

Abstract
The nuclear function of the c–Abl tyrosine kinase is not well understood. In order to identify nuclear substrates of Abl, we constructed a constitutively active and nuclear form of the protein. We found that active nuclear Abl efficiently phosphorylate c–Jun, a transcription factor not previously known to be tyrosine phosphorylated. After phosphorylation of c–Jun by Abl on Tyr170, both proteins interacted via the SH2 domain of Abl. Surprisingly, elevated levels of c–Jun activated nuclear Abl, resulting in activation of the JNK serine/threonine kinase. This phosphorylation circuit generates nuclear tyrosine phosphorylation and represents a reversal of previously known signalling models.
Keywords: Abl/JNK/Jun/nuclear tyrosine phosphorylation/phosphorylation circuit
Introduction
The tyrosine kinase Abl is involved in human leukaemias (Sawyers, 1999). Leukaemogenic forms of Abl show high constitutive activity and autophosphorylate in the activation loop (Superti-Furga and Courtneidge, 1995; Raitano et al., 1997). In contrast, the cellular form of Abl is tightly regulated in vertebrate cells (Wang, 1993; Van Etten, 1999). Overexpression of c–Abl does not lead to elevated tyrosine phosphorylation. It is thought that a combination of cellular factors and intramolecular interactions act in concert to ensure inhibition of c–Abl activity (Barilá and Superti-Furga, 1998; Van Etten, 1999).
Among tyrosine kinases, Abl has an unusual dual nuclear and cytoplasmic localization (Wang, 1993; Raitano et al., 1997; Van Etten, 1999). Both positive and negative regulation of growth and apoptosis have been ascribed to Abl, depending on its subcellular location, state of activity and cellular context (Wang, 1993; Raitano et al., 1997; Van Etten, 1999). Genotoxic stress, such as the damage produced by ionizing radiation, has been shown to induce a moderate activation of c–Abl in the nucleus (Kharbanda et al., 1998). Nuclear partners of Abl include proteins involved in the regulation of growth, apoptosis and DNA repair, such as the ataxia telangiectasia gene product (ATM), the RB and p53 tumour suppressor gene products, as well as p73, the p53-related protein (Kharbanda et al., 1998; Van Etten, 1999). The available evidence points to nuclear c–Abl as an important contributor to cellular fate decisions, in particular in cellular responses to DNA damage (Kharbanda et al., 1998; Van Etten, 1999). The nuclear circuitry in which Abl operates is still largely uncharted.
c–Jun, a component of the AP-1 family of leucine zipper transcription factors, acts as a convergence point of many signalling pathways (Karin et al., 1997; Ip and Davis, 1998). Its activity is regulated in a cell type-dependent manner by a variety of signals that are relayed through transcriptional and post-transcriptional mechanisms. Serine/threonine phosphorylation, and changes in the redox state and in protein–protein interactions have all been implicated in the latter mode of c–Jun regulation (Treisman, 1996; Karin et al., 1997; Ip and Davis, 1998). In spite of this wealth of knowledge regarding the regulatory mechanisms impinging on c–Jun, not all the biological functions of the protein, e.g. its role in transformation and apoptosis, are accounted for satisfactorily.
c–Jun is phosphorylated on Ser63, Ser73, Thr91 and/or Thr93 within the N–terminal portion of the molecule by MAP kinases of the ERK and JNK family (Treisman, 1996; Karin et al., 1997; Ip and Davis, 1998). Phosphorylation at these sites induces an increase in the DNA-binding and transactivation potential of the protein, as well as an increase in the stability of c–Jun. JNKs (Jun N–terminal kinases, also known as SAPKs, stress-activated protein kinases) are activated by a variety of stimuli, including various forms of stress and exposure to inflammatory cytokines (Kyriakis and Avruch, 1996; Treisman, 1996; Ip and Davis, 1998). Constitutively active forms of Abl cause an increase in JNK activity (Raitano et al., 1995; Renshaw et al., 1996) and Abl has been shown to be necessary to activate JNK following genotoxic stress (Kharbanda et al., 1998). Identification of nuclear substrates of Abl should help to understand the role of Abl in the nucleus and identify the phosphorylation networks to which it contributes.
Results
The substrate specificity of the Abl catalytic domain in vitro has been determined using random peptide libraries (Songyang et al., 1995). A database search of putative Abl substrates using a modification of the consensus sequence of Songyang et al. (1995) identified Tyr170 in the transcription factor c–Jun as a potential phosphorylation site (Figure 1). The substrate specificity of non-receptor tyrosine kinases overlaps the binding specificity of the SH2 domain of these enzymes (Songyang et al., 1995) for reasons that are not fully understood yet but may involve cooperative effects (Mayer et al., 1995). A potential binding site for the Abl SH2 domain had indeed already been identified in the c–Jun-related protein JunB (Songyang et al., 1993).

Fig. 1. Cartoon of the human c–Jun primary structure and domain arrangement. The sequence surrounding Tyr170 (residues 166–174) is shown in an enlargement. An asterisk marks the tyrosine that is a site of putative phosphorylation. A vertical arrow indicates the alanine residue following the tyrosine that is typical of good Abl substrates. These are human c-Crk, Tyr221 (Feller et al., 1994); human p73, Tyr99 (Yuan et al., 1999); human Rad51, Tyr54 (Yuan et al., 1998); and optimal peptide (Songyang et al., 1995). The following regions are highlighted: the delta domain (δ), the DNA-binding domain (DB) and the leucine zipper dimerization domain (LZ). The location of the NLS and of the N–terminal phosphorylation sites (JNK sites) is shown. N, N–terminus; C, C–terminus.
We decided to test whether Jun family members were substrates of the Abl tyrosine kinase. A double proline mutation in the regulatory ‘linker’ region between the SH2 and catalytic domain (P242E/P249E) confers constitutive activity to Abl, as in the Src family kinases Src and Lck (Gonfloni et al., 1997; Barilá and Superti-Furga, 1998). We combined the double proline mutation in the linker with mutation of a single leucine in the nuclear export signal (NES) at the C–terminal end of c–Abl (L1116A) (Taagepera et al., 1998). The resulting form of Abl, Abl-PP-Nuc, was constitutively active (see below) and predominantly nuclear in transfected human embryo kidney (HEK) 293 cells (Figure 2). A large fraction of transfected cells rounded up and died soon after transfection. In these cells, Abl was mainly cytoplasmic and perinuclear (data not shown). Abl-PP-Nuc was transiently transfected into HEK-293 cells along with hemagglutinin (HA) epitope-tagged human c–Jun. Abl-PP-Nuc, but not its kinase-inactive counterpart (bearing the additional K290R mutation), caused strong tyrosine phosphorylation of co-transfected c–Jun, as shown in immunoprecipitation and immunoblotting experiments (Figure 3A). v–Jun as well as the other Jun family members, JunB and JunD, were not phosphorylated on tyrosine in the same assay (data not shown).

Fig. 2. Subcellular localization of various Abl forms by immuno- fluorescence. HEK-293 cells were plated onto glass coverslips 16 h before transfection with several forms of Abl (5 μg) as shown in each panel. Cells were fixed 48 h after transfection. The Abl proteins were stained with mouse monoclonal anti–Abl antibody.

Fig. 3. Abl phosphorylates and interacts with Jun. (A) Tyrosine phosphorylation of Jun. HEK-293 cells were transiently transfected with CMV-HA-c–Jun (5 μg) and CMV–Abl-PP-Nuc or CMV–Abl-PP-Nuc-Kin– (2 μg) as a control. A 500 μg aliquot of total protein extract was immunoprecipitated with anti–Jun antibodies. Immunoblots were probed with anti-phosphotyrosine antibodies (upper panel) or antibodies against HA (middle panel). Abl protein levels are shown in the lower panel. (B) GST binding. HEK-293 cells were transiently transfected with CMV–Abl-PP-Nuc (2 μg) and CMV-HA-c–Jun (5 μg). A 500 μg aliquot of total protein extract was incubated in the presence of the various GST fusions pre-bound to glutathione beads. Pull-downs were analysed by immunoblotting. c–Jun was revealed using antibodies against the HA tag. (C) Co-immunoprecipitation of Abl and Jun from transiently transfected HEK-293 cells. Cells were transfected with CMV-HA-c–Jun (5 μg) and CMV–Abl-PP-Nuc (2 μg), or its catalytically inactive version, CMV–Abl-PP-Nuc-Kin–, as a control. A 1 mg aliquot of total protein extract was immunoprecipitated with antibodies against Abl (left panels) or with the anti–Jun antibody (right panels). The membranes were probed with anti–Abl antibodies (upper panels), anti-HA antibodies (middle panels) and anti-phosphotyrosine antibodies (lower panels). Migration of the Abl and Jun proteins is indicated by the arrows.
As mentioned above, there is an overlap between the substrate specificity and the SH2 domain-binding preference of Abl. Thus, we examined the possibility that the SH2 domain of Abl could bind tyrosine-phosphorylated c–Jun. An Abl glutathione S-transferase (GST)–SH2 domain fusion interacted strongly with c–Jun from total lysates derived from Abl- and Jun-transfected HEK-293 cells in a pull-down assay (Figure 3B). GST alone did not interact, while the N–terminal SH2 domain of the p85 phosphatidylinositol 3-kinase (PI3 kinase) regulatory subunit interacted weakly, indicating that tyrosinephosphorylated c–Jun bound preferentially to the Abl SH2 domain.
We asked whether the full-length Abl protein could also interact with c–Jun. We immunoprecipitated Abl from cell lysates obtained from HEK-293 cells that had been transfected with kinase-competent or kinase-inactive forms of Abl together with c–Jun. When Abl immunoprecipitates were probed with anti-phosphotyrosine antibodies, a reactive band of the size of c–Jun was observed only in extracts in which c–Jun was co-transfected. The band was not seen in lysates from cells transfected with kinase-inactive Abl (Figure 3C, left lower panel). However, c–Jun was not detected using the HA antibody on the anti–Abl precipitates (Figure 3C, left central panel). This suggests that immunoprecipitation of Abl enriched for tyrosine-phosphorylated c–Jun that was detected by the sensitive anti-phosphotyrosine antibodies. In the reciprocal experiment, kinase-active, but not inactive Abl was present in anti–Jun immunoprecipitate (Figure 3C, right top panel). Together, these data demonstrate that c–Jun and a constitutively active nuclear form of Abl interact in a manner that depends on tyrosine phosphorylation of Jun.
To test whether Abl could also phosphorylate endogenous c–Jun, we transiently transfected NIH-3T3 cells. Several tyrosine-phosphorylated proteins appear in nuclear extracts from cells transfected with Abl-PP-Nuc (Figure 4A). Immunoprecipitation using anti–Jun antibodies allowed the detection of a tyrosine-phosphorylated band that migrated slightly more slowly than the bulk of immunoprecipitated c–Jun (Figure 4B). The different electrophoretic mobility may result from phosphorylation at the N–terminal Ser/Thr sites. Our data showed that endogenous c–Jun can be phosphorylated on tyrosine in the presence of active nuclear Abl.

Fig. 4. Phosphorylation of endogenous Jun. (A) Abl can induce phosphorylation of several proteins in the nucleus. NIH-3T3 cells were transfected with CMV–Abl-Nuc or CMV–Abl-PP-Nuc (5 μg). A 50 μg aliquot of nuclear protein extracts was separated, blotted and probed with anti-phosphotyrosine antibodies. (B) Abl can induce phosphorylation of endogenous c–Jun. NIH-3T3 cells were transfected with CMV–Abl-Nuc or CMV–Abl-PP-Nuc (5 μg). A 500 μg aliquot of nuclear protein extract was immunoprecipitated with anti-c–Jun antibodies. Blots of immunoprecipitates were probed with antibodies against Jun (left panel) or anti-phosphotyrosine (right panel).
We then investigated whether Tyr170 in c–Jun could be the site of phosphorylation by Abl as speculated based on the sequence comparison discussed above. Human c–Jun contains five tyrosines. These were mutated individually to phenylalanine and tested in transient co-transfection assays with Abl-PP, an active form of Abl that is expressed in both the nucleus and the cytoplasm. Mutation of Tyr170 severely impaired tyrosine phosphorylation of Jun (Figures 1 and 5A). In contrast, mutation of any of the other tyrosines did not affect tyrosine phosphorylation significantly. This confirms that Tyr170 is an Abl phosphorylation site. Tyr170 lies in the centre of the molecule, at the C–terminal end of the A1 transcriptional activation region (Angel et al., 1989; Baichwal and Tjian, 1990) (Figure 1). To obtain evidence for Tyr170 phosphorylation independent of the use of anti-phosphotyrosine antibodies, HEK-293 cells were transfected with c–Jun or Jun-Y170F along with Abl-PP and labelled with [32P]orthophosphate. After anti–Jun immunoprecipitation, the c–Jun band was excised from the gel and its phosphoamino acid content analysed (Figure 5B). Under these conditions, c–Jun incorporated a significant proportion of the label into phosphotyrosine, while Jun-Y170F did not, confirming that Tyr170 is the major site of tyrosine phosphorylation by Abl. Consistent with this, a GST fusion protein containing residues 102–256 was a very good substrate of purified recombinant Abl in vitro, with an apparent Km of <0.2 μM, whereas a GST fusion of Jun residues 1–101 was not phosphorylated by Abl (data not shown). Because Abl induced c–Jun phosphorylation efficiently in vivo and in vitro, and because it associated with c–Jun, we believe that Abl phosphorylates c–Jun directly in the cell.
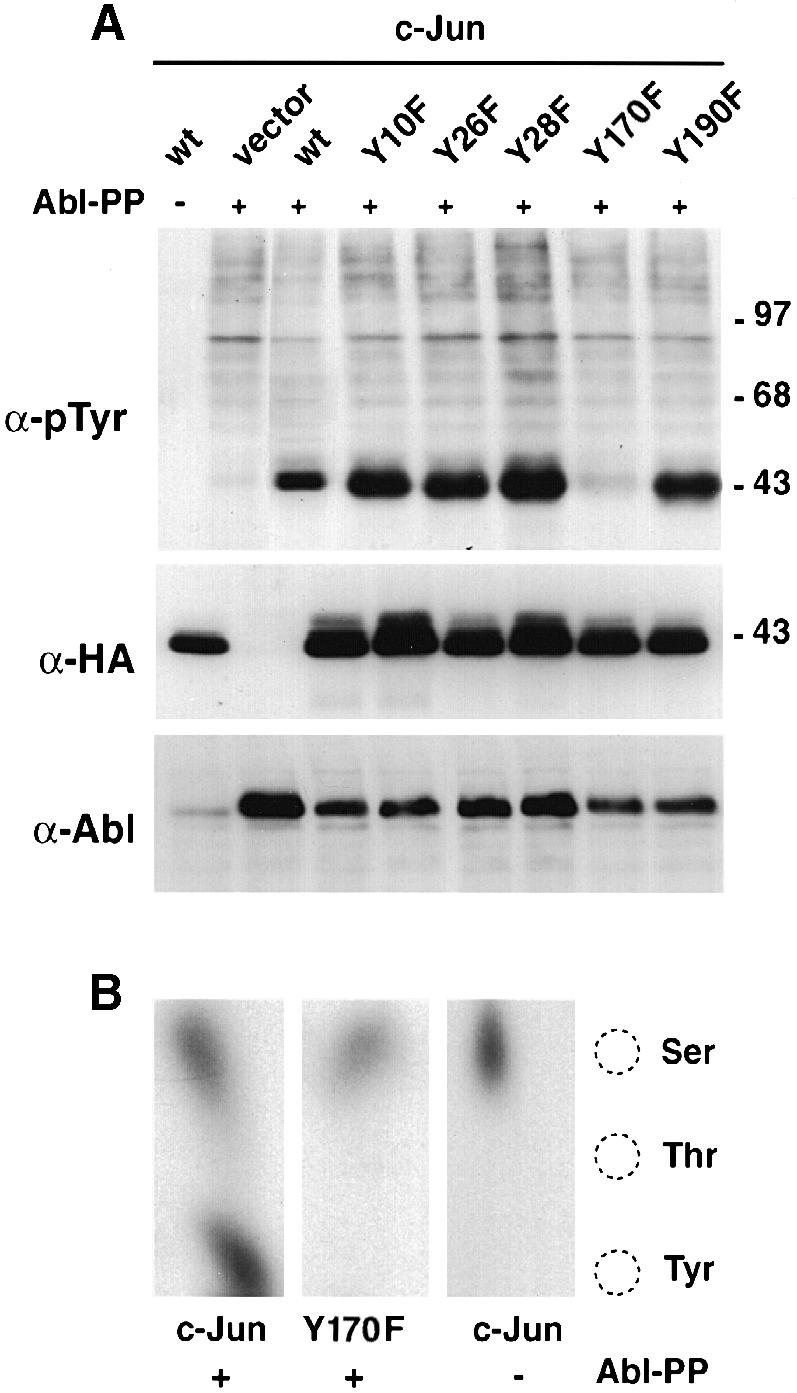
Fig. 5. Mapping of the tyrosine phosphorylation site in c–Jun. (A) HEK-293 cells were transfected with c–Jun tyrosine mutants (CMV-HA constructs, 5 μg) and pSGT–Abl-PP (5 μg). Total protein extracts were separated, blotted and probed with anti-phosphotyrosine (top panel), anti-HA (middle panel) and anti–Abl antibodies (lower panel). (B) In vivo labelling. CMV-HA-c–Jun or CMV-HA–Jun-Y170F (5 μg) were transfected in HEK-293 cells along with pSGT–Abl-PP (5 μg) in the presence of [32P]orthophosphate. Total protein extracts were immunoprecipitated with anti–Jun antibodies, separated and blotted. The band corresponding to Jun was revealed by auto- radiography, excised and subjected to phosphoamino acid analysis using thin-layer chromatography. Migration of the phosphoaminoacid markers is shown on the right.
The linker proline mutant forms of Abl that have been used to study c–Jun phosphorylation thus far were constitutively active. We wanted to test the ability of the regulated and localized forms of Abl to phosphorylate c–Jun (Taagepera et al., 1998). c–Jun requires a nuclear localization signal (NLS) in order to accumulate in the nucleus (Chida and Vogt, 1992). A C–terminal truncation of c–Jun (Jun1–223) that lacks the DNA-binding and nuclear localization domain as well as the leucine zipper was used (Treier et al., 1994). As a control, we used a C–terminal fusion of the NLS of the SV40 large T nuclear antigen to the same truncated Jun. Wild-type c–Jun and the truncations were expressed at similar levels in transfected HEK-293 cells, as were the different Abl forms (Figure 6A, left panels). Abl-PP, deregulated and localizing in both the nucleus and the cytoplasm, caused tyrosine phosphorylation of all three forms of Jun, as seen in an anti-phosphotyrosine immunoblot (Figure 6A, right panels). In contrast, cytoplasmic and regulated Abl (Abl-Cyt) did not. When using the nuclear and regulated form of Abl (Abl-Nuc, see Figure 2), only phosphorylation of the Jun forms containing an NLS was observed (Figure 6A, lower right panel). These results indicated that Jun can be a target of regulated Abl only in the nucleus.

Fig. 6. Jun is a substrate and an activator of Abl in the nucleus. (A) HEK-293 cells were transiently co-transfected with various Abl and Jun forms (5 μg each). Abl forms were: Abl-PP, bearing the activating double proline mutation; Abl-Cyt, mutated in the NLS; and Abl-Nuc, mutated in the NES. Jun forms were: wild-type c–Jun; Jun1–223, a C–terminal truncation lacking the DNA-binding, NLS and leucine zipper domains; and Jun1–223-NLS, identical to the previous form but bearing an added NLS from the SV40 large T antigen. A 50 μg aliquot of total protein extract was processed for immunoblotting. Samples were separated by 7.5% (Abl) and 10% (Jun) SDS–PAGE. Left panels: protein levels as indicated. Right panels: phosphotyrosine levels. Migration of the Abl and Jun proteins is indicated by the arrows. (B) HEK-293 cells were transiently transfected with Abl-Nuc (5 μg) and increasing amounts of c–Jun (0.25, 0.5, 1, 2 and 4 μg). A 50 μg aliquot of total protein extract was processed for immunoblotting with anti-HA antibodies (bottom panel). Abl activation was measured by anti-phosphotyrosine immunoblotting after immunoprecipitation (top panel). Levels of immunoprecipitated Abl are shown below (middle panel).
Testing the state of phosphorylation of Abl, we found, very unexpectedly, that the nuclear forms of Jun caused considerable tyrosine phosphorylation of nuclear Abl, indicative of effective activation of Abl (Figure 6A, top right panel, see the arrow). In contrast, Abl was not tyrosine phosphorylated when transfected along with the form of Jun lacking the NLS. This suggested that nuclear Jun could be both a target and an activator of nuclear Abl.
Possibly, an increase of c–Jun protein levels in the nucleus could lead to activation of Abl. To test this hypothesis, we transfected Abl-Nuc along with increasing amounts of c–Jun. As shown in Figure 6B, c–Jun led to a dose-dependent increase in tyrosine phosphorylation of Abl.
We next examined the structural requirements of c–Jun for Abl activation. Mutation of Ser73 (Jun-S73A) or of all N–terminal phosphorylation sites to alanine (Jun-Pan-Ala) strongly increased the ability of c–Jun to activate co-transfected nuclear Abl in HEK-293 cells (Figure 7A, top panel). At the same time, these Jun mutants were much better Abl substrates than wild-type c–Jun (Figure 7A, third panel from the top). While the reason for this is unclear, it is possible that mutation of the N–terminal sites to alanine impairs JNK or CBP/p300 binding to c–Jun (Bannister et al., 1995), which can be imagined to interfere with Abl binding. Alternatively, it could confer a conformational change on c–Jun (Papavassiliou et al., 1995), increasing its affinity for Abl. Mutation of Tyr170 impaired, but did not abolish, Abl activation both alone and in the context of the N–terminal JNK phosphorylation site mutations (Figure 7A, top panel). c–Jun lacking the δ region was not tyrosine phosphorylated efficiently but was capable of activating Abl (Figure 7A, first and third panels from the top). Together, these results suggested that the DNA-binding and leucine zipper region as well as the δ domain were not critical for Abl activation, while Tyr170 was important. All Jun forms that were good substrates of nuclear Abl were also efficient activators. This suggests that initial Abl activation may occur through interactions that are independent of Tyr170 and that phosphorylation of Tyr170 may be required for the subsequent more robust (and possibly processive) activation, once an initial Abl activity is triggered.

Fig. 7. Structural requirements for Abl activation. (A) HEK-293 cells were transiently transfected with CMV–Abl-Nuc (5 μg) and different Jun mutants (5 μg each). Abl activation was measured by anti-phosphotyrosine immunoblotting after immunoprecipitation (top panel). Levels of immunoprecipitated Abl are shown below. Levels and the tyrosine phosphorylation of the different Jun proteins were detected using anti-HA antibodies or anti-phosphotyrosine antibodies, respectively (lower two panels). (B) Ability of several Abl mutants to phosphorylate Jun. HEK-293 cells were transiently transfected with various Abl forms (5 μg) as indicated, with our without CMV-HA-c–Jun-S73A (5 μg). Lysates were analysed by immunoblotting using specific antibodies as indicated. (C) JNK negatively affects Jun phosphorylation by Abl. HEK-293 cells were transfected with CMV–Abl-Nuc (2 μg) and CMV-HA-c–Jun or CMV-HA-c–JunS73A (2 μg) in the presence or absence of SV40-HA-JNK (6 μg). Total protein was immunoblotted and probed with anti-phosphotyrosine antibodies (top panel), anti-HA antibodies (HA–Jun and HA-JNK, two central panels) or by anti–Abl antibodies (bottom panel). (D) Expression of the Jun δ region enhances the ability of Jun to be phosphorylated by Abl. HEK-293 cells were transfected with CMV–Abl-Nuc (2 μg) and CMV-HA-c–Jun (2 μg) in the presence of pEBG or pEBG-δ–Jun (6 μg). Phosphorylation of c–Jun protein was measured by anti-phosphotyrosine antibodies (top panel) and protein levels were detected by specific antibodies (lower panels).
To test the structural requirements for c–Jun phosphorylation and Abl activation within Abl, we co-expressed different Abl forms together with Jun-S73A, the Jun form that activated Abl-Nuc most efficiently (Figure 7B). Co-expression of Jun-S73A with wild-type c–Abl, which localizes in both the nucleus and the cytoplasm in transfected HEK-293 cells, led to tyrosine phosphorylation of Jun as seen in total extracts (Figure 7B, top panel), but only barely detectable phosphorylation of Abl after immunoprecipitation (Figure 7B, third panel), suggesting that Jun-S73A phosphorylation was a more sensitive measurement of Abl activity. As expected, nuclear Abl was activated very efficiently by Jun-S73A (Figure 7B, top and third panels). A nuclear form of Abl bearing a point mutation in the FLVRES motif of the SH2 domain (Abl-Nuc-SH2–), on the other hand, was not efficiently activated by Jun (Figure 7B, top and third panels), suggesting that interaction of the SH2 domain with phosphorylated Jun is required for efficient activation of Abl.
Efficient activation of Abl by c–Jun mutated at the N–terminal sites prompted us to test the potential role of JNK in the Abl and c–Jun relationship. JNK is known to bind the c–Jun δ domain in unstimulated cells (Hibi et al., 1993). We expressed JNK along with c–Jun and Abl in HEK-293 cells and monitored tyrosine phosphorylation of c–Jun. Ectopic expression of JNK caused a decrease in activation of nuclear Abl and c–Jun phosphorylation (Figure 7C). JNK expression also reduced tyrosine phosphorylation of Jun mutated at the N–terminal phosphorylation sites, suggesting that it was JNK binding, rather than phosphorylation, that negatively affected the ability of Jun to activate Abl and become a substrate (Figure 7C). This result raised the possibility that occupancy of the c–Jun δ domain by JNK was interfering with the c–Jun–Abl interaction. Expression of a protein fragment spanning the c–Jun δ region increased the ability of c–Jun to activate Abl and caused enhanced c–Jun phosphorylation on tyrosine (Figure 7D). In summary, these results are compatible with a negative role for JNK in the ability of c–Jun to activate Abl.
Expression of active forms of Abl has been shown to lead to JNK activation (Raitano et al., 1995; Renshaw et al., 1996). To test whether Abl activation by c–Jun resulted in an increase in JNK activity, we performed JNK kinase assays on GST–Jun pull-downs using extracts derived from HEK-293 cells transfected with nuclear Abl with or without Jun. Indeed, activation of nuclear Abl by c–Jun resulted in an increase in endogenous JNK activity over the basal levels (Figure 8A). JNK was present in all lysates at similar levels (data not shown). JNK activity induced by Abl and c–Jun expression was similar to the activity of cells transfected with v–Abl (Figure 8A). The increase in JNK activity correlated with an increase in c–Jun phosphorylation at the N–terminal sites as measured with phosphospecific antibodies directed against Ser73 of c–Jun (data not shown).

Fig. 8. Activation of JNK by Abl and Jun. (A) JNK kinase assay on GST–Jun pull-downs from HEK-293 cells transfected with nuclear Abl (CMV–Abl-Nuc, 5 μg) in the presence of CMV-HA-c–Jun or control vector (5 μg). Positive controls were extracts from cells transfected with pSGT-v–Abl (5 μg) or cells treated with 0.5 μg/ml anisomycin for 40 min. Total proteins were incubated with ∼10 μg of GST–Jun1–101 pre-bound to glutathione beads. After the kinase reaction, phosphorylation was analysed by SDS–PAGE and revealed by autoradiography. To calculate JNK activation, bands were excised and incorporated radioactivity measured by scintillation counting. The values represent the average from at least two independent experiments. (B) JNK kinase assay on GST–Jun pull-downs from HEK-293 cells transfected with CMV-HA-c–Jun or with CMV-HA-c–JunY170F (10 μg) was performed as described in (A). (C) Jun+/+ and Jun–/– mouse fibroblasts (Schreiber et al., 1999) were kept in normal serum and treated with 34 nM TPA for 14 h or with 0.5 μg/ml anisomycin for 40 min. Jun levels in nuclear extracts were detected by immunoblotting, and the JNK kinase assay was performed as described above. (D) Abl/Arg+/+ and Abl/Arg–/– mouse embryo (Koleske et al., 1998) fibroblasts were kept in normal serum treated with increasing amounts of TPA for 14 h. Detection of Jun levels and JNK kinase assay were performed as described above.
We went on to test whether high c–Jun levels obtained by transfection of HEK-293 cells could also lead to activation of JNK in the absence of co-transfected Abl. We found that, indeed, Jun transfection led to a modest but consistent stimulation of JNK activity (Figure 8B). Jun mutated at Tyr170 showed a reduced stimulation compared with wild-type Jun, suggesting the involvement of endogenous Abl in the process.
We wanted to investigate whether an increase in endogenous c–Jun levels could also induce JNK activity in an Abl-dependent manner. To this end, we used fibroblasts established from mice in which either the c-jun gene (Hilberg et al., 1993; Schreiber et al., 1999) or the genes encoding both Abl kinase family members Abl and Arg had been inactivated (Koleske et al., 1998), or cells derived from wild-type sibling animals, and treated them with the phorbol ester TPA, a known inducer of c–Jun (Brenner et al., 1989). The increase in endogenous c–Jun protein levels correlated with a 6-fold induction of JNK activity in Jun+/+ cells. TPA treatment of Jun–/– fibroblasts failed to activate JNK, demonstrating a close relationship between JNK activation following TPA treatment and c–Jun accumulation (Figure 8C). This was not due to a general defect in the ability of these cells to stimulate JNK because the response to anisomycin was normal. Abl/Arg–/– and Abl/Arg+/+ cells showed a lower accumulation of Jun in response to TPA. This resulted in a 2-fold activation of JNK in Abl/Arg+/+ but not in Abl/Arg–/– cells (Figure 8D). This points to a role for Abl family kinases in JNK activation by elevated c–Jun levels.
Discussion
Tyrosine phosphorylation is commonly thought to be associated with cytoplasmic processes. Very little is known about nuclear tyrosine phosphorylation circuits. The c–Jun transcription factor is a central transcriptional mediator of cellular signalling in all metazoans (Karin et al., 1997). Using various forms of c–Abl and c–Jun in transient cell transfections, we have detected activation of the c–Abl tyrosine kinase by c–Jun- and Abl-dependent phosphorylation of c–Jun. No effect on the transcriptional potential of c–Jun was found to depend on tyrosine phosphorylation. Abl activation is accompanied by an increase in the activity of the JNK serine/threonine kinase and an increase in Jun phosphorylation at the N–terminal sites. Elevation of the c–Jun levels, either by transfection or by phorbol ester treatment, leads to a significant increase in the activity of JNK. c–Jun and either or both of the two kinases of the Abl family are required for JNK activation.
Our data can be summarized in a model, according to which elevated c–Jun levels can activate Abl in the nucleus (Figure 9). Activation requires the central part of the c–Jun protein and the presence of Tyr170 in order to be efficient. In turn, consistent with previous reports, activation of nuclear Abl results in an increase in JNK activity. This may occur through a mechanism that involves a positive effect on a JNK activator, a negative effect on a JNK repressor, or could even be direct. Once activated, JNK may then phosphorylate c–Jun at the N–terminal sites and increase c–Jun stability, DNA binding and transcriptional activity (Treisman, 1996; Karin et al., 1997; Ip and Davis, 1998). If JNK then dissociates from Jun after phosphorylation, as proposed (Hibi et al., 1993), this would allow more JNK-free Jun to activate Abl and result in a positive feedback loop.

Fig. 9. Diagram illustrating the possible functional relationship between Jun, Abl and JNK. Elevated Jun levels cause an increase in the activity of nuclear Abl, possibly by a direct mechanism (solid forward arrow). Abl phosphorylates Jun on tyrosine (solid backward arrow), increasing the ability of Jun to activate Abl further (positive feedback loop). Abl activity results in an increase in JNK activity, most likely by an indirect mechanism (dashed arrow). Active JNK in turn phosphorylates Jun (solid arrow). Elevated JNK levels negatively affect Jun phosphorylation by Abl (blunt arrow).
The mechanism by which c–Jun could achieve activation of nuclear Abl is not clear. One possibility is that c–Jun activates Abl allosterically, by direct binding to Abl. Our data show that nuclear Abl is regulated by intramolecular interactions involving the SH2–catalytic domain linker, as we have shown previously for the bulk cellular Abl protein and for Abl synthesized in reticulocyte lysate (Barilá and Superti-Furga, 1998). We assume that c–Jun binding to Abl may interfere with the regulatory intramolecular interactions. Initially, tyrosine phosphorylation of c–Jun may not be required for this activation and could occur, for example, via interaction of the proline-rich sequences in c–Jun with the Abl SH3 domain. Alternatively, basal phosphorylation of c–Jun by Abl may suffice to initiate the positive feedback loop. Although Abl and/or Arg seem to be required for c–Jun activation of JNK, we cannot exclude the possibility that c–Jun, or other AP-1 family members, can activate JNK by a mechanism that does not involve Abl in other cell types or under different physiological circumstances. It will be interesting to test whether some of the severe defects in mice lacking c–Jun can be attributed to defects in activating JNK or related kinases (Hilberg et al., 1993; Johnson et al., 1993).
We propose that activation of nuclear Abl and JNK via c–Jun may occur when newly synthesized c–Jun protein accumulates in the absence of a specific modification or in excess to specific binding partners, be they JNK or AP-1 family members. Because of the necessity to synthesize and accumulate Jun, JNK activation by this mechanism may follow slow kinetics and be sustained. Moreover, it may result in specific activation of only the nuclear populations of Abl and JNK. Interestingly, TPA led to an increase in c–Jun levels and JNK activation without prior serum starvation of the cells. It will be interesting to test whether simultaneous stimulation of different pathways interferes with the process. Genotoxic stress causes activation of the c-jun gene and accumulation of newly synthesized c–Jun protein (Kharbanda et al., 1991; Rubin et al., 1992). Genotoxic stress also leads to activation of nuclear JNK, and it may occur independently of cytoplasmic activators and without causing nuclear translocation of cytoplasmic JNK (Lee et al., 1998; Sanchez-Perez et al., 1998). On the other hand, several reports have placed JNK downstream of Abl. JNK is activated by oncogenic forms of Abl (Raitano et al., 1995; Renshaw et al., 1996). Moreover, Abl is required for JNK activation by genotoxic stresses in cycling cells (Kharbanda et al., 1995a,b), but probably not in confluent, growth-arrested cells (Liu et al., 1996). Interestingly, cell density was found to be inhibitory to JNK activity (Lallemand et al., 1998). Abl, JNK and c–Jun have been implicated in apoptosis (Karin et al., 1997; Ip and Davis, 1998; Kharbanda et al., 1998). In particular, it has been proposed that de novo synthesis of c–Jun may be required to allow apoptosis to occur in certain cell types (Karin et al., 1997). JNK activation, in the absence of ERK activation, may be an apoptotic clue (Xia et al., 1995). We believe that c–Jun activation of Abl and JNK may form part of a phosphorylation circuit that integrates growth and stress-related stimuli for cellular fate decisions.
Our data propose c–Jun as a nuclear tyrosine-phosphorylated protein and Abl substrate. To our knowledge, only the p53-related p73 protein may represent another case of a transcription factor becoming tyrosine phosphorylated in the nucleus, while the STAT family of transcription factors is tyrosine phosphorylated in the cytoplasm before entering the nucleus (Karin and Hunter, 1995; Agami et al., 1999; Gong et al., 1999; Yuan et al., 1999). The nucleus may contain conspicuous tyrosine phosphorylation, especially in oncogene-transformed cells (Bell et al., 1987). The discovery of the circuitry presented here suggests the existence of a larger nuclear tyrosine phosphorylation network.
Materials and methods
Computer searches
Computer searches were carried out using the program FINDPATTERNS of the Wisconsin Package version 10.0 [Genetics Computer Group (GCG), Madison, WI] and the human entries of Swissprot as database. A search using the profile (V,I,L)YA, the basic consensus identified by Songyang et al. (1995), yielded ∼800 matches. Of these matches, <100 were of nuclear proteins, among which were several members of the AP1 family. A more stringent search including the phosphorylation sites of Crk and Rad51, (E,A)XX(I,V,L,P,A)YAXX(F,S,K), identified 27 entries, of which seven were of nuclear proteins, including c–Jun and JunD.
DNA constructs
Murine Abl-wt, Abl mutated in the NES and Abl mutated in the NLS cloned into the expression vector MSCV were obtained from J.Y.Wang (Taagepera et al., 1998) and used to generate pCMV–Abl-wt, pCMV–Abl-Nuc and pCMV–Abl-Cyt, respectively, by NotI restriction and subcloning into pcDNA3. Human pSGT–Abl-PP (P242E,P249E) and pSGT-v–Abl have been described previously (Barilá and Superti-Furga, 1998). Abl-PP-Nuc, Abl-Nuc-SH2–, Abl-Nuc-Kin– and Abl-PP-Nuc-Kin– were produced by combining into the pcDNA3 vector the EcoRI–BclI fragment obtained from pSGT–Abl-PP, pGEM3–Abl-SH2-R171L, pSGT–Abl-Kin– (K290R) and pSGT–Abl-PP-Kin–, respectively, with the BclI–NotI fragment obtained from the murine Abl mutated in the NES. Plasmids for mammalian cell expression of cytomegalovirus (CMV)-driven HA-tagged c–Jun-wt, c–Jun-Pan-Ala, c–Jun-Δδ, c–Jun1–223 and c–Jun1–223 SV40-NLS have been described previously (Treier et al., 1994). Tyrosine mutants of c–Jun (Y10F, Y26F, Y28F, Y170F and Y190F) were obtained by a two-step PCR procedure using human c–Jun in pBluescript as a template and cloning the PCR products into the CMV-driven HA-tagged expression vector using BamHI–HindIII. c–JunS73A was generated in the human CMV-HA-c–Jun vector by PCR, using the QuickChange site-directed mutagenesis system (Stratagene). All mutants were verified by sequencing. The expression vector for HA-tagged JNK was kindly provided by S.Gutkind (Coso et al., 1995). The pEBG vector was a kind gift of B.J.Mayer (Tanaka et al., 1995). pEBG-δ–Jun represents the fusion of human Jun residues P29–D59 to GST, and was a kind gift of E.Chierici. GST–c–Jun1–101 was produced by PCR using human c–Jun and was cloned in pGEX2T (Pharmacia Biotech).
Cell culture and transfection
HEK-293 cells and Jun+/+ and Jun–/– fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). NIH-3T3 cells were cultured in DMEM supplemented with 10% FCS, while Abl/Arg+/+ and Abl/Arg–/– fibroblasts were cultured in DMEM supplemented with 15% FCS. HEK-293 cells were transfected with the calcium phosphate method, while all other cells were transfected using Lipofectamine (Gibco-BRL). In general, 70% confluent cells in 10 cm dishes were transfected with the different expression constructs or empty vectors for a total of 10 μg of DNA. At 40 h post-transfection, total cell lysates were obtained using lysis buffer [50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP-40, 5 mM EGTA, 5 mM EDTA, 20 mM NaF, 0.1 mM orthovanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml TPCK, 5 μg/ml TLCK, 1 μg/ml leupeptin, 10 μg/ml soybean trypsin inhibitor, 1 μg/ml aprotinin]. To obtain nuclear proteins, cell extracts were harvested in cold phosphate-buffered saline (PBS) and lysed in hypotonic buffer (10 mM HEPES pH 7.9, 1 mM MgCl2, 10 mM KCl, 0.5% NP-40, containing all the protease and phosphatase inhibitors as above) for 5 min on ice. Nuclei were collected by centrifugation at 14 000 r.p.m. for 30 s. Nuclear proteins were extracted by incubating the pellet in nuclei buffer (20 mM HEPES pH 7.9, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, containing all the protease and phosphatase inhibitors as above) for 1 h at 4°C on a rotating wheel.
Immunofluorescence microscopy
HEK-293 cells were plated on glass coverslips 16 h before transfection by calcium phosphate precipitation. Cells were fixed 48 h after transfection for 10 min in PBS containing 4% paraformaldehyde. The Abl proteins were stained with mouse monoclonal anti–Abl antibody 24-21 (Abi-3; Oncogene Sciences) diluted 1:50 in PBS, 0.1% Triton X-100, 10% goat serum and subsequent labelling with secondary Texas red anti-mouse antibody (Jackson Laboratories) diluted 1:50 in PBS, 0.1% Triton X-100, 10% goat serum. Images were collected using a Cohu CCD 4910 camera with NIH image on a Zeiss Axiophot microscope equipped with a Zeiss 63× objective.
Immunoblotting, immunoprecipitation and GST pull-downs
For immunoblotting, 50 μg of total protein extract were separated by SDS–PAGE, blotted onto nitrocellulose and probed with specific antibodies. The following antibodies have been used: mouse monoclonal anti–Abl antibody 24-21 [1:500 in 3% bovine serum albumin (BSA), Abi-3; Oncogene Sciences]; mouse monoclonal anti-HA antibody 12CA5 (1:300 in 1% milk; Boehringer Mannheim); mouse monoclonal anti-phosphotyrosine 4G10 (1:1500 in 3% BSA; UBI); and rabbit polyclonal anti-GST antibody (1:2000 in 1% milk, sc-459; Santa Cruz). Endogenous Jun was detected using a rabbit polyclonal anti–Jun antibody (1:3000 in 1% milk, kindly provided by M.Nicklin). In all cases, horseradish peroxidase (HRP)-coupled secondary antibodies and the ECL chemiluminescence method were used (Amersham Pharmacia). Abl and Jun proteins were immunoprecipitated from 500 μg of protein extracts in lysis buffer using 10 μl of mouse monoclonal anti–Abl antibody 24-21 or 5 μl of a rabbit polyclonal antibody anti-c-jun (sc-45; Santa Cruz), respectively. Immunocomplexes were recovered using protein G–Sepharose or protein A–Sepharose beads (Amersham Pharmacia) according to the first antibody preference. For GST pull-down experiments, 10–20 μg of bacterially produced GST fusion proteins were pre-bound to glutathione–Sepharose beads and incubated with 500 μg of total protein extracts. After 1 h rotating at 4°C, the beads were processed for SDS–PAGE and immunoblotting.
In vivo labelling and phosphoamino acid analysis
HEK-293 cells were transfected as above. After 40 h, cells were washed with phosphate-free DMEM for 1 h and then incubated in the same medium containing [32P]orthophosphate (0.6 mCi/ml final concentration, carrier free; ICN) for 2 h. After labelling, the cells were lysed in lysis buffer and Jun protein was immunoprecipitated, as described above. Proteins were resolved by SDS–PAGE, transferred to PVDF membrane (Immobilon; Millipore) and the bands corresponding to Jun were visualized by autoradiography and excised. Phosphoamino acids were analysed by HCl hydrolysis of the protein followed by thin-layer chromatography using standard procedures (Kamps, 1991).
Kinase assays
JNK activity was determined by incubation of 400 μg of total cell extracts with 10 μg of GST–c–Jun1–101, pre-bound to glutathione–Sepharose beads, for 2 h at 4°C. The beads were washed three times with PBS containing 1% NP-40 and 2 mM orthovanadate, once with 100 mM HEPES pH 7.5, 0.5 M LiCl and once with 1× kinase buffer (12.5 mM HEPES pH 7.5, 12.5 mM glycerophosphate, 0.5 mM EGTA, 0.5 mM NaF, 0.5 mM orthovanadate). The kinase reaction was initiated by addition of 30 μl of kinase buffer plus 5 μCi of [γ-32P]ATP (3000 Ci/mmol; Amersham); 13 μM ATP, 3.3 μM dithiothreitol (DTT), and carried out for 30 min at 30°C. The reaction was terminated by addition of 10 μl of SDS-containing sample buffer and boiled for 5 min. The samples were analysed by SDS–PAGE and autoradiography.
Acknowledgments
Acknowledgements
We are very grateful to J.Y.Wang for the generous gift of the Abl NES and NLS mutants, to T.Koleske for the generous gift of Abl/Arg-deficient cells, and to P.Angel and E.Wagner for the generous gift of c–Jun-deficient cells. We also thank E.Chierici for the GST–Jun δ region construct, M.Treier for numerous plasmids, M.Nicklin for the anti–Jun serum, B.J.Mayer for pEBG, P.Angel, A.Nebreda, R.Klein, K.Dorey, H.Pluk, T.Graf, C.Nerlov, F.Ruberti and R.Chenna for suggestions, and M.Way and A.Nebreda for critical reading of the manuscript. D.B. and S.G. were recipients of fellowships from the Antonio Castelnuovo Foundation and the Boncompagni-Ludovisi Foundation, respectively. R.M. was supported by the Italian Foundation for Cancer Research (FIRC).
References
- Agami R., Blandino, G., Oren, M. and Shaul, Y. (1999) Interaction of c–Abl and p73α and their collaboration to induce apoptosis. Nature, 399, 809–813. [DOI] [PubMed] [Google Scholar]
- Angel P., Smeal, T., Meek, J. and Karin, M. (1989) Jun and v-jun contain multiple regions that participate in transcriptional activation in an interdependent manner. New Biol., 1, 35–43. [PubMed] [Google Scholar]
- Baichwal V.R. and Tjian, R. (1990) Control of c–Jun activity by interaction of a cell-specific inhibitor with regulatory domain δ: differences between v- and c–Jun. Cell, 63, 815–825. [DOI] [PubMed] [Google Scholar]
- Bannister A.J., Oehler, T., Wilhelm, D., Angel, P. and Kouzarides, T. (1995) Stimulation of c–Jun activity by CBP: c–Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene, 11, 2509–2514. [PubMed] [Google Scholar]
- Barilá D. and Superti-Furga, G. (1998) An intramolecular SH3-domain interaction regulates c–Abl activity. Nature Genet., 18, 280–282. [DOI] [PubMed] [Google Scholar]
- Bell J.C., Mahadevan, L.C., Colledge, W.H., Frackelton, A.R., Jr, Sargent, M.G., and Foulkes, J.G. (1987) Abelson-transformed fibroblasts contain nuclear phosphotyrosyl-proteins which preferentially bind to murine DNA. Nature, 325, 552–554. [DOI] [PubMed] [Google Scholar]
- Brenner D.A., O'Hara, M., Angel, P., Chojkier, M. and Karin, M. (1989) Prolonged activation of jun and collagenase genes by tumour necrosis factor-α. Nature, 337, 661–663. [DOI] [PubMed] [Google Scholar]
- Chida K. and Vogt, P.K. (1992) Nuclear translocation of viral Jun but not of cellular Jun is cell cycle dependent. Proc. Natl Acad. Sci. USA, 89, 4290–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coso O.A., Chiariello, M., Yu, J.C., Teramoto, H., Crespo, P., Xu, N., Miki, T. and Gutkind, J.S. (1995) The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell, 81, 1137–1146. [DOI] [PubMed] [Google Scholar]
- Feller S.M., Knudsen, B. and Hanafusa, H. (1994) c–Abl kinase regulates the protein binding activity of c-Crk. EMBO J., 13, 2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonfloni S., Williams, J.C., Hattula, K., Weijland, A., Wierenga, R.K. and Superti-Furga, G. (1997) The role of the linker between the SH2 domain and catalytic domain in the regulation and function of Src. EMBO J., 16, 7261–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J.G., Costanzo, A., Yang, H.Q., Melino, G., Kaelin, W.G., Jr, Levrero, M. and Wang, J.Y. (1999) The tyrosine kinase c–Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature, 399, 806–809. [DOI] [PubMed] [Google Scholar]
- Hibi M., Lin, A., Smeal, T., Minden, A. and Karin, M. (1993) Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c–Jun activation domain. Genes Dev., 7, 2135–2148. [DOI] [PubMed] [Google Scholar]
- Hilberg F., Aguzzi, A., Howells, N. and Wagner, E.F. (1993) c-jun is essential for normal mouse development and hepatogenesis. Nature, 365, 179–181. [DOI] [PubMed] [Google Scholar]
- Ip Y.T. and Davis, R.J. (1998) Signal transduction by the c–Jun N–terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol., 10, 205–219. [DOI] [PubMed] [Google Scholar]
- Johnson R.S., van Lingen, B., Papaioannou, V.E. and Spiegelman, B.M. (1993) A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev., 7, 1309–1317. [DOI] [PubMed] [Google Scholar]
- Kamps M.P. (1991) Determination of phosphoamino acid composition by acid hydrolysis of protein blotted to Immobilon. Methods Enzymol., 201, 21–27. [DOI] [PubMed] [Google Scholar]
- Karin M. and Hunter, T. (1995) Transcriptional control by protein phosphorylation: signal transmission from the cell surface to the nucleus. Curr. Biol., 5, 747–757. [DOI] [PubMed] [Google Scholar]
- Karin M., Liu, Z. and Zandi, E. (1997) AP-1 function and regulation. Curr. Opin. Cell Biol., 9, 240–246. [DOI] [PubMed] [Google Scholar]
- Kharbanda S., Datta, R. and Kufe, D. (1991) Regulation of c-jun gene expression in HL-60 leukemia cells by 1-β-d-arabinofuranosylcytosine. Potential involvement of a protein kinase C dependent mechanism. Biochemistry, 30, 7947–7952. [DOI] [PubMed] [Google Scholar]
- Kharbanda S., Pandey, P., Ren, R., Mayer, B., Zon, L. and Kufe, D. (1995a) c–Abl activation regulates induction of the SEK1/stress-activated protein kinase pathway in the cellular response to 1-β-d-arabinofuranosylcytosine. J. Biol. Chem., 270, 30278–30281. [DOI] [PubMed] [Google Scholar]
- Kharbanda S., Ren, R., Pandey, P., Shafman, T.D., Feller, S.M., Weichselbaum, R.R. and Kufe, D.W. (1995b) Activation of the c–Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature, 376, 785–788. [DOI] [PubMed] [Google Scholar]
- Kharbanda S., Yuan, Z.M., Weichselbaum, R. and Kufe, D. (1998) Determination of cell fate by c–Abl activation in the response to DNA damage. Oncogene, 17, 3309–3318. [DOI] [PubMed] [Google Scholar]
- Koleske A.J., Gifford, A.M., Scott, M.L., Nee, M., Bronson, R.T., Miczek, K.A. and Baltimore, D. (1998) Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron, 6, 1259–1272. [DOI] [PubMed] [Google Scholar]
- Kyriakis J.M. and Avruch, J. (1996) Protein kinase cascades activated by stress and inflammatory cytokines. BioEssays, 18, 567–577. [DOI] [PubMed] [Google Scholar]
- Lallemand D., Ham, J., Garbay, S., Bakiri, L., Traincard, F., Jeannequin, O., Pfarr, C.M. and Yaniv, M. (1998) Stress-activated protein kinases are negatively regulated by cell density. EMBO J., 17, 5615–5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.A., Dritschilo, A. and Jung, M. (1998) Impaired ionizing radiation-induced activation of a nuclear signal essential for phosphorylation of c–Jun by dually phosphorylated c–Jun amino–terminal kinases in ataxia telangiectasia fibroblasts. J. Biol. Chem., 273, 32889–32894. [DOI] [PubMed] [Google Scholar]
- Liu Z.-G., Baskaran, R., Lea-Chou, E.T., Wood, L.D., Chen, Y., Karin, M. and Wang, J.Y.J. (1996) Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature, 384, 273–276. [DOI] [PubMed] [Google Scholar]
- Mayer B.J., Hirai, H. and Sakai, R. (1995) Evidence that the SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr. Biol., 5, 296–305. [DOI] [PubMed] [Google Scholar]
- Papavassiliou A.G., Treier, M. and Bohmann, D. (1995) Intramolecular signal transduction in c–Jun. EMBO J., 14, 2014–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitano A.B., Halpern, J.R., Hambuch, T.M. and Sawyers, C.L. (1995) The Bcr–Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc. Natl Acad. Sci. USA, 92, 11746–11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitano A.B., Whang, Y.E. and Sawyers, C.L. (1997) Signal transduction by wild-type and leukemogenic Abl proteins. Biochim. Biophys. Acta, 1333, F201–F216. [DOI] [PubMed] [Google Scholar]
- Renshaw M.W., Lea-Chou, E. and Wang, J.Y.J. (1996) Rac is required for v–Abl tyrosine kinase to activate mitogenesis. Curr. Biol., 6, 76–83. [DOI] [PubMed] [Google Scholar]
- Rubin E., Kharbanda, S., Gunji, H., Weichselbaum, R. and Kufe, D. (1992) cis-Diamminedichloroplatinum (II) induces c-jun expression in human myeloid leukemia cells: potential involvement of a protein kinase C-dependent signaling pathway. Cancer Res., 52, 878–882. [PubMed] [Google Scholar]
- Sanchez-Perez I., Murguia, J.R. and Perona, R. (1998) Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene, 16, 533–540. [DOI] [PubMed] [Google Scholar]
- Sawyers C.L. (1999) Chronic myeloid leukemia. N. Engl. J. Med., 340, 1330–1340. [DOI] [PubMed] [Google Scholar]
- Schreiber M., Kolbus, A., Piu, F., Szabowski, A., Mohle-Steinlein, U., Tian, J., Karin, M., Angel, P. and Wagner, E.F. (1999) Control of cell cycle progression by c–Jun is p53 dependent. Genes Dev., 13, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z. et al. (1993) SH2 domains recognize specific phosphopeptide sequences. Cell, 72, 767–778. [DOI] [PubMed] [Google Scholar]
- Songyang Z. et al. (1995) Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature, 373, 536–539. [DOI] [PubMed] [Google Scholar]
- Superti-Furga G. and Courtneidge, S.A. (1995) Structure–function relationships in Src family and related protein tyrosine kinases. BioEssays, 17, 321–330. [DOI] [PubMed] [Google Scholar]
- Taagepera S., McDonald, D., Loeb, J.E., Whitaker, L.L., McElroy, A.K., Wang, J.Y. and Hope, T.J. (1998) Nuclear–cytoplasmic shuttling of C-ABL tyrosine kinase. Proc. Natl Acad. Sci. USA, 95, 7457–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Gupta, R. and Mayer, B.J. (1995) Differential inhibition of signaling pathways by dominant-negative SH2/SH3 adapter proteins. Mol. Cell. Biol., 15, 6829–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treier M., Staszewski, L.M. and Bohmann, D. (1994) Ubiquitin-dependent c–Jun degradation in vivo is mediated by the δ domain. Cell, 78, 787–798. [DOI] [PubMed] [Google Scholar]
- Treisman R. (1996) Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell Biol., 8, 205–215. [DOI] [PubMed] [Google Scholar]
- Van Etten R.A. (1999) Cycling, stressed-out and nervous: cellular functions of c–Abl. Trends Cell Biol., 9, 179–186. [DOI] [PubMed] [Google Scholar]
- Wang J.Y.J. (1993) Abl tyrosine kinase in signal transduction and cell-cycle regulation. Curr. Opin. Genet. Dev., 3, 35–43. [DOI] [PubMed] [Google Scholar]
- Xia Z., Dickens, M., Raingeaud, J., Davis, R.J. and Greenberg, M.E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science, 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Yuan Z.M. et al. (1998) Regulation of Rad51 function by c–Abl in response to DNA damage. J. Biol. Chem., 273, 3799–3802. [DOI] [PubMed] [Google Scholar]
- Yuan Z.M. et al. (1999) p73 is regulated by tyrosine kinase c–Abl in the apoptotic response to DNA damage. Nature, 399, 814–817. [DOI] [PubMed] [Google Scholar]