

Cardiac arrest due to torsade de pointes (TdP) in the acquired form of drug-induced long-QT syndrome (LQTS) is a rare but potentially catastrophic event in hospital settings. Administration of a QT-prolonging drug to a hospitalized population may be more likely to cause TdP than administration of the same drug to an outpatient population, because hospitalized patients often have other risk factors for a proarrhythmic response. For example, hospitalized patients are often elderly people with underlying heart disease who may also have renal or hepatic dysfunction, electrolyte abnormalities, or bradycardia and to whom drugs may be administered rapidly via the intravenous route.
In hospital units where patients’ electrocardiograms (ECGs) are monitored continuously, the possibility of TdP may be anticipated by the detection of an increasing QT interval and other premonitory ECG signs of impending arrhythmia. If these ECG harbingers of TdP are recognized, it then becomes possible to discontinue the culprit drug and manage concomitant provocative conditions (eg, hypokalemia, bradyarrhythmias) to reduce the occurrence of cardiac arrest.
The purpose of this scientific statement is to raise awareness among those who care for patients in hospital units about the risk, ECG monitoring, and management of drug-induced LQTS. Topics reviewed include the ECG characteristics of TdP and signs of impending arrhythmia, cellular mechanisms of acquired LQTS and current thinking about genetic susceptibility, drugs and drug combinations most likely to cause TdP, risk factors and exacerbating conditions, methods to monitor QT intervals in hospital settings, and immediate management of marked QT prolongation and TdP.
Characteristic Pattern of TdP
The term torsade de pointes was coined by Dessertenne in 1966 as a polymorphic ventricular tachycardia characterized by a pattern of twisting points.1 Several ECG features are characteristic of TdP and are illustrated in Figure 1. First, a change in the amplitude and morphology (twisting) of the QRS complexes around the isoelectric line is a typical feature of the arrhythmia; however, this characteristic twisting morphology may not be evident in all ECG leads. Second, episodes of drug-induced TdP usually start with a short-long-short pattern of R-R cycles consisting of a short-coupled premature ventricular complex (PVC) followed by a compensatory pause and then another PVC that typically falls close to the peak of the T wave.2 However, because of the underlying long-QT interval, this R-on-T PVC does not have the short coupling interval that is characteristic of idiopathic ventricular fibrillation. On the basis of experiments performed in isolated canine ventricular wedge preparations, this short-long-short sequence is thought to promote TdP by increasing heterogeneity of repolarization across the myocardial wall. Third, TdP episodes usually show a warm-up phenomenon, with the first few beats of ventricular tachycardia exhibiting longer cycle lengths than subsequent arrhythmia complexes. The rate of TdP ranges from 160 to 240 beats per minute, which is slower than ventricular fibrillation. Fourth, in contrast to ventricular fibrillation that does not terminate without defibrillation, TdP frequently terminates spontaneously, with the last 2 to 3 beats showing slowing of the arrhythmia. However, in some cases, TdP degenerates into ventricular fibrillation and causes sudden cardiac death.
Figure 1.

Onset of TdP during the recording of a standard 12-lead ECG in a young male with a history of drug addiction treated with chronic methadone therapy who presented to a hospital emergency department after ingesting an overdose of prescription and over-the-counter drugs from his parent’s drug cabinet. Classic ECG features evident in this rhythm strip include a prolonged QT interval with distorted T-U complex, initiation of the arrhythmia after a short-long-short cycle sequence by a PVC that falls near the peak of the distorted T-U complex, “warm-up” phenomenon with initial R-R cycles longer than subsequent cycles, and abrupt switching of QRS morphology from predominately positive to predominately negative complexes (asterisk).
The term torsade de pointes has also been used to describe polymorphic ventricular arrhythmias in which QT intervals are not prolonged. However, the term is better confined to those polymorphic tachycardias with marked (>500 ms) QT-interval prolongation and QT-U deformity, because they appear to be a distinct mechanistic and therapeutic entity.
Premonitory ECG Signs of TdP
Lessons learned from research in large cohorts of individuals with congenital LQTS indicate that there is a gradual increase in risk for TdP as the heart rate–corrected QT interval (QTc) increases. Each 10-ms increase in QTc contributes approximately a 5% to 7% exponential increase in risk for TdP in these patients.3,4 Therefore, a patient with a QTc of 540 ms has a 63% to 97% higher risk of developing TdP than a patient with a QTc of 440 ms. There is no threshold of QTc prolongation at which TdP is certain to occur. Data from congenital LQTS studies5,6 indicate that a QTc >500 ms is associated with a 2- to 3-fold higher risk for TdP. Likewise, case reports and small series of patients with drug-induced TdP show similar increased risk when the threshold of QTc >500 ms is exceeded.7–9
Although research in congenital LQTS indicates that the risk for syncope and sudden death varies directly with the duration of the QT interval,5 monitoring the QT/QTc intervals alone may be inadequate to accurately predict TdP.10 One reason QT monitoring alone may be inadequate is that it is difficult to measure this interval accurately in clinical practice and in clinical trials. Automated systems and human observers are reasonably adept at measuring QT intervals that have normal duration and morphology; however, establishing the end of the QT interval that is morphologically distorted is much more challenging and prone to interrater differences. The typical short-long-short sequence of R-R intervals seen before the initiation of TdP is associated with marked QT prolongation and T-U–wave distortion in the last sinus beat (terminating the long pause) before the episode. Distortion often involves changes in T-wave morphology such as T-wave flattening, bifid T waves, prominent U waves that are fused with T waves, and an extended and gradual sloping of the descending limb of the T wave, which makes it difficult to determine the end of the T wave. Some reports indicate that TdP is especially likely when the QT interval is prolonged because of an increase in the terminal portion of the T wave, from the peak of the T wave to its end (Tpeak-Tend).11,12
In a patient with drug-induced LQTS, the QT interval may be prolonged during normal sinus rhythm without adverse effect, but after a pause (eg, after an ectopic beat or during transient atrioventricular block), QT-interval prolongation and T-U deformity become markedly exaggerated, and TdP is triggered. This beat-to-beat instability of the QT interval not only appears likely to influence the accuracy of measurement, but it may also be related to the underlying mechanism of the arrhythmia.13 In addition to an ever-increasing and distorted QT interval, another rare but ominous premonitory ECG sign of impending TdP is macroscopic T-wave alternans,14 as illustrated in Figure 2. In the future, it may be possible to assess risk by use of sophisticated T-U–wave morphology analysis; however, until such analysis becomes available, exaggerated QT-interval prolongation with T-U distortion after a pause should be considered a strong marker of risk for TdP.
Figure 2.

Top rhythm strip, TdP degenerating into ventricular fibrillation in an 83-year-old female hospitalized in the intensive care unit for pneumonia. She was started on intravenous erythromycin several hours before cardiac arrest. A ventricular couplet followed by a pause provided the short-long-short cycle sequence that triggered TdP. Bottom rhythm strip, ECG 1 hour before the onset of TdP shows extreme prolongation of the QT interval (QTc in cycles with larger T waves=730 ms), a ventricular couplet (asterisk), and macroscopic T-wave alternans (vertical arrows). If these signs of impending TdP had been recognized, discontinuation of the culprit drug and administration of magnesium most likely would have prevented the subsequent cardiac arrest.
Cellular Mechanisms of Acquired LQTS
Prolongation of the QT interval, changes in T-U wave morphology, and subsequent TdP are results of abnormal function (and structure) of ion channels and related proteins involved in the repolarization process in ventricular myocytes. These abnormalities can be caused by mutations of genes that encode ion channels or associated proteins in congenital forms of LQTS; however, they can also be caused by the action of drugs in acquired LQTS. Drugs with the potential to cause TdP most frequently inhibit the rapid component of the delayed rectifier potassium current (IKr), which causes a reduction in the net repolarizing current and results in prolongation of the ventricular action potential duration and a prolonged QT interval on the ECG.15
Experiments in canine ventricular wedge preparations have shown that in normal circumstances, there are differences in repolarization in the various layers of the myocardium, with the subepicardium having the shortest action potential duration, the subendocardium having an intermediate duration, and the mid myocardium (M cells) having the longest action potential duration.16,17 However, because the myocardial layers are tightly coupled in the intact human heart, such differences are small. Many reports indicate that the QT interval on the ECG represents the longest repolarization in the M-cell region. This physiological transmural dispersion of repolarization usually does not lead to TdP; however, proarrhythmic states may arise as a result of specific gene mutations or actions of medications that cause selective action potential prolongation in certain layers of the myocardium (usually the M-cell region) that lead to increased transmural repolarization gradients.17 This increased transmural gradient is thought to create the conditions for reentry and subsequent TdP.
The trigger for TdP is thought to be a PVC that results from an early afterdepolarization generated during the abnormally prolonged repolarization phase of the affected myocardium.18 A long preceding pause increases the amplitude of early afterdepolarizations, which makes them more likely to reach the threshold necessary to produce a PVC or ventricular couplet. Because of the marked delay of repolarization in certain areas of the myocardium, conduction of the PVC is blocked initially in some directions but not in others, which sets up reentry that perpetuates TdP.
Not all QT-prolonging drugs are associated with risk for TdP. Therefore, it appears that QT prolongation alone is insufficient and that heterogeneity of repolarization may also be necessary to produce an arrhythmogenic response. However, the mechanisms whereby not all QT prolongation confers the same degree of risk are not well established.
Experts in electrocardiography, including members of this writing group, have been curious about the peculiar pattern of sine-wave QRS changes with TdP. El-Sherif et al19 provided an electrophysiological mechanism for the characteristic periodic transition of the QRS axis during TdP. In an experimental setting, they demonstrated that the initial beat of TdP arose as a subendocardial focal activity, whereas subsequent beats were due to reentrant excitation in the form of rotating scrolls. The arrhythmia ended when reentrant excitation was terminated. The transition in the QRS axis coincided with a transient bifurcation of the predominantly single rotating scroll into 2 simultaneous scrolls that involved both the right ventricle and left ventricle separately. The common mechanism for the initiation or termination of this bifurcation was the development of functional conduction block between the anterior or posterior right ventricular free wall and the ventricular septum.
Genetic Susceptibility to Drug-Induced TdP
It is becoming increasingly evident that genetic susceptibility, whether due to the presence of rare LQTS-causing mutations or the presence of functional common polymorphisms, must be considered in the patient who manifests drug-induced QT prolongation and TdP. Since the sentinel discovery of congenital LQTS as a channelopathy with mutations identified in genes encoding voltage-gated potassium and sodium channels in 1995,20,21 nearly 1000 individually rare LQTS-causing mutations have now been detected in 12 distinct LQTS-susceptibility genes. Three of the 12 LQTS-susceptibility genes (KCNQ1-encoded IKs α-subunit [LQT1], KCNH2-encoded IKr α-subunit [LQT2], and SCN5A-encoded Nav1.5 α-subunit [LQT3]) are the major LQTS-susceptibility genes, accounting for nearly 75% of all congenital LQTS cases.22
Approximately two thirds of LQTS stems from loss-of-function mutations in either KCNQ1 or KCNH2 whereby there is a perturbation in phase 3 repolarization that results in a prolongation in the action potential duration and hence QT-interval prolongation. These defects provide the pathogenic substrate on which an ill-timed PVC and its cellular early afterdepolarization can precipitate TdP. Besides the predominant mechanism of potassium channel loss of function, approximately 5% to 10% of LQTS stems from gain-of-function mutations in the sodium channel whereby the mutations (mostly missense, ie, single amino acid substitutions) produce a sodium channel with a marked accentuation in late sodium current. Rather than shutting down within the first 5 ms of a cardiac action potential, this persistent but relatively small influx of inward sodium current disrupts phase 2 of the fine-tuned balance of the action potential, which prolongs the cellular action potential duration and confers the substrate for TdP. In addition to these 3 major LQTS-susceptibility genes that account for 75% of congenital LQTS, 9 minor LQTS-susceptibility genes account for an additional 5%. The remaining 20% of congenital LQTS cases remain genotype negative.
From 1995 to 2004, research-based LQTS genetic testing revealed a plethora of genotype-phenotype relationships, including genotype-suggestive ECG patterns, arrhythmogenic triggers, and genetically determined responses to pharmacotherapy. In 2004, LQTS genetic testing matured into a clinically available test because of its established diagnostic, prognostic, and therapeutic implications. Just as a period of time (eg, during swimming or during the postpartum period) can suggest the presence of congenital LQTS, drug-induced long QT and TdP may also signal the presence of an LQTS genetic defect. In fact, the yield from LQTS genetic testing with respect to the 3 major LQTS-susceptibility genes is approximately 10% to 15% in individuals with isolated drug-induced acquired LQTS.23–25
In addition to these individually rare mutations that confer susceptibility for the primary channelopathy known as congenital LQTS, which affects approximately 1 in 2500 persons, numerous common polymorphisms in these same cardiac channel genes have been identified, and some are now known to contribute to a reduced repolarization reserve and confer a modifier effect.26 For example, SCN5A-S1103Y is one of the most common polymorphisms in black Africans, 10% to 15% of whom may be heterozygous for this common, nonsynonymous single-nucleotide polymorphism. SCN5A-S1103Y is now known to produce or acquire a cellular phenotype of accentuated late sodium current (LQT3-like) when exposed to cellular acidosis and confer clinical susceptibility to proarrhythmia and premature sudden death as early as infancy in African Americans.27–30 In addition, KCNE2-Q9E was published originally as a rare, LQT6-causing missense mutation after its identification in a 76-year-old African American female with profound QT prolongation and TdP who required defibrillation after 7 doses of intravenous erythromycin and 2 doses of oral clarithromycin, both of which are known IKr blockers.31 Functional studies demonstrated that an IKr complex containing Q9E in the KCNE2-encoded β-subunit resulted in a potassium channel with a marked increase in sensitivity to hERG (human ether-a-go-go) block by clarithromycin. However, in contrast to its initial impression of rarity (absence in more than 2000 control alleles of unspecified ethnicity), KCNE2-Q9E is a relatively black-specific common polymorphism present in approximately 3% to 5% of African Americans.32
Case series of drug-induced TdP (usually involving antiarrhythmic agents) identify subclinical congenital LQTS in 5% to 20% of cases.23–25 However, the extent to which the congenital LQTS confers risk during administration of drugs is not well understood. To illustrate the lack of clarity about genetic susceptibility and drug risk, moxifloxacin is a drug that very rarely causes TdP; however, the risk does not appear to increase even in the presence of congenital LQTS for the following reason. The incidence of TdP with moxifloxacin is very low, 1:100 000 to 1:1 000 000 exposures. Moxifloxacin is pharmacokinetically well behaved, with no known drug interactions or organ dysfunction that severely alters plasma concentrations. Given the fact that the mutations associated with congenital LQTS occur in 1 in 2500 individuals in the population,33 it appears irrefutable that many patients with congenital LQTS have been exposed to the drug without adverse effects.
This kind of logic points to a likely distinction between high- and low-risk drugs. For example, the high-risk drugs, such as antiarrhythmic agents, methadone, and haloperidol, may increase risk for TdP in individuals with genetic mutations, whereas the low-risk drugs, such as moxifloxacin, may require other risk factors such as electrolyte disorders.
Drugs That Cause TdP: Incidence and Other Features
When sudden death occurs without autopsy evidence for an explainable cause of death, an arrhythmic death is assumed. However, the proportion of sudden arrhythmic deaths that are due to TdP is unclear, because few individuals are being monitored at the time of death. When TdP occurs in outpatient settings, the first responders who arrive on the scene with portable monitor-defibrillators are likely to observe ventricular fibrillation. In this situation, it is impossible to determine whether ventricular fibrillation was preceded by QT prolongation and TdP. In hospital settings, the same lack of clarity about the arrhythmia mechanism that caused the cardiac arrest may occur if a patient is not undergoing continuous ECG monitoring at the time of arrest. Postarrest ECG changes are not uncommon, and a link to LQTS may not be made. For example, the postarrest QT interval may be prolonged because of the hypoxic/anoxic insult, or it may be quite short, presumably due to elevated potassium in this setting.
Preclinical and early-phase clinical testing of new drugs may reveal a QT-prolongation signal that may be identified by consulting the drug label. Use of a QT-prolonging drug must be based on risk-benefit analysis in individual patients, and where efficacy of alternatives is equivalent, the non–QT-prolonging agent should be preferred. Where benefit clearly outweighs risk, QT prolongation should not limit necessary therapy. QT prolongation is not necessarily equivalent to arrhythmogenicity. The only class of drugs for which reasonable TdP incidence data are available is the antiarrhythmic agents. Those known to prolong the QT interval and block sodium and potassium channels (older drugs such as quinidine, disopyramide, and procainamide), as well as those that block potassium channels (sotalol, dofetilide, ibutilide), appear to have a TdP incidence of 1% to 10%.34 For the older drugs, the numbers are derived from uncontrolled case series, whereas for the newer agents, summary data from clinical trials and new drug applications are available.35–40
Many non-antiarrhythmic drugs have also been associated with TdP. For some drugs, multiple case reports and small case series confirm that the drug causes the arrhythmia. High-profile examples include methadone,41 thioridazine,42 and haloperidol.43 Although the absolute TdP incidence is difficult to establish from these reports of non-antiarrhythmic agents, it is generally believed to be less than that reported for antiarrhythmic agents.
IKr inhibition is a very common effect of many drugs, and case reports and small series implicate the involvement of many such drugs with TdP. Some of these are widely and commonly used, such as erythromycin and droperidol. In addition, a number of drugs have been withdrawn from the market or relabeled because of the risk for TdP.26 The absolute incidence figures are difficult to establish but appear to be very small, even in these cases of banned drugs. Thus, for example, nearly one hundred million prescriptions for the antihistamine terfenadine had been written before a very small risk for TdP was recognized. The overall incidence of TdP with terfenadine is exceedingly small and appears to be confined largely to patients with specific risk factors related to metabolism of the drug.44
For virtually all QT-prolonging drugs, risk increases as a function of dose and, more specifically, plasma drug concentration, with the exception of quinidine. Quinidine is a potent IKr blocker,45 so at low concentrations it may prolong action potentials, whereas this effect may be blunted (by the drug’s sodium channel–blocking properties) at higher concentrations, which explains the clinical observation that quinidine-induced TdP often occurs at low concentrations.
The high-potency IKr blocker terfenadine undergoes near-complete presystemic metabolism, mediated largely by a specific hepatic cytochrome P450 (CYP3A4). Both terfenadine and its metabolite fexofenadine are potent antihistamines, but fexofenadine does not block IKr8; nevertheless, there is 1 case report of fexofenadine-related TdP.46 The vast majority of cases of terfenadine-associated TdP were associated with inhibition of CYP3A4 due to advanced liver disease, overdose, or ingestion of specific inhibitor drugs, notably erythromycin and ketoconazole. Erythromycin itself can also cause TdP, almost always with high doses or with use of the intravenous route and often in patients with other risk factors.47
The problem of dramatic drug accumulation due to use of high doses, dysfunction of organs of elimination, or interacting drugs applies to other situations. Dofetilide and sotalol are cleared by the kidneys, and the use of ordinary doses in patients with renal failure increases TdP risk with these drugs. Procainamide undergoes hepatic clearance to an active metabolite, N-acetylprocainamide (NAPA), which has IKr-blocking properties. NAPA itself is eliminated by the kidneys, so patients with renal dysfunction may develop NAPA-related TdP during procainamide therapy.37 Thioridazine is bioinactivated by CYP2D6, and subjects with deficient activity of this enzyme due to genetic factors (5% to 10% of white and black populations) or the use of CYP2D6-inhibiting drugs such as quinidine, fluoxetine, or paroxetine have higher plasma drug concentrations.48
Case series of methadone-related TdP indicate that the use of high doses and/or recent dose increases are common clinical features of affected patients.41 Methadone is cleared by multiple pathways; although inhibiting drugs have been implicated, their precise role is unclear at this time. Nearly 1 million Americans use methadone for narcotic dependence or for chronic pain therapy.49 Recently published methadone clinical guidelines recommend a pretreatment ECG for QTc interval screening and a follow-up ECG within 30 days and then annually.49
The risk for TdP should be evaluated in any patient who presents to the emergency department with an overdose of a QT-prolonging drug. However, because it is often unclear what drug or combination of drugs the patient may have taken, the ECG of all drug overdose victims should be assessed for signs of prolonged QT, QT-U distortion, and other signs of impending TdP (Figure 2). The tricyclic antidepressants such as amitriptyline can cause TdP, although the incidence is not well established and other arrhythmias due to sodium channel blocker toxicity (eg, wide QRS and sinusoidal ventricular tachycardia) may also be present. Less frequent use of these antidepressants for outpatient treatment of depression has decreased the presentation of patients with an overdose of these agents. Because depressed patients are the most susceptible to purposeful drug overdoses, pharmaceutical manufacturers have attempted to create multiple new antidepressants such as selective serotonin reuptake inhibitors for use in depression.50 Despite this, TdP has been reported in patients with overdoses of these medications, such as citalopram.50 Other nontricyclic antidepressants, such as trazodone, have also been implicated in TdP in patients with purposeful overdose.51 Moreover, a recent analysis of current users of older typical versus newer atypical antipsychotic agents revealed that both groups had a similar dose-related increased risk of sudden cardiac death compared with matched nonusers of antipsychotic drugs.52
Chronic administration of amiodarone markedly prolongs the QT interval, yet it is very rarely associated with TdP.53 It has been postulated (although as yet unproven) that unlike high-risk drugs that selectively prolong repolarization in myocytes located in the mid myocardium (M cells), amiodarone uniformly delays repolarization in all layers of the myocardial wall. As a result, there is only QT prolongation and no transmural heterogeneity of repolarization, which is the necessary substrate for the development of a reentrant arrhythmia. Another theory regarding the low TdP risk nature of amiodarone suggests that the drug also inhibits the physiological late sodium currents that ultimately produce the arrhythmia.54
This theory also applies to verapamil, a relatively potent IKr blocker55 that has never been associated with TdP, probably because it is a much more potent blocker of L-type calcium channels. The newer antianginal agent ranolazine also blocks IKr,56,57 but the extent of the QT prolongation appears limited during long-term therapy, probably because the drug also blocks the physiological late sodium current. In a large clinical trial, ranolazine was not associated with an increased incidence of TdP.58
Intravenous administration can be associated with higher drug concentrations and greater cardiac exposure than corresponding oral dosing. Thus, the intravenous route may be a risk factor for TdP. In addition, there are provocative data from an animal model of TdP that suggest that rapid infusion may be more likely to cause the arrhythmia than slower infusion (of higher drug doses).59 The mechanism underlying this effect is unknown but may reflect differential drug delivery to various sites within the myocardium.
The Arizona Center for Education & Research on Therapeutics maintains an updated list of drugs that have a risk of causing TdP on their World Wide Web site at www.qtdrugs.org. Table 1 shows a drug list from this World Wide Web site that has been modified to exclude amiodarone (regarded as low risk) and drugs that are no longer available in the United States. Table 1 represents the most common drugs that can be implicated in TdP, but it is not a complete list of all reported possible contributing substances. Importantly, the drugs listed in Table 1 are not equipotent in their risk of causing TdP. For example, the risk of TdP ranges from approximately 0.001% for Propulsid (cisapride) to approximately 8% for the antiarrhythmic quinidine. We also reemphasize that the use of these medications may be clearly indicated from a risk-benefit perspective despite the presence of the possibility of drug-induced TdP. For example, a recent analysis of a large number of surgical patients (>290 000) revealed no change in the incidence of TdP in patients who received antiemetic therapy with low-dose droperidol versus those without droperidol therapy.60 Therefore, in an individual patient, a drug listed in Table 1 may provide a therapeutic benefit that outweighs its risk of causing TdP.
Table 1.
Drugs that Have a Risk of Causing Torsade de Pointes*
| Generic Name | Brand Name(s) |
Clinical Use |
|---|---|---|
| Arsenic trioxide | Trisenox | Cancer/leukemia |
| Bepridil | Vascor | Antianginal |
| Chloroquine | Aralen | Antimalarial |
| Chlorpromazine | Thorazine | Antipsychotic, schizophrenia, antiemetic |
| Cisapride | Propulsid | Gastrointestinal stimulant |
| Clarithromycin | Biaxin | Antibiotic |
| Disopyramide | Norpace | Antiarrhythmic |
| Dofetilide | Tikosyn | Antiarrhythmic |
| Droperidol | Inapsine | Sedative, antiemetic |
| Erythromycin | E.E.S., Erythrocin |
Antibiotic, increase gastrointestinal motility |
| Halofantrine | Halfan | Antimalarial |
| Haloperidol | Haldol | Antipsychotic, schizophrenia, agitation |
| Ibutilide | Corvert | Antiarrhythmic |
| Levomethadyl | Orlaam | Opiate agonist, pain control, narcotic dependence |
| Mesoridazine | Serentil | Antipsychotic, schizophrenia |
| Methadone | Dolophine, Methadose |
Opiate agonist, pain control, narcotic dependence |
| Pentamidine | NebuPent, Pentam |
Antiinfective, pneumocystis pneumonia |
| Pimozide | Orap | Antipsychotic, Tourette tics |
| Procainamide | Pronestyl, Procan |
Antiarrhythmic |
| Quinidine | Quinaglute, Cardioquin |
Antiarrhythmic |
| Sotalol | Betapace | Antiarrhythmic |
| Sparfloxacin | Zagam | Antibiotic |
| Thioridazine | Mellaril | Antipsychotic, schizophrenia |
Drugs with low risk and drugs no longer available in the United States are not included in this table. Modified from the Arizona CERT World Wide Web site at www.qtdrugs.org on April 18, 2009.
TdP Risk Factors and Exacerbating Conditions in Hospital Settings
Risk factors for the development of TdP in hospitalized patients are listed in Table 2, along with references to clinical data, reviews and meta-analyses, and selected experimental studies. Clinically recognizable historical, ECG, and laboratory risk factors are emphasized,61–65 but the potential role for analyses of predictive genetic polymorphisms as risk markers is also noted.26,66–69 The term repolarization reserve was introduced by Roden,70 who explained that normal cardiac repolarization depends critically on the interplay of multiple ion currents, and these provide some redundancy, or reserve, to protect against excessive QT prolongation by drugs. Roden proposed that lesions in these repolarizing mechanisms that result in reduced repolarization reserve can remain subclinical but nevertheless increase risk on drug exposure.
Table 2.
Risk Factors for Torsade de Pointes in Hospitalized Patients
| Clinically recognizable risk factors 61-65 |
| QTc >500 ms 71-74 |
| LQT2-type repolarization: notching, long Tpeak–Tend 11,12 |
| Use of QT-prolonging drugs 75-77 |
| Concurrent use of more than 1 QT-prolonging drug 78-80 |
| Rapid infusion by intravenous route 59 |
| Heart disease 64,73,75,76 |
| Congestive heart failure 39 |
| Myocardial infarction 39,73 |
| Advanced age 75,77,86 |
| Female sex 64,72,73,75-77,79,81-85 |
| Hypokalemia 46,74,87-90 |
| Hypomagnesemia 89,91-94 |
| Hypocalcemia 95,96 |
| Treatment with diuretics 72,74,97 |
| Impaired hepatic drug metabolism (hepatic dysfunction or drug-drug interactions) 76,79 |
| Bradycardia 65,87 |
| Sinus bradycardia, heart block, incomplete heart block with pauses 98,99 |
| Premature complexes leading to short-long-short cycles 65,72 |
| Multiple clinically recognizable risk factors 64,65,76,79,84 |
| Clinically silent risk factors |
| Occult (latent) congenital LQTS 23,64 |
| Genetic polymorphisms (reduced repolarization reserve) 26,27,31,66-69 |
In hospitalized patients, TdP is commonly associated with acquired prolongation of the uncorrected or rate-corrected QT interval,71–74 with or without underlying genetic predisposition,24 often in the presence of a noncardiac drug that is known (or not known) to prolong the QT interval.75–77 Of note, the risk for TdP increases significantly with concurrent use of more than 1 QT prolonging drug,78–80 and concomitant medication or intrinsic disease that alters liver metabolism of 1 or more of these drugs also increases risk.76,79 Accordingly, careful examination of administered drugs can identify patients who should have continuous ECG monitoring.
Multiple studies have shown that risk for TdP among hospitalized patients is strikingly greater in women than in men, by a factor of approximately 2-fold.72,73,75–77,79,81–85 TdP is more common in patients older than 65 years than in younger patients.75,77,86 Predisposition to the development of TdP has been associated with underlying heart disease of multiple origins.39,64,65,73,75,76 Hypokalemia, perhaps by modifying the function of the IKr channel to prolong the QT interval in a manner that results in heterogeneity and dispersion of repolarization, is a well-established predisposing risk factor for TdP,46,74,87–90 as is hypomagnesemia.89,91–94 Hypocalcemia, which also prolongs the QT interval, has been associated with TdP only in rare cases.95,96 The association of diuretic use with in-hospital TdP may be explained by its correlation with congestive heart failure, hypokalemia, and hypomagnesemia.72,74 In addition, some diuretics directly block potassium currents and may therefore reduce repolarization reserve.97 Intravenous potassium has been shown to reverse the QT-prolonging effects of quinidine in humans,90 but the value of acute potassium repletion is less well documented than that of intravenous magnesium for the acute treatment of drug-induced TdP.64,65,91,92 Normal levels of both potassium and magnesium should be maintained aggressively in hospitalized patients at risk.
Bradycardia is an additional important risk factor for TdP in patients when other predisposing findings are present.65,87 Prolonged ventricular cycle length can take the form of simple sinus bradycardia, complete atrioventricular block, or any rhythm in which sudden long cycles may lead to arrhythmogenic early afterdepolarizations.98,99 Premature beats that lead to short-long-short cycles may foster the development of TdP.65,72 In contrast, isoproterenol infusion or overdrive pacing can suppress TdP in these circumstances. Interestingly, and despite the association with short-long-short cycles, the risk for TdP appears to be decreased when the underlying rhythm is atrial fibrillation, unless there is also complete heart block.100
At present, no quantitative multivariate risk index exists for the prediction of TdP in the hospital-based population. Perhaps the greatest risk for the development of TdP in the hospital setting occurs with the clustering of multiple recognizable risk factors in a single patient.64,65,76,79,84 Accordingly, an elderly woman with diuretic-treated heart failure taking more than 1 potentially QT-prolonging drug with sinus bradycardia and occasional ventricular bigeminy would be a good candidate for ECG/QTc monitoring and electrolyte repletion.
Methods to Monitor QT/QTc in Hospital Settings
For many years, periodically recorded standard 12-lead ECGs have been relied on in hospital settings to measure QT intervals and to provide a heart-rate–corrected QT interval (QTc). In hospital units with continuous ECG monitoring, manual measurement of QT intervals with handheld calipers using rhythm strips from bedside cardiac monitors has also been performed. Traditionally, the Bazett correction has been used to adjust measured QT for cycle length by dividing the observed, uncorrected QT interval by the square root of the R-R interval (in seconds). When not otherwise stated, QTc generally refers to the Bazett correction. It has become increasingly well recognized that the Bazett correction tends to produce overlong QTc values at faster heart rates, particularly above 85 beats per minute, as is commonly found in hospitalized patients. Alternative QTc calculation methods are available, including both linear and nonlinear corrections that adjust more appropriately at faster rates. Alternative QT corrections, such as the Fridericia formula (which divides observed QT by the cube root of cycle length), are likely to find increasing roles in hospital monitoring settings in the future.101–103
Several monitor manufacturers now provide electronic calipers, which can be used to measure the QT and R-R intervals in a computer-assisted fashion. Most recently, a fully automated hospital monitor system for real-time QT/QTc monitoring has been introduced.104 There are no research studies to indicate the best method for monitoring QT/QTc intervals in hospital settings; thus, what follows is a description of measurement strategies used in current clinical practice, with comments about their benefits and pitfalls.
Definition of Prolonged QT Interval
The expert writing group recommends that a QTc over the 99th percentile should be considered abnormally prolonged. Approximate 99th percentile QTc values for otherwise healthy postpubertal individuals are 470 ms for males and 480 ms for females (Figure 3). For both males and females, a QTc >500 ms is considered highly abnormal.7–9 It must be kept in mind, however, that some standard 12-lead ECG algorithms label a QTc >440 ms as borderline QT prolongation, despite the fact that this value is exceeded by approximately 10% to 20% of the population.
Figure 3.
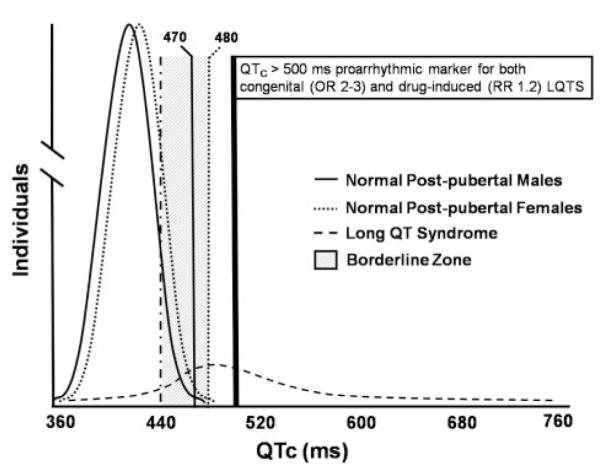
QTc distribution curves in normal males and females and in a cohort of patients with congenital LQTS. Upper limits of normal (99th percentile) for QTc are 470 ms in males and 480 ms in females. For both males and females, a QTc >500 ms is considered dangerous. OR indicates odds ratio; RR, relative risk.
Manual Measurement
The QT interval is measured from the beginning of the QRS complex to the end of the T wave and approximates the time it takes the ventricles to repolarize (ie, a body-surface estimation of the cellular action potential duration). If the patient develops a wide QRS complex (eg, due to a new bundle-branch block), this will increase the total QT interval. Such an increase of the QT interval due to a new conduction block should not be considered indicative of acquired LQTS and risk for TdP.105 One method to adjust the QT measurement after the development of a bundle-branch block is to subtract the difference in QRS widths before and after the block. Another method is to measure a J-T interval from the end of the QRS complex to the end of the T wave, which eliminates the QRS in the measurement altogether. The important point, however, is that if an adjustment method is used, it needs to be applied consistently when a patient is being monitored over time.
Although the onset of the QRS complex is usually readily apparent, the end of the T wave is often obscure, especially when T waves are of low amplitude or T-U distortion is present in drug-induced states. The lead recommended for manual QT measurement is the one from the patient’s 12-lead ECG that has a T-wave amplitude of at least 2 mm and a well-defined T-wave end. Thus, the lead choice will vary among patients. Because of the variation in QT interval durations across the 12 leads, it is important to measure the QT interval in the same lead in a given patient over time and to document the lead being used. In situations in which the end of the T wave may be difficult to determine (eg, biphasic or notched T waves, T waves with superimposed U waves), the end of the T wave can be determined by drawing a line from the peak of the T wave following the steepest T-wave downslope.106 The intersection of this line with the isoelectric baseline is considered the end of the T wave.
Calculation of QTc in the setting of atrial fibrillation is challenging, because the QT interval varies from beat to beat depending on the varying RR intervals. One way to deal with the irregularity of the rhythm is to identify the shortest and longest R-R intervals, calculate the QTc for each, and average the 2 QTc values. Alternatively, a long rhythm strip can be printed to determine whether, on average, the interval from R wave to the peak (or nadir) of the T wave is more than 50% of the R-R interval. This does not give an exact QTc value but provides an indication that it would be longer than the critical threshold of 500 ms if measured.
Electronic Calipers
The current generation of hospital ECG monitoring systems provides a computer-assisted tool (electronic calipers) for QT-interval measurement. When electronic calipers are used, increasing the size of waveforms from a standardization of 1 to 2, 3, or 4 and increasing the recording speed from 25 to 50 mm/s can enhance visualization. The electronic calipers are moved to the beginning of the QRS complex and the end of the T wave, and the resulting value is entered. The preceding R-R interval is then measured in the same fashion. Several monitor manufacturers have a QTc calculator built into their electronic caliper systems so when the QT and R-R intervals are entered, the system calculates the QTc and prints the value on the rhythm strip. Because electronic caliper systems depend on humans to select the appropriate ECG lead and to identify the measurement onset and offset points, measurement of the QT interval with electronic calipers is prone to the same error as manual measurement.
Fully Automated QT/QTc Monitoring
The current standard practice of periodic manual measurement of the QT interval, and even the use of electronic calipers, has drawbacks. For example, error can occur in determining the beginning or end of the QT interval, in the application of a heart rate–correction formula, and from inconsistency in the choice of lead for QT measurement.107 In addition, random selection of 1 beat in 1 lead is likely not to be representative, because significant beat-to-beat variation exists not only because of manual measurement error but also due to actual QT-interval changes. Moreover, development of bundle-branch blocks or irregular rhythms, such as atrial fibrillation, compounds the problem of QT measurement.
Because of the difficulty and unreliability of manual measurement, Helfenbein et al104 reported the development and laboratory testing of an algorithm to measure QT/QTc intervals continuously in real-time using bedside monitors. The algorithm measures QT/QTc intervals every 5 minutes. An audible alarm sounds if there is an increase in QTc >60 ms from baseline (first measurement unless reset manually before drug administration) or a QTc >500 ms for at least 3 consecutive measurements (≈15 minutes).
Differences in QT Measurements Between Standard 12-Lead Electrocardiographs
Manufacturers of electrocardiographs have proprietary and often substantially different computer algorithms for QT-interval measurement.108 In addition, 2 standard 12-lead electrocardiographs may differ substantially in their QT measurement depending on when they were manufactured.109 Newer electrocardiographs typically use global QT measurements derived from simultaneous multilead acquisition, whereas older electrocardiographs typically use single-lead measurement. Therefore, if serial comparisons of QT intervals are being made with standard 12-lead ECGs, the same electrocardiograph instrument should be used so that any observed QT-interval increase is truly due to prolongation of ventricular repolarization rather than a change in computer algorithm.
Practical Considerations in QT/QTc Monitoring
According to the American Heart Association’s practice standards for ECG monitoring in hospital settings,110 indications for QT-interval monitoring include the following: (1) Initiation of a drug known to cause TdP; (2) overdose from potentially proarrhythmic agents; (3) new-onset bradyarrhythmias; and (4) severe hypokalemia or hypomagnesemia. Because there is often a lack of clarity with regard to the types and amounts of drugs taken in an intentional overdose situation, it is prudent to monitor QT intervals in all overdose victims.
Until fully automated QT/QTc monitoring is validated and widely available in clinical settings, a reasonable strategy is to document the QTc interval before and at least every 8 to 12 hours after the initiation, increased dose, or overdose of QT-prolonging drugs. If QTc prolongation is observed, more frequent measurements should be documented.111 How long QTc measurement should be continued depends on the drug half-life, how long it takes for the drug to be eliminated from the body (which may depend on renal or hepatic function), whether the drug is given once versus as ongoing therapy, how long it takes for the QTc to return to the predrug baseline, and whether the ECG shows QT-related arrhythmias. For example, the drug ibutilide, which is administered as a 1-time treatment for termination of atrial fibrillation or flutter, was reported to cause TdP in 4.3% of 586 patients; however, all but 1 arrhythmia episode occurred within 1 hour of the end of infusion, and none occurred after 3 hours.112 Thus, it is unnecessary to monitor QTc after 3 hours in patients who receive a 1-time ibutilide dose.
Summary and Recommendations for Monitoring QT/QTc in Hospital Settings
Because hospitals differ with respect to their cardiac monitoring equipment, there is no one-size-fits-all strategy that can be recommended. For example, Hospital A may have a fully automated QT-monitoring system, whereas Hospital B has only the computer-assisted electronic caliper feature. Of utmost importance, however, is that a hospital protocol be established so that a single consistent method is used by all healthcare professionals charged with the responsibility for cardiac monitoring. The protocol should stipulate the equipment to use for QT measurement, the method to determine the end of the T wave, the formula for heart rate correction, lead-selection criteria, (eg, the lead that has a visible T wave with a clear-cut ending), and the importance of measuring the same lead in the same patient over time.
Management of Drug-Induced QT Prolongation and TdP in Hospital Settings
Drug-Induced Prolonged QT
The 2006 American College of Cardiology/American Heart Association/European Society of Cardiology guidelines for management of patients with ventricular arrhythmias34 make relatively few recommendations on prevention of TdP in the hospital setting. The guidelines do recommend removal of the offending agent in patients with drug-induced LQTS (Class I, Level of Evidence: A); however, they do not define what QTc value should prompt such discontinuation.
Continuous QTc monitoring is appropriate for drugs deemed most at risk to cause not only QT prolongation but also TdP. After administration of an at-risk drug, if the QTc exceeds 500 ms or there has been an increase of at least 60 ms compared with the predrug baseline value, especially when accompanied by other ECG signs of impending TdP, prompt action is indicated. Appropriate actions include alternative pharmacotherapy; assessment of potentially aggravating drug-drug interactions, bradyarrhythmias, or electrolyte abnormalities; and the ready availability of an external defibrillator. Patients should not be transported from the unit for diagnostic or therapeutic procedures, and they should be in a unit with the highest possible ECG monitoring surveillance.
Nonsustained and Sustained TdP
For patients with TdP that does not terminate spontaneously or that degenerates into ventricular fibrillation, immediate direct-current cardioversion should be performed. The guideline34 states that intravenous magnesium sulfate is reasonable for patients taking QT-prolonging drugs who present with episodes of TdP and a prolonged QT interval (Class IIa, Level of Evidence: B). Magnesium sulfate 2 g can be infused intravenously as a first-line agent to terminate TdP irrespective of the serum magnesium level.113 If episodes of TdP persist, it may be necessary to repeat infusions of magnesium sulfate 2 g. The mechanism underlying the protective effect of magnesium is unknown. An increase in heart rate to prevent pauses that may trigger TdP may be attempted with temporary transvenous atrial or ventricular pacing at rates >70 beats per minute.114 Repletion of potassium to supratherapeutic levels of 4.5 to 5 mmol/L may also be considered, although there is little evidence to support this practice (Class IIb, Level of Evidence: C).34
Hospital Discharge
When discharged, the patient should be educated about avoiding the culprit drug, other related drugs, and potential drug-drug interactions. A list of possible QT-prolonging drugs (available at www.qtdrugs.org) should be provided to the patient and appropriate documentation made in the medical record. If drug-induced TdP has occurred, a careful review of the patient’s personal and family history should be obtained, because it may be the sentinel event heralding the presence of congenital LQTS.23 If a personal/family history of unexplained syncope or premature sudden death emerges, a 12-lead ECG should be recommended for all first-degree relatives, and consideration should be given to clinically available genetic testing for congenital LQTS.
Summary
TdP is an uncommon but potentially fatal arrhythmia that can be caused by drugs that cause selective prolongation of action potential durations in certain layers of the ventricular myocardium, which creates dispersion of repolarization and a long, distorted QT-U interval on the ECG. A summary of key points to remember is provided in Table 3.
Table 3.
Summary of Key Points
|
For patients who receive QT-prolonging drugs in hospital units with continuous ECG monitoring, TdP should be avoidable if there is an awareness of individual risk factors and the ECG signs of drug-induced LQTS. Particularly important are the ECG risk factors for TdP, including marked QTc prolongation to >500 ms (with the exception of amiodarone- or verapamil-induced QT prolongation), marked QT-U prolongation and distortion after a pause, onset of ventricular ectopy and couplets, macroscopic T-wave alternans, or episodes of polymorphic ventricular tachycardia that are initiated with a short-long-short R-R cycle sequence (typically, PVC– compensatory pause–PVC). Recognition of these ECG harbingers of TdP allows for treatment with intravenous magnesium, removal of the offending agent, and correction of electrolyte abnormalities and other exacerbating factors, including the prevention of bradycardia and long pauses with temporary pacing if necessary.
Disclosures
Writing Group Disclosures
| Writing Group Member |
Employment | Research Grant | Other Research Support |
Speakers’ Bureau/ Honoraria |
Expert Witness |
Ownership Interest |
Consultant/Advisory Board |
Other |
|---|---|---|---|---|---|---|---|---|
| Barbara J. Drew |
University of California San Francisco |
GE Healthcare†; Philips† |
None | GE Healthcare*; Philips* |
None | None | None | None |
| Michael J. Ackerman |
Mayo Clinic | None | None | None | None | None | Boston Scientific*; Medtronic*; PGxHealth†; St. Jude Medical Inc.* |
Royalty payments from Mayo Clinic from the licensing of technology to PGxHealth for their FAMILION genetic tests† |
| Marjorie Funk | Yale University |
Philips Healthcare* | None | GE HealthCare*; Philips Healthcare* |
None | None | None | None |
| W. Brian Gibler |
University of Cincinnati College of Medicine |
Abbott POC/i-STAT†; Bristol-Myers Squibb†; Sanofi-Aventis†; Schering Plough† |
None | None | None | Inovise*; Siloam* |
ArgiNOx*; Astellas*; AstraZeneca*; Daiichi Sankyo/ Lilly*; HeartScape Technologies*; Schering Plough*; Sanofi-Aventis/ Bristol-Myers Squibb* |
Unrestricted educational grant support from Abbott POC/i-STAT†; ArgiNOx†; Biosite†; Bristol-Myers Squibb†; Daiichi Sankyo/Lilly†; Inovise†; The Medicines Co†; Millennium Pharmaceuticals, Inc†; PDL BioPharma†; Roche Diagnostics†; Sanofi- Aventis†; Scios† |
| Paul Kligfield | Cornell Medical Center |
None | GE Healthcare*; Mortara Instrument*; Philips Medical* |
None | None | None | Cardiac Science*; MDS Pharma* |
None |
| Venu Menon | Cleveland Clinic Foundation |
None | None | Roche Datascope* |
None | None | Medicure* | None |
| George J. Philippides |
Boston University Medical Center |
None | None | None | None | None | None | None |
| Dan M. Roden |
Vanderbilt University School of Medicine |
None | None | St. Jude* | None | None | Adolor*; ARCA*; AstraZeneca*; Avanir*; Cardiome*; CardioDx*; Eli Lilly*; Novartis†; Ortho Diagnostics*; Sanofi* |
Patent payment (royalty) from Vanderbilt/Clinical Data (formerly Genaissance)* |
| Wojciech Zareba |
University of Rochester |
None | None | None | None | None | Biogen*; Durect*; Genzyme†; Cardiac Technologies† (offshoot of University of Rochester) |
None |
This table represents the relationships of writing group members that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all members of the writing group are required to complete and submit. A relationship is considered to be “significant” if 1) the person receives $10 000 or more during any 12-month period or 5% or more of the person’s gross income; or 2) the person owns 5% or more of the voting stock or share of the entity or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Significant.
Reviewer Disclosures
| Reviewer | Employment | Research Grant | Other Research Support |
Speakers’ Bureau/Honoraria |
Expert Witness | Ownership Interest | Consultant/ Advisory Board |
Other |
|---|---|---|---|---|---|---|---|---|
| Eric R. Bates |
University of Michigan |
None | None | None | None | None | None | None |
| N.A. Mark Estes III |
New England Medical Center |
None | None | None | None | None | None | None |
| Leonard S. Gettes |
University of North Carolina at Chapel Hill |
None | None | None | None | None | Philips Electronics* |
None |
| Federico Gentile |
Centro Medico Diagnostico |
None | None | None | None | None | None | None |
| Robert A. Harrington |
Duke University |
None | None | None | None | None | None | None |
| Barry J. Maron |
Minneapolis Heart Institute Foundation |
None | None | None | None | None | None | None |
| Debabrata Mukherjee |
University of Kentucky |
Abbott Vascular*; Schering-Plough* |
None | None | None | None | None | None |
| Robert S. Rosenson |
University of Michigan |
Abbott†; Anthera†; AstraZeneca† |
None | Abbott†; AstraZeneca†; Daiichi Sankyo† |
None | LipoScience† | AstraZeneca†; Abbott*; Anthera*; Daiichi Sankyo*; LipoScience*; Novo Nordisk*; Roche† |
None |
| Andrea M. Russo |
Cooper University |
Medtronic*; St. Jude*; Biotronic*; Boston Scientific* |
Medtronic*; St. Jude*; Boston Scientific* |
|||||
| Melvin Scheinman |
University of California, San Francisco |
None | None | None | None | None | None | None |
| Kathryn Wood |
Duke University |
None | None | None | None | None | None | None |
This table represents the relationships of reviewers that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all reviewers are required to complete and submit. A relationship is considered to be “significant” if 1) the person receives $10 000 or more during any 12-month period or 5% or more of the person’s gross income; or 2) the person owns 5% or more of the voting stock or share of the entity or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Significant.
Footnotes
The American Heart Association and the American College of Cardiology Foundation make every effort to avoid any actual or potential conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the writing panel. Specifically, all members of the writing group are required to complete and submit a Disclosure Questionnaire showing all such relationships that might be perceived as real or potential conflicts of interest.
Expert peer review of AHA Scientific Statements is conducted at the AHA National Center. For more on AHA statements and guidelines development, visit http://www.americanheart.org/presenter.jhtml?identifier=3023366.
Permissions: Multiple copies, modification, alteration, enhancement, and/or distribution of this document are not permitted without the express permission of the American Heart Association. Instructions for obtaining permission are located at http://www.americanheart.org/presenter.jhtml?identifier=4431. A link to the “Permission Request Form” appears on the right side of the page.
References
- 1.Dessertenne F. La tachycardie ventriculaire á deux foyers opposes variables. Arch Mal Coeur Vaiss. 1966;59:263–272. [PubMed] [Google Scholar]
- 2.Kay GN, Plumb VJ, Arciniegas JG, Henthorn RW, Waldo AL. Torsade de pointes: the long-short initiating sequence and other clinical features: observations in 32 patients. J Am Coll Cardiol. 1983;2:806–817. doi: 10.1016/s0735-1097(83)80226-5. [DOI] [PubMed] [Google Scholar]
- 3.Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A, Jr, Robinson JL, Benhorin J, Choi S. The long QT syndrome: prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 4.Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH, Hall WJ. Influence of genotype on the clinical course of the long-QT syndrome: International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 6.Sauer AJ, Moss AJ, McNitt S, Peterson DR, Zareba W, Robinson JL, Qi M, Goldenberg I, Hobbs JB, Ackerman MJ, Benhorin J, Hall WJ, Kaufman ES, Locati EH, Napolitano C, Priori SG, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–337. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 7.Roden DM, Woosley RL, Primm RK. Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am Heart J. 1986;111:1088–1093. doi: 10.1016/0002-8703(86)90010-4. [DOI] [PubMed] [Google Scholar]
- 8.Woosley RL, Chen Y, Freiman JP, Gillis RA. Mechanism of the cardiotoxic actions of terfenadine. JAMA. 1993;269:1532–1536. [PubMed] [Google Scholar]
- 9.De Bruin ML, Langendijk PN, Koopmans RP, Wilde AA, Leufkens HG, Hoes AW. In-hospital cardiac arrest is associated with use of non-antiarrhythmic QTc-prolonging drugs. Br J Clin Pharmacol. 2007;63:216–223. doi: 10.1111/j.1365-2125.2006.02722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hondeghem LM. Thorough QT/QTc not so thorough: removes torsadogenic predictors from the T-wave, incriminates safe drugs, and misses profibrillatory drugs. J Cardiovasc Electrophysiol. 2006;17:337–340. doi: 10.1111/j.1540-8167.2006.00347.x. [DOI] [PubMed] [Google Scholar]
- 11.Topilski I, Rogowski O, Rosso R, Justo D, Copperman Y, Glikson M, Belhassen B, Hochenberg M, Viskin S. The morphology of the QT interval predicts torsade de pointes during acquired bradyarrhythmias. J Am Coll Cardiol. 2007;49:320–328. doi: 10.1016/j.jacc.2006.08.058. [DOI] [PubMed] [Google Scholar]
- 12.Kawabata M, Hirao K, Takeshi S, Sakurai K, Inagaki H, Hachiya H, Isobe M. Torsades de pointes related to transient marked QT prolongation following successful emergent percutaneous coronary intervention for acute coronary syndrome. J Electrocardiol. 2008;41:117–122. doi: 10.1016/j.jelectrocard.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, Volders PG, Vos MA. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation. 2004;110:2453–2459. doi: 10.1161/01.CIR.0000145162.64183.C8. [DOI] [PubMed] [Google Scholar]
- 14.Zareba W, Moss AJ, le Cessie S, Hall WJ. T wave alternans in idiopathic long QT syndrome. J Am Coll Cardiol. 1994;23:1541–1546. doi: 10.1016/0735-1097(94)90653-x. [DOI] [PubMed] [Google Scholar]
- 15.Zareba W. Drug induced QT prolongation. Cardiol J. 2007;14:523–533. [PubMed] [Google Scholar]
- 16.Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsades de pointes in the long-QT syndrome. Circulation. 2002;105:1247–1253. doi: 10.1161/hc1002.105231. [DOI] [PubMed] [Google Scholar]
- 17.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003;24:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 19.El-Sherif N, Chinushi M, Caref EB, Restivo M. Electrophysiological mechanism of the characteristic electrocardiographic morphology of torsade de pointes tachyarrhythmias in the long-QT syndrome: detailed analysis of ventricular tridimensional activation patterns. Circulation. 1997;96:4392–14399. doi: 10.1161/01.cir.96.12.4392. [DOI] [PubMed] [Google Scholar]
- 20.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 22.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Effect of clinical phenotype on yield of long QT syndrome genetic testing. J Am Coll Cardiol. 2006;47:764–768. doi: 10.1016/j.jacc.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 23.Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, Hohnloser SH, Shimizu W, Schwartz PJ, Stanton M, Murray KT, Norris K, Gerorge AL, Jr, Roden DM. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 24.Lehtonen A, Fodstad H, Laitinen-Forsblom P, Toivonen L, Kontula K, Swan H. Further evidence of inherited long QT syndrome gene mutations in antiarrhythmic drug-associated torsades de pointes. Heart Rhythm. 2007;4:603–607. doi: 10.1016/j.hrthm.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 25.Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, Schulze-Bahr E, Haverkamp W, Breithardt G, Cohen N, Aerssens J. Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J Mol Med. 2004;82:182–188. doi: 10.1007/s00109-003-0522-z. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgerald PT, Ackerman MJ. Drug-induced torsades de pointes: the evolving role of pharmacogenetics. Heart Rhythm. 2005;2(suppl):S30–S37. doi: 10.1016/j.hrthm.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 27.Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, Cappuccio FP, Sagnella GA, Kass RS, Keating MT. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–1336. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 28.Burke A, Creighton W, Mont E, Li L, Hogan S, Kutys R, Fowler D, Virmani R. Role of SCN5A Y1102 polymorphism in sudden cardiac death in blacks. Circulation. 2005;112:798–802. doi: 10.1161/CIRCULATIONAHA.104.482760. [DOI] [PubMed] [Google Scholar]
- 29.Plant LD, Bowers PN, Liu QY, Morgan T, Zhang T, State MW, Chen W, Kittles RA, Goldstein SA. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest. 2006;116:430–435. doi: 10.1172/JCI25618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Norstrand DW, Tester DJ, Ackerman MJ. Overrepresentation of the proarrhythmic, sudden death predisposing sodium channel polymorphism S1103Y in a population-based cohort of African-American sudden infant death syndrome. Heart Rhythm. 2008;5:712–715. doi: 10.1016/j.hrthm.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 32.Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479–1487. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases? The intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1120–1121. doi: 10.1046/j.1540-8167.2003.03339.x. [DOI] [PubMed] [Google Scholar]
- 34.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C, Smith SC, Jr, Jacobs AK, Adams CD, Antman EM, Anderson JL, Hunt SA, Halperin JL, Nishimura R, Ornato JP, Page RL, Riegel B, Priori SG, Blanc JJ, Budaj A, Camm AJ, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo JL, Zamorano JL. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) J Am Coll Cardiol. 2006;48:e247–e346. doi: 10.1016/j.jacc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Lown B, Wolf M. Approaches to sudden death from coronary heart disease. Circulation. 1971;44:130–142. doi: 10.1161/01.cir.44.1.130. [DOI] [PubMed] [Google Scholar]
- 36.Wald RW, Waxman MB, Colman JM. Torsades de pointes ventricular tachycardia: a complication of disopyramide shared with quinidine. J Electrocardiol. 1981;14:301–307. doi: 10.1016/s0022-0736(81)80013-1. [DOI] [PubMed] [Google Scholar]
- 37.Chow MJ, Piergies AA, Bowsher DJ, Murphy JJ, Kushner W, Ruo TI, Asada A, Talano JV, Atkinson AJ., Jr Torsade de pointes induced by N-acetylprocainamide. J Am Coll Cardiol. 1984;4:621–624. doi: 10.1016/s0735-1097(84)80111-4. [DOI] [PubMed] [Google Scholar]
- 38.Soyka LF, Wirtz C, Spangenberg RB. Clinical safety profile of sotalol in patients with arrhythmias. Am J Cardiol. 1990;65:74A–81A. doi: 10.1016/0002-9149(90)90207-h. [DOI] [PubMed] [Google Scholar]
- 39.Torp-Pedersen C, Møller M, Bloch-Thomsen PE, Køber L, Sandøe E, Egstrup K, Agner E, Carlsen J, Videbaek J, Marchant B, Camm AJ, Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group Dofetilide in patients with congestive heart failure and left ventricular dysfunction. N Engl J Med. 1999;341:857–865. doi: 10.1056/NEJM199909163411201. [DOI] [PubMed] [Google Scholar]
- 40.Murray KT. Ibutilide. Circulation. 1998;97:493–497. doi: 10.1161/01.cir.97.5.493. [DOI] [PubMed] [Google Scholar]
- 41.Krantz MJ, Lewkowiez L, Hays H, Woodroffe MA, Robertson AD, Mehler PS. Torsade de pointes associated with very-high-dose methadone. Ann Intern Med. 2002;137:501–504. doi: 10.7326/0003-4819-137-6-200209170-00010. [DOI] [PubMed] [Google Scholar]
- 42.Hennessy S, Bilker WB, Knauss JS, Margolis DJ, Kimmel SE, Reynolds RF, Glasser DB, Morrison MF, Strom BL. Cardiac arrest and ventricular arrhythmia in patients taking antipsychotic drugs: cohort study using administrative data. BMJ. 2002;325:1070. doi: 10.1136/bmj.325.7372.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilt JL, Minnema AM, Johnson RF, Rosenblum AM. Torsade de pointes associated with the use of intravenous haloperidol. Ann Intern Med. 1993;119:391–394. doi: 10.7326/0003-4819-119-5-199309010-00007. [DOI] [PubMed] [Google Scholar]
- 44.Honig PK, Wortham DC, Zamani K, Conner DP, Mullin JC, Cantilena LR. Terfenadine-ketoconazole interaction: pharmacokinetic and electrocardiographic consequences. JAMA. 1993;269:1513–1518. [PubMed] [Google Scholar]
- 45.Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr: implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:407–411. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]
- 46.Pinto YM, Van Gelder IC, Heeringa M, Crijns HJ. QT lengthening and life-threatening arrhythmias associated with fexofenadine. Lancet. 1999;353:980. doi: 10.1016/s0140-6736(99)01009-0. [DOI] [PubMed] [Google Scholar]
- 47.Gitler B, Berger LS, Buffa SD. Torsades de pointes induced by erythromycin. Chest. 1994;105:368–372. doi: 10.1378/chest.105.2.368. [DOI] [PubMed] [Google Scholar]
- 48.Von Bahr C, Movin G, Nordin C, Lidén A, Hammarlund-Udenaes M, Hedberg A, Ring H, Sjöqvist F. Plasma levels of thioridazine and metabolites are influenced by the debrisoquin hydroxylation phenotype. Clin Pharmacol Ther. 1991;49:234–240. doi: 10.1038/clpt.1991.22. [DOI] [PubMed] [Google Scholar]
- 49.Krantz MJ, Martin J, Stimmel B, Mehta D, Haigney MC. QTc interval screening in methadone treatment. Ann Intern Med. 2009;150:387–395. doi: 10.7326/0003-4819-150-6-200903170-00103. [DOI] [PubMed] [Google Scholar]
- 50.Tarabar AF, Hoffman RS, Nelson LS. Citalopram overdose: late presentation of torsades de pointes (TdP) with cardiac arrest. J Med Toxicol. 2008;4:101–105. doi: 10.1007/BF03160963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chung KJ, Wang YC, Liu BM, Supernaw RB. Management of ventricular dysrhythmia secondary to trazadone overdose. J Emerg Med. 2008;35:171–174. doi: 10.1016/j.jemermed.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 52.Ray WA, Chung CP, Murray KT, Hall K, Stein CM. Atypical antipsychotic drugs and the risk of sudden cardiac death [published correction appears in N Engl J Med. 2009;361:1814] N Engl J Med. 2009;360:225–235. doi: 10.1056/NEJMoa0806994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazzara R. Amiodarone and torsade de pointes. Ann Intern Med. 1989;111:549–551. doi: 10.7326/0003-4819-111-7-549. [DOI] [PubMed] [Google Scholar]
- 54.Mason JW, Hondeghem LM, Katzung BG. Block of inactivated sodium channels and of depolarization-induced automaticity in guinea pig papillary muscle by amiodarone. Circ Res. 1984;55:278–285. doi: 10.1161/01.res.55.3.278. [DOI] [PubMed] [Google Scholar]
- 55.Yang T, Snyders D, Roden DM. Drug block of I(kr): model systems and relevance to human arrhythmias. J Cardiovasc Pharmacol. 2001;38:737–744. doi: 10.1097/00005344-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 56.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, DiDiego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- 58.Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, Molhoek P, Verheugt FW, Gersh BJ, McCabe CH, Braunwald E. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non–ST-segment–elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non–ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 59.Carlsson L, Abrahamsson C, Andersson B, Duker G, Schiller-Linhardt G. Proarrhythmic effects of the class III agent almokalant: importance of infusion rate, QT dispersion, and early afterdepolarisations. Cardiovasc Res. 1993;27:2186–2193. doi: 10.1093/cvr/27.12.2186. [DOI] [PubMed] [Google Scholar]
- 60.Nuttall GA, Eckerman KM, Jacob KA, Pawlaski EM, Wigersma SK, Marienau ME, Oliver WC, Narr BJ, Ackerman MJ. Does low-dose droperidol administration increase the risk of drug-induced QT prolongation and torsade de pointes in the general surgical population? Anesthesiology. 2007;107:531–536. doi: 10.1097/01.anes.0000281893.39781.64. [DOI] [PubMed] [Google Scholar]
- 61.Fenichel RR, Malik M, Antzelevitch C, Sanguinetti M, Roden DM, Priori SG, Ruskin JN, Lipicky RJ, Cantilena LR. Independent Academic Task Force. Drug-induced torsades de pointes and implications for drug development. J Cardiovasc Electrophysiol. 2004;15:475–495. doi: 10.1046/j.1540-8167.2004.03534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kannankeril PJ, Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol. 2007;22:39–43. doi: 10.1097/HCO.0b013e32801129eb. [DOI] [PubMed] [Google Scholar]
- 63.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 64.Zeltser D, Justo D, Halkin A, Prokhorov V, Heller K, Viskin S. Torsade de pointes due to noncardiac drugs: most patients have easily identifiable risk factors. Medicine (Baltimore) 2003;82:282–290. doi: 10.1097/01.md.0000085057.63483.9b. [DOI] [PubMed] [Google Scholar]
- 65.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 66.Mank-Seymour AR, Richmond JL, Wood LS, Reynolds JM, Fan YT, Warnes GR, Milos PM, Thompson JF. Association of torsades de pointes with novel and known single nucleotide polymorphisms in long QT syndrome genes. Am Heart J. 2006;152:1116–1122. doi: 10.1016/j.ahj.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 67.Modell SM, Lehmann MH. The long QT syndrome family of cardiac ion channelopathies: a HuGE review. Genet Med. 2006;8:143–155. doi: 10.1097/01.gim.0000204468.85308.86. [DOI] [PubMed] [Google Scholar]
- 68.Napolitano C, Schwartz PJ, Brown AM, Ronchetti E, Bianchi L, Pinnavaia A, Acquaro G, Priori SG. Evidence for a cardiac ion channel mutation underlying drug-induced QT prolongation and life-threatening arrhythmias. J Cardiovasc Electrophysiol. 2000;11:691–696. doi: 10.1111/j.1540-8167.2000.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 69.Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005;115:2025–2032. doi: 10.1172/JCI25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 71.Elming H, Brendorp B, Kober L, Sahebzadah N, Torp-Petersen C. QTc interval in the assessment of cardiac risk. Card Electrophysiol Rev. 2002;6:289–294. doi: 10.1023/a:1016345412555. [DOI] [PubMed] [Google Scholar]
- 72.Houltz B, Darpö B, Edvardsson N, Blomström P, Brachmann J, Crijns HJ, Jensen SM, Svernhage E, Vallin H, Swedberg K. Electrocardiographic and clinical predictors of torsades de pointes induced by almokalant infusion in patients with chronic atrial fibrillation or flutter: a prospective study. Pacing Clin Electrophysiol. 1998;21:1044–1057. doi: 10.1111/j.1540-8159.1998.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 73.Pedersen HS, Elming H, Seibaek M, Burchardt H, Brendorp B, Torp-Pedersen C, Køber L. Risk factors and predictors of torsade de pointes ventricular tachycardia in patients with left ventricular systolic dysfunction receiving Dofetilide. Am J Cardiol. 2007;100:876–880. doi: 10.1016/j.amjcard.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 74.Singh BN. Safety profile of bepridil determined from clinical trials in chronic stable angina in the United States. Am J Cardiol. 1992;69:68D–74D. doi: 10.1016/0002-9149(92)90962-x. [DOI] [PubMed] [Google Scholar]
- 75.Aström-Lilja C, Odeberg JM, Ekman E, Hägg S. Drug-induced torsades de pointes: a review of the Swedish pharmacovigilance database. Pharmacoepidemiol Drug Saf. 2008;17:587–592. doi: 10.1002/pds.1607. [DOI] [PubMed] [Google Scholar]
- 76.Justo D, Zeltser D. Torsades de pointes induced by antibiotics. Eur J Intern Med. 2006;17:254–259. doi: 10.1016/j.ejim.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 77.Shaffer D, Singer S, Korvick J, Honig P. Concomitant risk factors in reports of torsades de pointes associated with macrolide use: review of the United States Food and Drug Administration Adverse Event Reporting System. Clin Infect Dis. 2002;35:197–200. doi: 10.1086/340861. [DOI] [PubMed] [Google Scholar]
- 78.LaPointe NM Allen, Curtis LH, Chan KA, Kramer JM, Lafata JE, Gurwitz JH, Raebel MA, Platt R. Frequency of high-risk use of QT-prolonging medications. Pharmacoepidemiol Drug Saf. 2006;15:361–368. doi: 10.1002/pds.1155. [DOI] [PubMed] [Google Scholar]
- 79.Justo D, Zeltser D. Torsade de pointes induced by systemic antifungal agents: lessons from a retrospective analysis of published case reports. Mycoses. 2006;49:463–470. doi: 10.1111/j.1439-0507.2006.01278.x. [DOI] [PubMed] [Google Scholar]
- 80.Roe CM, Odell KW, Henderson RR. Concomitant use of antipsychotics and drugs that may prolong the QT interval. J Clin Psychopharmacol. 2003;23:197–200. doi: 10.1097/00004714-200304000-00013. [DOI] [PubMed] [Google Scholar]
- 81.Drici MD, Clément N. Is gender a risk factor for adverse drug reactions? The example of drug-induced long QT syndrome. Drug Saf. 2001;24:575–585. doi: 10.2165/00002018-200124080-00002. [DOI] [PubMed] [Google Scholar]
- 82.Drici MD, Knollmann BC, Wang WX, Woosley RL. Cardiac actions of erythromycin: influence of female sex. JAMA. 1998;280:1774–1776. doi: 10.1001/jama.280.20.1774. [DOI] [PubMed] [Google Scholar]
- 83.Ebert SN, Liu XK, Woosley RL. Female gender as a risk factor for drug-induced cardiac arrhythmias: evaluation of clinical and experimental evidence. J Womens Health. 1998;7:547–557. doi: 10.1089/jwh.1998.7.547. [DOI] [PubMed] [Google Scholar]
- 84.Justo D, Prokhorov V, Heller K, Zeltser D. Torsade de pointes induced by psychotropic drugs and the prevalence of its risk factors. Acta Psychiatr Scand. 2005;111:171–176. doi: 10.1111/j.1600-0447.2004.00469.x. [DOI] [PubMed] [Google Scholar]
- 85.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 86.Yasuda M, Nakazato Y, Sasaki A, Kawano Y, Nakazato K, Tokano T, Daida H. Clinical evaluation of adverse effects during bepridil administration for atrial fibrillation and flutter. Circ J. 2006;70:662–666. doi: 10.1253/circj.70.662. [DOI] [PubMed] [Google Scholar]
- 87.Roden DM. Early after-depolarizations and torsade de pointes: implications for the control of cardiac arrhythmias by prolonging repolarization. Eur Heart J. 1993;14(suppl H):56–61. doi: 10.1093/eurheartj/14.suppl_h.56. [DOI] [PubMed] [Google Scholar]
- 88.Eriksson JW, Carlberg B, Hillörn V. Life-threatening ventricular tachycardia due to liquorice-induced hypokalaemia. J Intern Med. 1999;245:307–310. doi: 10.1046/j.1365-2796.1999.00476.x. [DOI] [PubMed] [Google Scholar]
- 89.Roden DM, Iansmith DH. Effects of low potassium or magnesium concentrations on isolated cardiac tissue. Am J Med. 1987;82:18–23. doi: 10.1016/0002-9343(87)90128-8. [DOI] [PubMed] [Google Scholar]
- 90.Choy AM, Lang CC, Chomsky DM, Rayos GH, Wilson JR, Roden DM. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation. 1997;96:2149–2154. doi: 10.1161/01.cir.96.7.2149. [DOI] [PubMed] [Google Scholar]
- 91.Keren A, Tzivoni D. Torsades de pointes: prevention and therapy. Cardiovasc Drugs Ther. 1991;5:509–513. doi: 10.1007/BF03029778. [DOI] [PubMed] [Google Scholar]
- 92.White CM, Xie J, Chow MS, Kluger J. Prophylactic magnesium to decrease the arrhythmogenic potential of class III antiarrhythmic agents in a rabbit model. Pharmacotherapy. 1999;19:635–640. doi: 10.1592/phco.19.8.635.31528. [DOI] [PubMed] [Google Scholar]
- 93.Lüderitz B, Manz M. The value of magnesium in intensive care medicine [in German] Z Kardiol. 1994;83(suppl 6):121–126. [PubMed] [Google Scholar]
- 94.Nakasone H, Sugama R, Sakugawa H, Matayoshi R, Miyagi T, Maeshiro T, Yamashiro T, Higa F, Hokama A, Kinjo F, Saito A, Toda T. Alcoholic liver cirrhosis complicated with torsade de pointes during plasma exchange and hemodiafiltration. J Gastroenterol. 2001;36:564–568. doi: 10.1007/s005350170061. [DOI] [PubMed] [Google Scholar]
- 95.Daya SK, Gowda RM, Khan IA. Ciprofloxacin- and hypocalcemia-induced torsade de pointes triggered by hemodialysis. Am J Ther. 2004;11:77–79. doi: 10.1097/00045391-200401000-00014. [DOI] [PubMed] [Google Scholar]
- 96.Akiyama T, Batchelder J, Worsman J, Moses HW, Jedlinski M. Hypocalcemic torsades de pointes. J Electrocardiol. 1989;22:89–92. doi: 10.1016/0022-0736(89)90026-5. [DOI] [PubMed] [Google Scholar]
- 97.Fiset C, Drolet B, Hamelin BA, Turgeon J. Block of IKs by the diuretic agent indapamide modulates cardiac electrophysiological effects of the class III antiarrhythmic drug dl-sotalol. J Pharmacol Exp Ther. 1997;283:148–156. [PubMed] [Google Scholar]
- 98.Díiaz-Castro O, Puchol A, Almendral J, Torrecilla EG, Arenal A, Martínez-Selles M. Predictors of in-hospital ventricular fibrillation or torsades de pointes in patients with acute symptomatic bradycardia. J Electrocardiol. 2004;37:55–60. doi: 10.1016/j.jelectrocard.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 99.Raviña T, Raviña P, Gutierrez J. Acquired long QT syndrome: risperidone-facilitated triggered activity and torsades de pointes during complete AV block: I. Int J Cardiol. 2007;116:416–420. doi: 10.1016/j.ijcard.2006.04.084. [DOI] [PubMed] [Google Scholar]
- 100.Darbar D, Kimbrough J, Jawaid A, McCray R, Ritchie MD, Roden DM. Persistent atrial fibrillation is associated with reduced risk of torsades de pointes in patients with drug-induced long QT syndrome. J Am Coll Cardiol. 2008;51:836–842. doi: 10.1016/j.jacc.2007.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davey P. How to correct the QT interval for the effects of heart rate in clinical studies. J Pharmacol Toxicol Methods. 2002;48:3–9. doi: 10.1016/S1056-8719(03)00008-X. [DOI] [PubMed] [Google Scholar]
- 102.Funck-Brentano C, Jaillon P. Rate-corrected QT interval: techniques and limitations. Am J Cardiol. 1993;72:17B–22B. doi: 10.1016/0002-9149(93)90035-b. [DOI] [PubMed] [Google Scholar]
- 103.Batchvarov V, Malik M. Individual patterns of QT/RR relationship. Card Electrophysiol Rev. 2002;6:282–288. doi: 10.1023/a:1016393328485. [DOI] [PubMed] [Google Scholar]
- 104.Helfenbein ED, Zhou SH, Lindauer JM, Field DQ, Gregg RE, Wang JJ, Kresge SS, Michaud FP. An algorithm for continuous real-time QT interval monitoring. J Electrocardiol. 2006;39(suppl):S123–S127. doi: 10.1016/j.jelectrocard.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 105.Sommargren C, Drew BJ. Preventing torsades de pointes by careful cardiac monitoring in hospital settings. AACN Adv Crit Care. 2007;18:285–293. doi: 10.4037/15597768-2007-3007. [DOI] [PubMed] [Google Scholar]
- 106.Postema PG, De Jong JS, Van der Bilt IA, Wilde AA. Accurate electrocardiographic assessment of the QT interval: teach the tangent. Heart Rhythm. 2008;5:1015–1018. doi: 10.1016/j.hrthm.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 107.Viskin S, Rosovski U, Sands AJ, Chen E, Kistler PM, Kalman JM, Chavez L Rodriguez, Torres P Iturralde, Cruz FFE, Centurión OA, Fujiki A, Maury P, Chen X, Krahn AD, Roithinger F, Zhang L, Vincent GM, Zeltser D. Inaccurate electrocardiographic interpretation of long QT: the majority of physicians cannot recognize a long QT when they see one. Heart Rhythm. 2005;2:569–574. doi: 10.1016/j.hrthm.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 108.Kligfield P, Gettes LS, Bailey JJ, Childers R, Deal BJ, Hancock EW, van Herpen G, Kors JA, Macfarlane P, Mirvis DM, Pahlm O, Rautaharju P, Wagner GS. Recommendations for the standardization and interpretation of the electrocardiogram: part I: the electrocardiogram and its technology: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology; and the Heart Rhythm Society. Circulation. 2007;115:1306–1324. doi: 10.1161/CIRCULATIONAHA.106.180200. [DOI] [PubMed] [Google Scholar]
- 109.Kligfield P, Hancock EW, Helfenbein ED, Dawson EJ, Cook MA, Lindauer JM, Zhou SH, Xue J. Relation of QT interval measurements to evolving automated algorithms from different manufacturers of electrocardiographs. Am J Cardiol. 2006;98:88–92. doi: 10.1016/j.amjcard.2006.01.060. [DOI] [PubMed] [Google Scholar]
- 110.Drew BJ, Califf RM, Funk M, Kaufman ES, Krucoff MW, Laks MM, Macfarlane PW, Sommargren C, Swiryn S, Van Hare GF. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young. Circulation. 2004;110:2721–2746. doi: 10.1161/01.CIR.0000145144.56673.59. [DOI] [PubMed] [Google Scholar]
- 111.Drew BJ, Funk M. Practice standards for ECG monitoring in hospital settings: executive summary and guide for implementation. Crit Care Nurs Clin North Am. 2006;18:157–168. doi: 10.1016/j.ccell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 112.Kowey PR, Vanderlugt JT, Luderer JR. Safety and risk/benefit analysis of ibutilide for acute conversion of atrial fibrillation/flutter. Am J Cardiol. 1996;78(8A):46–52. doi: 10.1016/s0002-9149(96)00566-8. [DOI] [PubMed] [Google Scholar]
- 113.Banai S, Tzivoni D. Drug therapy for torsade de pointes. J Cardiovasc Electrophysiol. 1993;4:206–210. doi: 10.1111/j.1540-8167.1993.tb01224.x. [DOI] [PubMed] [Google Scholar]
- 114.Pinski SL, Eguía LE, Trohman RG. What is the minimal pacing rate that prevents torsades de pointes? Insights from patients with permanent pacemakers. Pacing Clin Electrophysiol. 2002;25:1612–1615. doi: 10.1046/j.1460-9592.2002.01612.x. [DOI] [PubMed] [Google Scholar]