from M.Roussel (St Jude Children's Research Hospital, Memphis, TN). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum (FCS; Gibco-BRL). Subconfluent cells were serum-starved for 24–48 h in DMEM containing 0.1% serum before treatment with IFNs or PDGF, which were added directly to the serum-free medium.
RNA, protein and cell cycle analyses
Total RNA was prepared by using the TRIzol (Gibco-BRL) method according to the manufacturer's instructions. Northern transfers were analyzed with c–myc, c–fos, c–jun, jun–B and p21 cDNA probes (Langer et al., 1992; el-Deiry et al., 1993). The glyceraldehyde 3–phosphate dehydrogenase (GAPDH) probe was from Ambion (Austin, TX). The expression levels of c–myc and GAPDH were quantitated with a PhosphorImager (Molecular Dynamics). Western analyses were performed as described by Chernov et al. (1998). Antibodies to Stat1 and p21waf1 were purchased from Transduction Laboratories and Santa Cruz Biotechnology, respectively. Cell cycle analysis was performed according to Bromberg et al. (1996). The data were analyzed using the LYSIS II program.
Reporter constructs, transient transfections and luciferase assays
Luciferase constructs containing elements of the c–myc promoter (–1100 to +580 and –424 to +580, relative to the P1 transcription site) have been described by Wong et al. (1995). The BglII fragment (–1138 to +580) was purified from M Bg CAT (Yang et al., 1986) and ligated into the BamHI site of pGL2-Basic Luc (Promega) or pJ3–cd2 (a gift from I.Kerr, ICRF, London) to generate c–myc promoter-driven luciferase and cd2 reporters. The 8× consensus (con) GAS-Luc was described by Horvai et al. (1997), and 7× c–myc GAS-Luc was generated by replacing the 8× GAS in the above construct with a concatamer containing seven copies of the c–myc GAS(TTCTGGGAA), using NheI and PstI linkers. Dominant-negative p50cdc37 was described previously (Grammatikakis et al., 1999). Transient transfections were performed using Fugene 6 (Boehringer Mannheim), according to the manufacturer. Twenty-four hours after transfection, the cells were serum-starved overnight and treated with human or murine IFN–γ (1500 IU/ml) for 6 h. Luciferase assays were performed according to the manufacturer (Promega), and the activities were normalized to total protein concentrations, determined in Bio-Rad assays. Each experiment was performed in duplicate and the results shown are the average of three independent experiments (standard deviations shown). The c–myc promoter-cd2 expression vector was transfected stably into NIH 3T3 cells, individual clones expressing cd2 were isolated, and the induction of cd2 by PDGF was confirmed by FACScan analysis. Sixteen hours after treating serum-starved cells with PDGF or PDGF plus IFN–γ, the expression of cd2 was determined by staining the cells with a phycoerythrin-conjugated monoclonal antibody against cd2 (Dako), followed by FACScan analysis using a Becton Dickinson instrument and the LYSYS II software package.
Immunoprecipitations and immunoblot analyses
Wild-type and PKR-null cells were untreated or stimulated with IFN–γ or IFN–α (1000 IU/ml) for 20 min and cell extracts were prepared as described by Kumar et al. (1997a). Equal amounts of extracts were immunoprecipitated with anti-Stat1 (Transduction Laboratories), and the immune complexes were collected on Gamma-bind G–Sepharose beads (Pharmacia). Samples were run on SDS–7.5% polyacrylamide gels, transferred to polyvinyledine difluoride membranes and probed with the immunoprecipitating antibody or with an antibody to a Stat1 peptide containing phosphorylated Ser727 (from Upstate Biotechnology) followed by anti-rabbit IgG coupled to horseradish peroxidase (Bio-Rad). The transfers were analyzed by using the Renaissance chemiluminescence reagent (DuPont NEN).
Electrophoretic mobility shift assays (EMSAs)
Nuclear extracts were prepared and EMSAs were performed as described by Kumar et al. (1997a), using as probe a double-stranded oligonucleotide containing the c–myc promoter sequence 5′–CCCTTTCTGGGAAGTCCGGGT–3′ (the homology to GAS sequences is underlined). The high-affinity Stat binding site, SIE (m67), was described by Vignais et al. (1996).
Acknowledgments
Acknowledgements
We thank Drs R.Schreiber, K.Marcu, M.Roussel, J.Cleveland, I.Kerr, M.Ostrowski, K.Calame, P.Howe, C.Glass and D.Frank for reagents, and M.Roussel for sequence information. This work was supported by NIH grant P01 CA62220.
References
- Abril E., Real, L.M., Serrano, A., Jimenez, P., Garcia, A., Canton, J., Trigo, I., Garrido, F. and Ruiz-Cabello, F. (1998) Unresponsiveness to interferon associated with STAT1 protein deficiency in a gastric adenocarcinoma cell line. Cancer Immunol. Immunother., 47, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz N., Cherwinski, H. and McMahon, M. (1999) Complementation of defective colony-stimulating factor 1 receptor signaling and mitogenesis by Raf and v-Src. Mol. Cell. Biol., 19, 1101–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. and Taylor-Papadimitriou, J. (1978) Interferon affects both G1 and S+G2 in cells stimulated from quiescence to growth. Nature, 274, 798–801. [DOI] [PubMed] [Google Scholar]
- Bao J. and Zervos, A.S. (1996) Isolation and characterization of Nmi, a novel partner of Myc proteins. Oncogene, 12, 2171–2176. [PubMed] [Google Scholar]
- Bennett M.R., Evan, G.I. and Newby, A.C. (1994) Deregulated expression of the c–myc oncogene abolishes inhibition of proliferation of rat vascular smooth muscle cells by serum reduction, interferon-γ, heparin, and cyclic nucleotide analogues and induces apoptosis. Circ. Res., 74, 525–536. [DOI] [PubMed] [Google Scholar]
- Blackwood E.M. and Eisenman, R.N. (1991) Max: a helix–loop–helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science, 251, 1211–1217. [DOI] [PubMed] [Google Scholar]
- Bouchard C., Staller, P. and Eilers, M. (1998) Control of cell proliferation by Myc. Trends Cell Biol., 8, 202–206. [DOI] [PubMed] [Google Scholar]
- Bromberg J.F., Horvath, C.M., Wen, Z., Schreiber, R.D., and Darnell J.E.,Jr (1996) Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon α and interferon γ. Proc. Natl Acad. Sci. USA, 93, 7673–7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg J.F., Fan, Z., Brown, C., Mendelsohn, J., and Darnell J.E.,Jr (1998) Epidermal growth factor-induced growth inhibition requires Stat1 activation. Cell Growth Differ., 9, 505–512. [PubMed] [Google Scholar]
- Caux C., Moreau, I., Saeland, S. and Banchereau, J. (1992) Interferon-γ enhances factor-dependent myeloid proliferation of human CD34+ hematopoietic progenitor cells. Blood, 79, 2628–2635. [PubMed] [Google Scholar]
- Cheng M., Wang, D. and Roussel, M.F (1999) Expression of c–myc in response to colony-stimulating factor-1 requires mitogen-activated protein kinase kinase-1. J. Biol. Chem., 274, 6553–6558. [DOI] [PubMed] [Google Scholar]
- Chernov M.V., Ramana, C.V., Adler, V.V. and Stark, G.R. (1998) Stabilization and activation of p53 are regulated independently by different phosphorylation events. Proc. Natl Acad. Sci. USA, 95, 2284–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernova O.B., Chernov, M.V., Ishizaka, Y., Agarwal, M.L. and Stark, G.R. (1998) MYC abrogates p53-mediated cell cycle arrest in N-(phosphonacetyl)-l-aspartate-treated cells, permitting CAD gene amplification. Mol. Cell. Biol., 18, 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin Y.E., Kitagawa, M., Su, W.C., You, Z.H., Iwamoto, Y. and Fu, X.-Y. (1996) Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21WAF1/CIP1 mediated by STAT1. Science, 272, 719–722. [DOI] [PubMed] [Google Scholar]
- Claesson-Welsh L. (1994) Platelet-derived growth factor receptor signals. J. Biol. Chem., 269, 32023–32026. [PubMed] [Google Scholar]
- Clemens M.J. and Elia, A. (1997) The double-stranded RNA-dependent protein kinase PKR: structure and function. J. Interferon Cytokine Res., 17, 503–524. [DOI] [PubMed] [Google Scholar]
- Cutforth T. and Rubin, G.M. (1994) Mutations in in HSP83 and cdc37 impair signaling by the sevenless receptor tyrosine kinase in Drosophila. Cell, 77, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Dang C.V. (1999) c–myc target genes involved in cell growth, apoptosis and metabolism. Mol. Cell. Biol., 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J.E. Jr (1997) STATs and gene regulation. Science, 277, 1630–1635. [DOI] [PubMed] [Google Scholar]
- David M., Petricoin, E., III, Benjamin, E., Pine, R., Weber, M.J. and Larner, A.C. (1995) Requirement for MAP kinase (ERK2) activity in interferon α- and interferon β-stimulated gene expression through STAT proteins. Science, 269, 1721–1723. [DOI] [PubMed] [Google Scholar]
- Der S.D., Zhou, A., Williams, B.R. and Silverman, R.H. (1998) Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl Acad. Sci. USA, 95, 15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin J.E., Hackenmiller, R., Simon, M.C. and Levy, D.E. (1996) Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell, 84, 443–450. [DOI] [PubMed] [Google Scholar]
- Einat M., Resnitzky, D. and Kimchi, A. (1985) Inhibitory effects of interferon on the expression of genes regulated by platelet-derived growth factor. Proc. Natl Acad. Sci. USA, 82, 7608–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry W.S. et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Fambrough D., McClure, K., Kazlauskas, A. and Lander, E. (1999) Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell, 97, 724–741. [DOI] [PubMed] [Google Scholar]
- Farrar M.A. and Schreiber, R.D. (1993) The molecular cell biology of interferon-γ and its receptor. Annu. Rev. Immunol., 11, 571–611. [DOI] [PubMed] [Google Scholar]
- Felsher D.W. and Bishop, J.M. (1999) Transient excess of Myc activity can elicit genomic instability and tumorigenesis. Proc. Natl Acad. Sci. USA, 96, 3940–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatikakis N., Lin, J.-H., Grammatikakis, A., Tsichlis, P.N. and Cochran, B.H. (1999) p50cdc37 acting in concert with HSP90 is required for Raf1 function. Mol. Cell. Biol., 19, 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori C. and Eisenman, R.N. (1997) Myc target genes. Trends Biochem. Sci., 22, 177–181. [DOI] [PubMed] [Google Scholar]
- Greenberg M.E. and Ziff, E.B. (1984) Stimulation of 3T3 cells induces transcription of the c–fos proto-oncogene. Nature, 311, 433–438. [DOI] [PubMed] [Google Scholar]
- Horvai A.E., Xu, L., Korzus, E., Brard, G., Kalafus, D., Mullen, T.M., Rose, D.W., Rosenfeld, M.G. and Glass, C.K. (1997) Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl Acad. Sci. USA, 94, 1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath C.M., Stark, G.R., Kerr, I.M. and Darnell, J.E.,Jr (1996) Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol. Cell. Biol., 16, 6957–6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan D.H., Shankaran, V., Dighe, A.S., Stockert, E., Aguet, M. and Schreiber, R.D. (1998) Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proc. Natl Acad. Sci. USA, 95, 7556–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkoff E., Houben, R., Loffler, S., Troppmair, J., Lee, J.-E. and Rapp, U.R. (1998) Regulation of c–myc expression by Ras/Raf signaling. Oncogene, 16, 211–216. [DOI] [PubMed] [Google Scholar]
- Kimchi A. (1992) Cytokine triggered molecular pathways that control cell cycle arrest. J. Cell. Biochem., 50, 1–9. [DOI] [PubMed] [Google Scholar]
- Kiuchi N., Nakajima, K., Ichiba, M., Fukada, T., Narimatsu, M., Mizuno, K., Hibi, M. and Hirano, T. (1999) Stat3 is required for the gp130-mediated full activation of the c–myc gene. J. Exp. Med., 189, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominsky S., Johnson, H.M., Bryan, G., Tanabe, T., Hobeika, A.C., Subramaniam, P.S. and Torres, B. (1998) IFNγ inhibition of cell growth in glioblastomas correlates with increased levels of the cyclin dependent kinase inhibitor p21WAF1/CIP1. Oncogene, 17, 2973–2979. [DOI] [PubMed] [Google Scholar]
- Koster R., Blatt, L.M., Streubert, M., Zietz, C., Hermeking, H., Brysch, W. and Sturzl, M. (1996) Consensus-interferon and platelet-derived growth factor adversely regulate proliferation and migration of Kaposi's sarcoma cells by control of c–myc expression. Am. J. Pathol., 149, 1871–1885. [PMC free article] [PubMed] [Google Scholar]
- Kovarik P., Stoiber, D., Novy, M. and Decker, T. (1998) Stat1 combines signals derived from IFN–γ and LPS receptors during macrophage activation. EMBO J., 17, 3660–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A. et al. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF–1 and NF-κB. EMBO J., 16, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Commane, M., Flickinger, T.W., Horvath, C.M. and Stark, G.R. (1997b) Defective TNF-α-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science, 278, 1630–1632. [DOI] [PubMed] [Google Scholar]
- Langer S.J., Bortner, D.M., Roussel, M.F., Sherr, C.J. and Ostrowski, M.C. (1992) Mitogenic signaling by colony-stimulating factor 1 and ras is suppressed by the ets-2 DNA-binding domain and restored by myc overexpression. Mol. Cell. Biol., 12, 5355–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaman D.W., Leung, S., Li, X. and Stark, G.R. (1996) Regulation of STAT-dependent pathways by growth factors and cytokines. FASEB J., 10, 1578–1588. [PubMed] [Google Scholar]
- Lebrun S.J., Shpall, R.L. and Naumovski, L. (1998) Interferon-induced upregulation and cytoplasmic localization of Myc-interacting protein Nmi. J. Interferon Cytokine Res., 18, 767–771. [DOI] [PubMed] [Google Scholar]
- Lin S.L., Kikuchi, T., Pledger, W.J. and Tamm, I. (1986) Interferon inhibits the establishment of competence in G0/S–phase transition. Science, 233, 356–359. [DOI] [PubMed] [Google Scholar]
- Lin Y., Wong, K.-K. and Calame, K. (1997) Repression of c–myc transcription by Blimp–1, an inducer of terminal B cell differentiation. Science, 276, 596–598. [DOI] [PubMed] [Google Scholar]
- Look D.C., Pelletier, M.R., Tidwell, R.M., Roswit, W.T. and Holtzman, M.J. (1995) Stat1 depends on transcriptional synergy with Sp1. J. Biol. Chem., 270, 30264–30267. [DOI] [PubMed] [Google Scholar]
- Lucas D.M., Lokuta, M.A., McDowell, M.A., Doan, J.E. and Paulnock, D.M. (1998) Analysis of the IFN–γ-signaling pathway in macrophages at different stages of maturation. J. Immunol., 160, 4337–4342. [PubMed] [Google Scholar]
- Marcu K.B., Bossone, S.A. and Patel, A.J. (1992) Myc function and regulation. Annu. Rev. Biochem., 61, 809–860. [DOI] [PubMed] [Google Scholar]
- Melamed D., Tiefenbrun, N., Yarden, A. and Kimchi, A. (1993) Interferons and interleukin-6 suppress the DNA-binding activity of E2F in growth-sensitive hematopoietic cells. Mol. Cell. Biol., 13, 5255–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz M.A. et al. (1996) Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK–STAT signaling pathway. Cell, 84, 431–442. [DOI] [PubMed] [Google Scholar]
- Mizuguchi R. and Hatakeyama, M. (1998) Conditional activation of Janus kinase (JAK) confers factor independence upon interleukin-3-dependent cells. Essential role of Ras in JAK-triggered mitogenesis. J. Biol. Chem., 273, 32297–32303. [DOI] [PubMed] [Google Scholar]
- Mohi M.G., Arai,Ki. and Watanabe,S. (1998) Activation and functional analysis of Janus kinase 2 in BA/F3 cells using the coumermycin/gyrase B system. Mol. Biol. Cell, 9, 3299–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obaya A.J., Mateyak, M.K. and Sedivy, J.M. (1999) Mysterious liaisons: the relationship between c–myc and the cell cycle. Oncogene, 18, 2934–2941. [DOI] [PubMed] [Google Scholar]
- Raveh T., Hovanessian, A.G., Meurs, E.F., Sonenberg, N. and Kimchi, A. (1996) Double-stranded RNA-dependent protein kinase mediates c–myc suppression induced by type I interferons. J. Biol. Chem., 271, 25479–25484. [DOI] [PubMed] [Google Scholar]
- Ray R. and Miller, D. (1991) Cloning and characterization of a human c–myc promoter-binding protein. Mol. Cell. Biol., 11, 2154–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S.I. (1980) The selection of S.cerevisiae mutants defective in the start event of cell division. Genetics, 95, 561–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B., Chee, K.J., Kim, T.H. and Maniatis, T. (1999) PRDI-BF1/Blimp–1 repression is mediated by corepressors of the Groucho family of proteins. Genes Dev., 13, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzsky D. and Kimchi, A. (1991) Deregulated c–myc expression abrogates the interferon- and interleukin 6-mediated G0/G1 cell cycle arrest but not other inhibitory responses in M1 myeloblastic cells. Cell Growth Differ., 2, 33–41. [PubMed] [Google Scholar]
- Roussel M.F., Davis, J.N., Cleveland, J.L., Ghysdael, J. and Hiebert, S.W. (1994) Dual control of Myc expression through a single DNA binding targeted by ets family proteins. Oncogene, 9, 405–415. [PubMed] [Google Scholar]
- Sacca R. and Cochran, B.H. (1990) Identification of a PDGF-responsive element in the murine c–myc gene. Oncogene, 5, 1499–1505. [PubMed] [Google Scholar]
- Schindler C. and Darnell, J.E.,Jr (1995) Transcriptional responses to polypeptide ligands: the JAK–STAT pathway. Annu. Rev. Biochem., 64, 621–651. [DOI] [PubMed] [Google Scholar]
- Sharma B. and Iozzo, R.V. (1998) Transcriptional silencing of perlecan gene expression by interferon-γ. J. Biol. Chem., 273, 4642–4646. [DOI] [PubMed] [Google Scholar]
- Shiohara M., Koike, K. and Nakahata, T. (1993) Synergism of interferon-γ and stem cell factor on the development of murine hematopoietic progenitors in serum-free culture. Blood, 81, 1435–1441. [PubMed] [Google Scholar]
- Sibinga N.E., Wang, H., Perrella, M.A., Endege, W.O., Patterson, C., Yoshizumi, M., Haber, E. and Lee, M.E. (1999) Interferon-γ-mediated inhibition of cyclin A gene transcription is independent of individual cis-acting elements in the cyclin A promoter. J. Biol. Chem., 274, 12139–12146. [DOI] [PubMed] [Google Scholar]
- Spencer C.A. and Groudine, M. (1991) Control of c–myc regulation in normal and neoplastic cells. Adv. Cancer. Res., 56, 1–48. [DOI] [PubMed] [Google Scholar]
- Stark G.R., Kerr, I.M., Williams, B.R.G., Silverman, R.H. and Schreiber, R.D. (1998) How cells respond to interferons. Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- Sun W.H., Pabon, C., Alsayed, Y., Huang, P.P., Jandeska, S., Uddin, S., Platanias, L.C. and Rosen, S.T. (1998) Interferon-α resistance in a cutaneous T-cell lymphoma cell line is associated with lack of STAT1 expression. Blood, 91, 570–576. [PubMed] [Google Scholar]
- Takaoka A., Tanaka, N., Mitani, Y., Miyazaki, T., Fujii, H., Sato, M., Kovarik, P., Decker, T., Schlessinger, J. and Taniguchi, T. (1999) Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN–γ, but not IFN–α, signaling. EMBO J., 18, 2480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K., Li, K., Silos, S., Rudnicka, L., Hashimoto, T., Nishikawa, T. and Uitto, J. (1995) Interferon-γ-mediated inactivation of transcription of the 230-kDa bullous pemphigoid antigen gene (BPAG1) provides novel insight into keratinocyte differentiation. J. Biol. Chem., 270, 392–396. [DOI] [PubMed] [Google Scholar]
- Tiefenbrun N., Melamed, D., Levy, N., Resnitzky, D., Hoffman, I., Reed, S.I. and Kimchi, A. (1996) α interferon suppresses the cyclin D3 and cdc25A genes, leading to a reversible G0-like arrest. Mol. Cell. Biol., 16, 3934–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairo G., Vadiveloo, P.K., Royston, A.K., Rockman, S.P., Rock, C.O., Jackowski, S. and Hamilton, J.A. (1995) Deregulated c–myc expression overrides IFN γ-induced macrophage growth arrest. Oncogene, 10, 1969–1976. [PubMed] [Google Scholar]
- Vignais M.-L., Sadowski, H.B., Watling, D., Rogers, N.C. and Gilman, M. (1996) Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol. Cell. Biol., 16, 1759–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z., and Darnell, J.E.,Jr (1997) Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res., 25, 2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z., Zhong, Z., and Darnell, J.E.,Jr (1995) Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell, 82, 241–250. [DOI] [PubMed] [Google Scholar]
- Williams B.R.G. (1995) The role of the dsRNA-activated kinase, PKR, in signal transduction. Semin. Virol., 6, 191–202. [Google Scholar]
- Wisdom R., Johnson, R.S. and Moore, C. (1999) c–jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J., 18, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A.H., Tam, N.W., Yang, Y.L., Cuddihy, A.R., Li, S., Kirchhoff, S., Hauser, H., Decker, T. and Koromilas, A.E. (1997) Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J., 16, 1291–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K.K., Zou, X., Merrell, K.T., Patel, A.J., Marcu, K.B., Chellappan, S. and Calame, K. (1995) v-Abl activates c–myc transcription through the E2F site. Mol. Cell. Biol., 15, 6535–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong L.H., Krauer, K.G., Hatzinisiriou, I., Estcourt, M.J., Hersey, P., Tam, N.D., Edmondson, S., Devenish, R.J. and Ralph, S.J. (1997) Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3γ. J. Biol. Chem., 272, 28779–28785. [DOI] [PubMed] [Google Scholar]
- Yang J.-Q., Remmers, E.F. and Marcu, K.B. (1986) The first exon of the c–myc proto-oncogene contains a novel positive control element. EMBO J., 5, 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.-L., Reis, L.F.L., Pavlovic, J., Aguzzi, A., Schaefer, R., Kumar, A., Williams, B.R., Aguet, M. and Weissmann, C. (1995) Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J., 14, 6095–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.J., Vinkemeier, U., Gu, W., Chakravarti, D., Horvath, C.M., and Darnell, J.E.,Jr (1996) Two contact regions between Stat1 and CBP/p300 in interferon γ signaling. Proc. Natl Acad. Sci. USA, 93, 15092–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.J., Zhao, Y., Chait, B.T., Lathem, W.W., Ritzi, M., Knippers, R., and Darnell, J.E.,Jr (1998) Ser727-dependent recruitment of MCM5 by Stat1α in IFN–γ-induced transcriptional activation. EMBO J., 17, 6963–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M., John, S., Berg, M. and Leonard, W.J. (1999) Functional association of Nmi with Stat5 and Stat1 in IL-2- and IFNγ-mediated signaling. Cell, 96, 121–130. [DOI] [PubMed] [Google Scholar]
- Zhu X., Wen, Z., Xu, L.Z., and Darnell, J.E.,Jr (1997) Stat1 serine phosphorylation occurs independently of tyrosine phosphorylation and requires an activated Jak2 kinase. Mol. Cell. Biol., 17, 6618–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]