

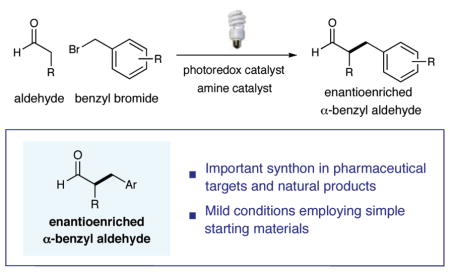
Abstract
The first enantioselective aldehyde α-benzylation using electron-deficient aryl and heteroaryl substrates has been accomplished. The productive merger of a chiral imidazolidinone organocatalyst and a commercially available iridium photoredox catalyst in the presence of household fluorescent light directly affords the desired homobenzylic stereogenicity in good to excellent yield and enantioselectivity. The utility of this methodology has been demonstrated via rapid access to an enantioen-riched drug target for angiogenesis suppression.
The benzylic alkylation of chiral enolates has become a mainstay transformation in chemical synthesis, mainly due to the seminal research efforts of Evans, Oppolzer, Seebach, and Myers.1 Surprisingly, however, catalytic enantioselective variants of this venerable reaction remain confined to a small but valuable group of substrate types. Phase transfer benzylation of glycine-derived imines,2 chiral triamine ligation of ketone-derived lithium enolates,3 and the use of Jacobsen’s Cr(salen) complex with preformed stannous enolates4 are among the most successful examples.5 Direct organocatalytic methods to form α-benzylated aldehydes have also been reported wherein the electrophilic partner requires multiple and/or electron-rich aryl rings (to enable the intermediate formation of stabilized benzylic carbocations).6 Recently, our laboratory introduced a new mode of activation termed photoredox organocatalysis,7,8 the mechanistic foundation of which relies on the propensity of electrophilic radicals (derived from photocatalytic reduction of alkyl halides) to combine with facially biased enamines (derived from aldehydes and a chiral amine catalyst). Inspired by this strategy, we hypothesized that the enantioselective α-benzylation of aldehydes might also be possible via the marriage of these activation pathways. Herein, we present a new direct and enantioselective catalytic protocol for the α-alkylation of aldehydes, a transformation that is successful with a wide array of simple monoaryl9 and monoheteroaryl methylene halides.
Design Plan
As detailed in Figure 1, it has been established that commercially available fac-Ir(ppy)3 (1) (ppy = 2-phenylpyridine), commonly used as a green triplet emitter in OLEDs, can readily accept a photon from weak fluorescent light to generate the strong reductant excited state fac-*Ir(ppy)3 (2) (E1/2 = −1.73 V vs saturated calomel electrode (SCE) in CH3CN).10 We hoped that single electron transfer (SET) from fac-*Ir(ppy)3 to a suitable benzylic bromide might render an aromatic radical anion that would rapidly undergo σ-bond cleavage to afford the bromide anion and an electrophilic benzyl radical 3. Within the same time frame, condensation of an aldehyde substrate with imidazolidinone catalyst 4 should form a highly π-nucleophilic enamine 5 which should then combine with the electron-deficient radical 3 to enantioselectively forge the crucial homobenzylic center. Rapid oxidation of the resultant α-amino radical11 6 by fac-Ir(ppy)3+ (7) (E1/2 = 0.77 V vs SCE in CH3CN)10 would then close the redox cycle while regenerating the photocatalyst fac-Ir(ppy)3 (1). Moreover, subsequent hydrolysis of iminium 8 would reconstitute the organocatalyst 4 while liberating the enantioenriched α-benzyl aldehyde adduct.
Figure 1.

Proposed catalytic cycle for aldehyde α-benzylation.
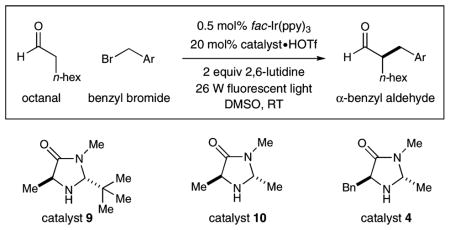
Evaluation of this tandem catalysis strategy was first examined with octanal, imidazolidinone catalyst 9, fac-Ir(ppy)3 (1), a 26 W compact fluorescent lamp, and 2,4-dinitrobenzyl bromide, benzyl bromide, or 4-(bromomethyl)pyridine as the alkylating reagent. As shown in Table 1, initial experiments revealed that the proposed benzylation was indeed possible; however, the coupling component was restricted to the most electron-deficient dinitro-aryl system (entry 2). Intriguingly, the use of benzyl bromide resulted in the complete recovery of starting materials12 while 4-(bromomethyl)-pyridine rapidly decomposed under photoredox conditions (entries 1, 5). These data suggest that electron-neutral benzylic halides do not readily undergo the initial reduction step, while the pyridinium radical is formed but does not couple with the enamine derived from amine 9. To address this latter limitation, we next examined amine catalysts of diminished steric demand with the presumption that a higher concentration and increased spatial exposure of the enamine intermediate should increase the rate of the critical radical-olefin addition step. Indeed, the pseudo-C2-symmetric catalyst 10 (wherein the tert-butyl substituent has been replaced by a methyl group) does enable the pyridinium methylene bromide to become a viable coupling partner, albeit with low selectivities (entry 6, 81% yield, 78% ee). Enantiocontrol is regained, however, when the benzyl, methyl disubstituted amine 4 is employed (entry 7, 90% ee), a catalyst that likely operates via the preferred E-enamine geometry with shielding of the π-nucleophilic Re-face by the benzylic moiety on the catalyst framework (DFT-5).13 The superior levels of scope, induction, and efficiency exhibited by the combination of catalysts 4 and fac-Ir(ppy)3 (1) in DMSO at 23 °C prompted us to select these conditions for further exploration.
Table 1.
Evaluation of Amine Catalyst for Benzylation
 | ||||
|---|---|---|---|---|
| entry | Ar | catalyst | % yielda | % eeb |
| 1 | Phenyl | 9 | 0 | ND |
| 2 | 2,4-(NO2)2C6H3 | 9 | 74 | 97 |
| 3 | 2,4-(NO2)2C6H3 | 10 | 93 | 82 |
| 4 | 2,4-(NO2)2C6H3 | 4 | 94 | 92 |
| 5c | 4-Pyridinyl | 9 | 0 | ND |
| 6c | 4-Pyridinyl | 10 | 81 | 78 |
| 7c | 4-Pyridinyl | 4 | 86 | 90 |
Isolated yields.
Enantiomeric excess determined by chiral HPLC.
4-(Bromomethyl)pyridine added as the hydrobromic acid salt with an additional equivalent of 2,6-lutidine.
We next performed a series of experiments to determine the scope of the aldehydic component in this asymmetric benzylation protocol. As revealed in Table 2, these mild redox conditions are compatible with a wide range of functional groups, including ethers, amines, imides, carbamates, and aromatic rings (72–91% yield, 87–90% ee). Moreover, significant variation in the steric demand of the aldehyde substituent can be accommodated without loss in enantiocontrol (entries 4 and 5, ≥73% yield, 90% ee).
Table 2.
Asymmetric Aldehyde α-Benzylation: Aldehyde Scope
 | |||||
|---|---|---|---|---|---|
| entry | producta | yield, eeb | entry | producta | yield, eeb |
| 1 |  |
86% yieldc 90% ee |
4 | 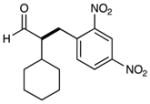 |
73% yield 90% ee |
| 2 | 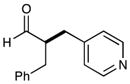 |
91%yieldc,d 90% ee |
5 | 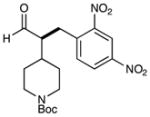 |
75% yield 90% ee |
| 3 | 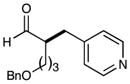 |
78% yieldc 87% ee |
6 | 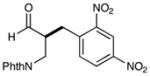 |
72% yieldd 90% ee |
Stereochemistry assigned by chemical correlation or by analogy.
Enantiomeric excess determined by chiral SFC or HPLC.
Substrate added as the hydrobromic acid salt with an additional equivalent of 2,6-lutidine. The free base organocatalyst was used.
Isolated yield of the corresponding alcohol.
We have found that a broad range of electron-deficient aryl and heteroaryl methylene bromides also participate in this enantioselective benzylation reaction (Table 3). For example, benzyl systems that incorporate a nitro-substituent with other electron-withdrawing groups such as nitriles and esters (Table 3, entries 1–3) are well-tolerated. Moreover the 1,2-nitro-fluoro aryl ring can serve to produce a suitably electrophilic radical without the intervention of SNAr byproducts (entry 3). Perhaps more notable with respect to medicinal agent synthesis, a large range of heteroaryl rings can be successfully employed such as pyridines, quinolines, benzimidazoles, pyrimidines, and triazines. As highlighted in entries 4 and 5, bromomethyl pyridines that have electron-donating substitution (entry 5, 2-methyl, 91% ee) or electron-withdrawing groups (entry 4, 5-nitro, 90% ee) are both competent in this enantioselective coupling. Moreover, fused bicycles such as 4-quinolinyl and 2-benzimidazolyl also perform well (81–90% yield, 82–88% ee, entries 6, 10). We postulate that, for substrates with a basic nitrogen (4-pyridinyl, and entries 5–6, 10),16 substrate protonation facilitates the initial reduction step17 and thereafter enhances the electrophilicity of the resulting radical intermediate. However, heterocycles containing two nitrogens, such as 2-pyrazinyl and 4-pyrimidinyl, and three nitrogens, such as 2-triazinyl—which all lack a basic nitrogen—also react with good efficiency and excellent enantioselectivity (68–78%, 87–91% ee, entries 7–9).18
Table 3.
Asymmetric Aldehyde α-Benzylation: Bromide Scope
 | |||||
|---|---|---|---|---|---|
| entry | producta | yield, eeb | entry | producta | yield, eeb |
| 1 | 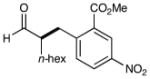 |
76% yield 93% ee |
6 | 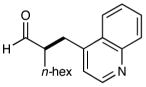 |
90% yielde 82% ee |
| 2 | 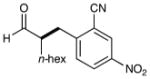 |
83% yield 90% ee |
7 | 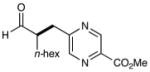 |
73% yieldf 90% ee |
| 3 | 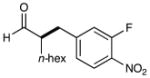 |
74%yieldc 90% ee |
8 | 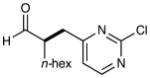 |
78% yield 87% ee |
| 4 | 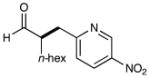 |
74% yieldd 90% ee |
9 | 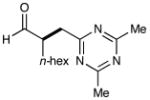 |
68% yieldg 91% ee |
| 5 | 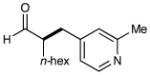 |
75%yielde 91% ee |
10 | 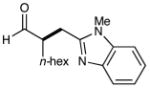 |
81% yielde,h 88% ee |
Stereochemistry assigned by chemical correlation or by analogy.
Enantiomeric excess determined by chiral SFC or HPLC.
30 mol% organocatalyst used.
Performed at 15 °C using Ru(bpy)3Cl2 as the photoredox catalyst; ref 14.
Substrate added as the hydrobromic acid salt with an additional equivalent of 2,6-lutidine. The free base organocatalyst was used.
Yield determined by 1H NMR.
Ir(dF(CF3)ppy)2(dtbbpy)PF6 was employed
as the photoredox catalyst; ref 15.
Isolated yield of the corresponding alcohol.
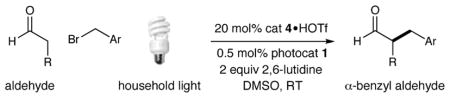
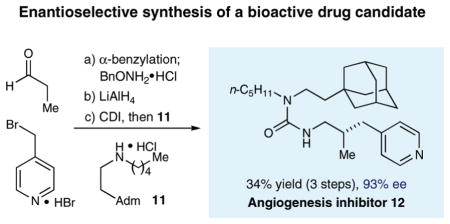
We fully expect that the α-heteroarylmethyl aldehyde products generated in this study will be of value for the generation and testing of medicinal agents. To highlight this possibility, we have applied our new catalytic enantioselective benzylation strategy to the synthesis of the angiogenesis inhibitor 12,19 a drug candidate that was developed for the treatment of diseases such as diabetic retinopathy and tumor proliferation. As illustrated above, exposure of propionaldehyde and 4-(bromomethyl)pyridine to our photoredox coupling protocol followed by in situ oxime formation yields the α-methylene pyridyl intermediate in 82% yield and 93% ee. Subsequent oxime reduction followed by urea coupling with amine 11 (available in two chemical steps) allows the rapid construction of angiogenesis inhibitor 12 in only three linear steps (34% overall yield, 93% ee).
In an effort to provide insight into the mechanistic details of this tandem catalysis coupling, we have performed several fluorescence quenching experiments (Stern–Volmer studies) with our fac-Ir(ppy)3 photoredox catalyst (Figure 2). Notably, fac-*Ir(ppy)3 oxidation of enamine 5 is not observed and, as such, cannot be the first step in the photocatalytic cycle (in contrast to previous studies from our laboratory that centered upon Ru(bpy)3Cl2).7 Instead, both aryl and heteroaryl methylene bromides display efficient quenching of the fac-*Ir(ppy)3 excited state, strongly suggesting that the electron transfer event occurs directly between fac-*Ir(ppy)3 and the aryl coupling partner. This is in accord with the catalytic cycle set out in Figure 1.
Figure 2.
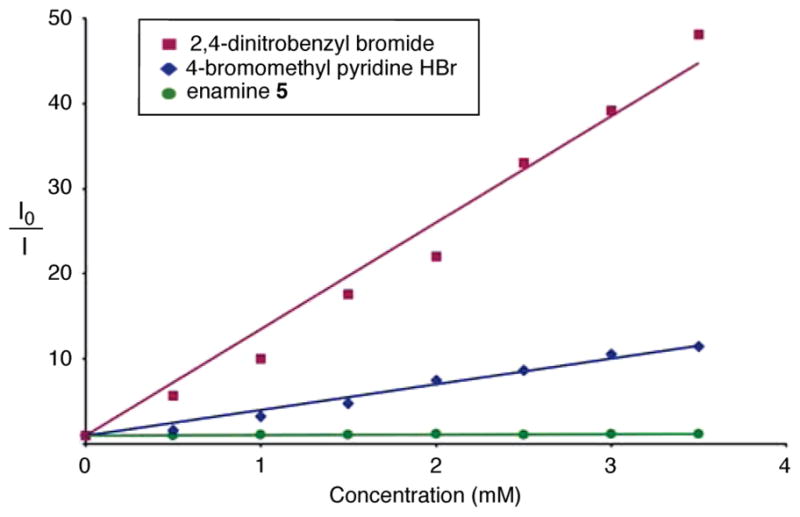
Fluorescence quenching by reaction substrates.
In conclusion, the first enantioselective aldehyde α-benzylation using electron-deficient aryl and heteroaryl substrates has been accomplished. The productive merger of a novel imidazolidinone organocatalyst and a commercially available iridium photoredox catalyst directly allows the stereocontrolled formation of homobenzylic stereogenicity in good to excellent yield. This new alkylation reaction, which exhibits broad scope and wide functional group tolerance, has been successfully utilized for the rapid and enantioselective construction of a previously developed angiogenesis inhibitor in only three linear chemical steps.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 01 GM093213-01), and kind gifts from Amgen and Abbott.
Footnotes
Supporting Information Available: Experimental procedures, structural proofs, and spectral data for all new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]; (b) Myers AG, Yang BH, Chen H, Gleason JL. J Am Chem Soc. 1994;116:9361. [Google Scholar]; (c) Seebach D, Wasmuth D. Helv Chim Acta. 1980;63:197. [Google Scholar]; (d) Oppolzer W, Moretti R, Thomi S. Tetrahedron Lett. 1989;30:5603. [Google Scholar]
- 2.(a) Maruoka K, Ooi T. Chem Rev. 2003;103:3013. doi: 10.1021/cr020020e. [DOI] [PubMed] [Google Scholar]; (b) Ooi T, Maruoka K. Angew Chem, Int Ed. 2007;46:4222. doi: 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]
- 3.Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. J Am Chem Soc. 1994;116:8829. [Google Scholar]
- 4.(a) Doyle AG, Jacobsen EN. J Am Chem Soc. 2005;127:62. doi: 10.1021/ja043601p. [DOI] [PubMed] [Google Scholar]; (b) Doyle AG, Jacobsen EN. Angew Chem, Int Ed. 2007;46:3701. doi: 10.1002/anie.200604901. [DOI] [PubMed] [Google Scholar]
- 5.An intramolecular enamine alkylation strategy has been demonstrated, see: Vignola N, List B. J Am Chem Soc. 2004;126:450. doi: 10.1021/ja0392566.
- 6.(a) Shaikh RR, Mazzanti A, Petrini M, Bartoli G, Melchiorre P. Angew Chem, Int Ed. 2008;47:8707. doi: 10.1002/anie.200803947. [DOI] [PubMed] [Google Scholar]; (b) Cozzi PG, Benfatti F, Zoli L. Angew Chem, Int Ed. 2009;48:1313. doi: 10.1002/anie.200805423. [DOI] [PubMed] [Google Scholar]; (c) Brown AR, Kuo WH, Jacobsen EN. J Am Chem Soc. 2010;132:9286. doi: 10.1021/ja103618r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For recent examples of photoredox catalysis, see: Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f.Koike T, Akita M. Chem Lett. 2009;38:166.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756. doi: 10.1021/ja9033582.Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875. doi: 10.1021/ja9053338.Du J, Yoon TP. J Am Chem Soc. 2009;131:14604. doi: 10.1021/ja903732v.Condie AG, González-Gómez JC, Stephenson CRJ. J Am Chem Soc. 2010;132:1464. doi: 10.1021/ja909145y.Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Org Lett. 2010;12:368. doi: 10.1021/ol902703k.Tucker JW, Nguyen JD, Narayanam JMR, Krabbe SW, Stephenson CRJ. Chem Commun. 2010;46:4985. doi: 10.1039/c0cc00981d.Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org Lett. 2010;12:3104. doi: 10.1021/ol101146f.Ischay MA, Lu Z, Yoon TP. J Am Chem Soc. 2010;132:8572. doi: 10.1021/ja103934y.
- 9.The racemic homolytic alkylation of stoichiometric enamines using p-nitrobenzyl chloride as radical precursor is known; see: Russell GA, Wang K. J Org Chem. 1991;56:3475.
- 10.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top Curr Chem. 2007;281:143. [Google Scholar]
- 11.α-Amino radicals possess oxidation potentials in the range of − 0.92 to −1.12 V vs SCE in CH3CN; see: Wayner DDM, Dannenberg JJ, Griller D. Chem Phys Lett. 1986;131:189.
- 12.We recognized from the outset that this strategy might not be successful with electron-neutral aryl systems due to the high reduction potential of systems such as benzyl bromide (E1/2 = −1.85 V vs SCE in CH3CN) and the nucleophilic nature of the unsubstituted benzylic radical. For benzyl bromide reduction potential, see: Koch DA, Henne BJ, Bartak DE. J Electrochem Soc. 1987;134:3062.For benzylic radical nucleophilicity, see: De Vleeschouwer F, Van Speybroeck V, Waroquier M, Geerlings P, De Proft F. Org Lett. 2007;9:2721. doi: 10.1021/ol071038k.
- 13.DFT calculations were performed with the use of B3LYP/6-311+G(2d,2p)//B3LYP/6-31G(d).
- 14.Reaction does proceed with Ir(III) catalyst, but with decreased yield. Ru(bpy)3Cl2 is a competent photoredox catalyst for these transformations but usually results in lower conversion; for photophysical properties, see: Kalyanasundaram K. Coord Chem Rev. 1982;46:159.
- 15.dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine; dtbbpy = 4,4′-di-tert-butyl-2,2′-dipyridyl; see: Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712.
- 16.These compounds have pKa values within 2 orders of magnitude of 2,6-lutidine, and the reduction potentials of the unprotonated species are too high to be reduced by the photocatalyst. For pKa values in DMSO, see: Bordwell FG. Acc Chem Res. 1988;21:456.For reduction potentials, see: Wiberg KB, Lewis TP. J Am Chem Soc. 1970;92:7154.
- 17.Thévenot D, Buvet R. J Electroanal Chem. 1972;39:429. [Google Scholar]
- 18.The parent bromomethyl-heterocycles lacking additional substitution (e.g., 2-(bromomethyl)pyrazine) are reported to be unstable; for pyrimidines, see: Brown DJ, Waring P. Aust J Chem. 1974;27:2251.For pyrazines, see: Sasaki K, Sugou K, Miyamoto K, Hirai J, Tsubouchi S, Miyasaka H, Itaya A, Kuroda Y. Org Biomol Chem. 2004;2:2852. doi: 10.1039/B408023H.For triazines, see: Von Angerer S. Sci Synth. 2003;17:449.
- 19.Matsuoka H, Nishimura K, Seike H, Aono H, Murai M. Preparation of pyridylalkylurea derivatives as angiogenesis inhibitors. 2008/0161270. US Patent. 2008 July 3;:A1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.