

Abstract
In hereditary hemochromatosis, mutations in HFE lead to iron overload through abnormally low levels of hepcidin. In addition, HFE potentially modulates cellular iron uptake by interacting with transferrin receptor, a crucial protein during erythropoiesis. However, the role of HFE in this process was never explored. We hypothesize that HFE modulates erythropoiesis by affecting dietary iron absorption and erythroid iron intake. To investigate this, we used Hfe-KO mice in conditions of altered dietary iron and erythropoiesis. We show that Hfe-KO mice can overcome phlebotomy-induced anemia more rapidly than wild-type mice (even when iron loaded). Second, we evaluated mice combining the hemochromatosis and β-thalassemia phenotypes. Our results suggest that lack of Hfe is advantageous in conditions of increased erythropoietic activity because of augmented iron mobilization driven by deficient hepcidin response. Lastly, we demonstrate that Hfe is expressed in erythroid cells and impairs iron uptake, whereas its absence exclusively from the hematopoietic compartment is sufficient to accelerate recovery from phlebotomy. In summary, we demonstrate that Hfe influences erythropoiesis by 2 distinct mechanisms: limiting hepcidin expression under conditions of simultaneous iron overload and stress erythropoiesis, and impairing transferrin-bound iron uptake by erythroid cells. Moreover, our results provide novel suggestions to improve the treatment of hemochromatosis.
Introduction
Iron metabolism and erythropoiesis are closely related. Delineating the mechanisms by which they cooperate and coregulate is crucial to understand a variety of vital physiologic functions. Erythropoiesis modulates iron absorption by participating in the regulation of hepcidin,1–3 a peptide hormone produced mainly in the liver.4,5 Hepcidin controls iron absorption and recycling by inducing internalization and degradation of ferroportin (Fpn1), the only known cellular iron exporter.6 Iron demand increases with erythropoietic activity, strongly down-regulating hepcidin expression1–3 and allowing for increased iron flow to the serum. In addition to being regulated by erythropoiesis, hepcidin expression also responds to hypoxia, iron levels, and inflammation.2 While erythropoiesis and hypoxia block hepcidin expression, inflammation and high iron levels increase it. Thus, modulation of this peptide hormone requires integration of opposing stimuli.7 Indeed, the effect of erythropoiesis can be partially impaired by iron overload and inflammation.7
HFE, the gene mutated in most cases of hereditary hemochromatosis (HH),8,9 is thought to participate in hepcidin regulation in response to iron.10 Iron overload in this disorder results from a failure to regulate iron absorption despite an increased iron load, leading to abnormal iron deposition in key organs if left untreated. Abnormally low hepcidin expression seems to be the major factor leading to iron overload in HH,11–13 although the exact molecular mechanism is still not fully understood. Association of Hfe and its partners, transferrin receptor-1 (Tfrc) and -2 (Tfr2), has been suggested to activate a regulatory pathway that controls hepcidin expression in response to serum iron levels.10,14 However, another study hypothesized that Hfe and Tfr2 modulate hepcidin by parallel pathways.15 Hfe has also been implicated in the regulation of transferring (Tf)-bound iron uptake. Several studies showed that Hfe competes with Tf for the same binding sites on Tfrc, thereby impairing iron uptake.16–19 Yet, despite all of the studies describing interaction of Hfe with Tfrc,19–21 few investigated their association within the erythroid compartment, where the latter is known to be essential.22 Earlier studies reported that Hfe could not be detected in nucleated erythroid cells23 and that liver-specific ablation of Hfe mirrors most of the HH phenotype, indicating that it plays a pivotal role in this organ.24 However, these studies did not include a thorough analysis of Hfe expression during erythroid maturation or directly investigate the effect of Hfe ablation on erythropoiesis. In contrast, several reports showed erythropoietic alterations in HH patients, including increased hemoglobin (Hb) levels, reticulocyte counts, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration.23,25,26 Additional observations demonstrated that the HFE promoter contains several binding sites for the erythroid transcription factor GATA-1, suggesting a direct role for HFE in erythropoiesis.27
We studied 2 potential functions of Hfe in erythropoiesis: (1) an indirect effect mediated by modulation of hepcidin expression under conditions of erythropoietic stress; and (2) a direct role involving modulation of erythroid iron homeostasis. Analysis of wild-type (wt), Hfe-KO, and thalassemic mice lacking Hfe indicates that this gene is a key player controlling hepcidin in response to iron overload at steady state, under conditions of stress erythropoiesis and even in β-thalassemia. As a result, we show that Hfe contributes indirectly to increased Hb production in erythroid cells. Moreover, we demonstrate that Hfe is expressed in erythroid progenitors and interacts with Tfrc in murine erythroleukemia (MEL) cells. In its absence, the amount of Tfrc on the surface of erythroid cells was reduced and, in this way, influenced iron intake. Further, transgenic expression of Hfe in the liver of Hfe-KO mice, although decreasing liver iron content, does not alter erythroid parameters, such as Hb and MCH. Finally, wt mice transplanted with Hfe-KO bone marrow (BM) responded better to erythroid stress than wt mice transplanted with wt BM, supporting the idea that Hfe has an autonomous function in erythroid iron metabolism.
Methods
Animal care and procedures
All mice used were on a C57BL/6 background. For BM transplantation, 5 × 106 BM cells were transplanted into lethally irradiated syngeneic recipients. Anemia was induced in 2- to 3-month-old animals either by injection of phenylhydrazine (100 mg/kg) or by phlebotomy (400 μL/25 g) performed under anesthesia on 3 consecutive days. An equal volume of normal saline replaced the blood removed. Complete blood counts were performed every 2 to 3 days. Some animals were fed an iron-deficient diet containing 2.5 ppm of iron (Harlan-Teklad), beginning 24 hours before the bloodletting. Iron overload in wt mice was achieved by feeding a diet supplemented with 2.5% carbonyl iron for one week. These mice were returned to a regular diet 24 hours before phlebotomy. Erythropoiesis was also stimulated by intraperitoneal administration of erythropoietin (Epo; 50 U/day per mouse) on 3 consecutive days. Animals were analyzed 24 hours after the last injection. For in vivo macrophage depletion studies, weekly intravenous injections of 0.2 mL of clodronate-containing liposomes were done for a total of 6 weeks. As a control, phosphate-buffered saline (PBS)–containing liposomes were injected into a cohort of similar animals. Liposomes were prepared as previously described.28
Lentiviral injections into the liver of pups
Aliquots of lentiviral concentrate (2 × 107 infectious particles)29 were injected directly into the livers of 3-day-old pups, using a microinjector consisting of a 100-μL nanofil syringe attached to SilFlex tubing connected to a 35-gauge beveled needle via a connector (World Precision Instruments). Pups were placed in a supine position to visualize their livers, and lentivirus was gently injected (supplemental Figure 1A-B, available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Immunoprecipitation and immunoblotting
Cells were lysed in rapid immunoprecipitation assay (150mM NaCl, 20mM Tris, pH 8.0, 1mM ethylenediaminetetraacetic acid, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) with protease and phosphatase inhibitors. After centrifugation, anti-green fluorescence protein (GFP) rabbit antibody (Invitrogen) was added and the lysates incubated for 2 hours at 4°C. The immunocomplex was then precipitated using protein A-Sepharose beads. After extensive washing, the complexes were eluted from the beads by boiling in 4 times sample buffer for 2 minutes. Samples were then loaded in a 10% Nupage Bis-Tris Gel (Invitrogen) and transferred to polyvinylidene difluoride membranes, followed by immunoblot analysis with anti-Tfrc antibody (Invitrogen). Immunoreactive proteins were visualized by enhanced chemiluminescence detection and film autoradiography. After stripping, the membranes were reprobed with mouse anti-GFP antibody (Abgent).
Hfe detection in erythroid cells
Erythroid cells were purified from the spleens of phlebotomized mice as described.30 Mouse erythroid colonies (colony-forming units-erythroid and mature burst-forming units-erythroid) were grown from BM in complete Methocult medium (M3334; StemCell Technologies) according to the manufacturer's instructions. Colonies were picked at day 4 or 5 of culture. Reverse-transcribed polymerase chain reaction on cDNA extracted from splenic erythroid cells or erythroid colonies was performed using specific primers for Hfe, Tfr2, Fpn1, and β-actin (supplemental Table 1).
Iron uptake experiments
Human apo-Tf (Sigma-Aldrich) was saturated with 59Fe (PerkinElmer Life and Analytical Sciences) by mixing Tf with a 2-fold excess of 59FeCl2, a 10-fold excess of nitrilotriacetate, and 80mM NaHCO3. This mixture was incubated for 1 hour at 20°C after which excess iron was removed using a Microcon YM-10 column. For iron uptake experiments, isolated erythroid cells were resuspended in serum-free Iscove medium, supplemented with 0.3 mg/mL of 59Fe-loaded Tf, at a concentration of 107 cells/mL. Cells were incubated for the desired time at 37°C. At each time point, cells were washed 3 times with ice-cold PBS and counted in a γ counter.
Statistical analysis
Statistical differences between means were calculated with Student t test (Microsoft Excel).
Results
Hfe-KO mice show alterations in steady- state hematologic parameters compared with wt mice
Several studies revealed altered red blood cell (RBC) parameters in HH patients25,26 but failed to analyze erythropoiesis in detail. As a first step toward determining whether a mouse model of HH exhibits changes in erythropoiesis, we performed complete blood counts on wt and Hfe-KO mice at different ages. Significant differences were observed, including increases in Hb, mean corpuscular volume, and MCH (Table 1). Hfe-KO mice at 12 months of age had unchanged Hb values and a slight reduction in RBC counts (Table 1). These data indicate increased Hb production in Hfe-KO RBCs, similar to the situation in HH patients. It is expected that iron overload alters erythropoiesis because iron is essential for Hb production in RBCs. Therefore, as a control for the effect of iron overload on hematologic parameters, Hamp-KO mice at 2 months of age were analyzed. These mice showed no change in Hb values and smaller differences than Hfe-KO mice in mean corpuscular volume and MCH compared with wt mice, despite having elevated liver iron contents (Table 1). These results indicated a potential role for Hfe in erythropoiesis apart from that in iron overload. We then looked at erythroid maturation in the BM and spleen by staining cell suspensions with an erythroid-specific marker (Ter119) in combination with markers for Tfrc (CD71) and the adhesion molecule CD44.31 Using these markers, no clear differences in erythroid maturation were observed comparing wt and Hfe-KO mice. However, we detected down-regulation of Tfrc in Hfe-KO erythroid cells as indicated by a decrease in the mean fluorescence intensity of Tfrc (supplemental Figure 2). Down-regulation of Tfrc expression was also observed in Hamp-KO mice (not shown), suggesting that this effect is somewhat associated with iron overload.
Table 1.
Hematologic parameters in wt, Hfe-KO, and Hamp-KO female mice
| Hb, g/dL | RBC, ×106/μL | Hct, % | MCV, fL | MCH, pg | Retic, ×109/L | LIC, μg/mg dry | SI, μg/dL | Tf Sat, % | |
|---|---|---|---|---|---|---|---|---|---|
| wt (2 months) | 15.5 ± 0.3 | 9.8 ± 0.5 | 47.0 ± 0.7 | 45.9 ± 0.3 | 15.2 ± 0.1 | 285 ± 14 | 0.29 ± 0.04 | 121 ± 39 | 53 ± 11 |
| wt (5 months) | 15.4 ± 0.2 | 10.0 ± 0.1 | 44.4 ± 0.5 | 44.3 ± 0.4 | 15.3 ± 0.1 | 246 ± 15 | 0.41 ± 0.09 | 123 ± 15 | 58 ± 14 |
| wt (12 months) | 15.2 ± 0.2 | 11.0 ± 0.1 | 49.7 ± 0.9 | 45.2 ± 0.6 | 13.9 ± 0.2 | 377 ± 28 | 0.43 ± 0.09 | 153 ± 25 | 53 ± 14 |
| Hfe-KO (2 months) | 16.6 ± 0.2† | 10.2 ± 0.1 | 48.8 ± 0.7 | 48.2 ± 0.3† | 16.4 ± 0.1‡ | 310 ± 10 | 1.36 ± 0.32‡ | 219 ± 20* | 89 ± 14* |
| Hfe-KO (5 months) | 16.2 ± 0.3* | 9.8 ± 0.2 | 46.9 ± 0.6† | 47.9 ± 0.5† | 16.5 ± 0.2‡ | 260 ± 17 | 1.59 ± 0.25‡ | 233 ± 20† | 91 ± 14† |
| Hfe-KO (12 months) | 15.7 ± 0.3 | 9.9 ± 0.2† | 47.3 ± 0.7* | 48.2 ± 0.7† | 16.0 ± 0.3‡ | 260 ± 35* | 1.87 ± 0.25‡ | 310 ± 10† | 86 ± 14‡ |
| Hamp-KO (2 months) | 15.7 ± 0.1§ | 9.9 ± 0.1 | 46.7 ± 0.5 | 47.0 ± 0.2*§ | 15.7 ± 0.2*§ | 305 ± 75 | 3.64 ± 0.48†§ | NA | NA |
Values are mean ± SEM of 5 to 20 female mice.
Hct indicates hematocrit; MCV, mean corpuscular volume; Retic, reticulocyte count; LIC, liver iron concentration; SI, serum iron; Tf Sat, transferrin saturation; and NA, not applicable.
P ≤ .05 relative to age-matched wt controls.
P ≤ .01 relative to age-matched wt controls.
P ≤ .001 relative to age-matched wt controls.
P ≤ .05 relative to 2-month-old Hfe-KO mice.
Hfe-KO mice respond better than wt mice to different erythropoietic stress conditions
To determine how Hfe-KO mice respond to erythropoietic stress, we induced anemia by administration of phenylhydrazine or by phlebotomy. Recovery from both phenylhydrazine-induced hemolytic anemia and phlebotomy was faster in Hfe-KO compared with wt mice (Figure 1A-B). This was associated with increased Hb synthesis by Hfe-KO RBCs, as noted by increased Hb content in RBC and reticulocytes throughout the treatment (not shown). As a control, iron-overloaded wt mice were used. Compared with Hfe-KO mice, their liver iron content was similar (not shown), their hepcidin expression was superior (Figure 1E), and their recovery from phlebotomy was reduced but similar to that seen in wt mice fed a regular diet (Figure 1B). We then determined the relative contributions of dietary and stored iron toward the response to phlebotomy in wt and Hfe-KO mice, by analyzing their recovery from anemia when fed a low-iron diet. The wt mice kept on an iron-deficient diet were unable to overcome the phlebotomy-induced anemia, whereas Hfe-KO mice fed a similar diet increased their Hb levels, although they did not fully recover (Figure 1B). Analysis of serum iron and Tf saturation during the first 48 hours after phlebotomy showed that wt mice on a standard diet were able to raise their serum iron parameters very rapidly to levels similar to those of Hfe-KO mice. In contrast, wt mice fed a low-iron diet showed a significant reduction of iron-related serum parameters during the same period (Figure 1C-D). These data suggest that: (1) gastrointestinal iron absorption in both wt and Hfe-KO mice is a major factor leading to recovery from anemia; (2) iron overload in wt mice increases hepcidin levels, thereby limiting use of stored iron in response to increased erythroid demand; and (3) low hepcidin levels in Hfe-KO mice contribute to better use of iron stored in the liver to increase Hb levels. Altogether, these observations support the argument that lack of Hfe improves erythropoiesis under stress conditions.
Figure 1.

Hfe-KO mice respond better to erythropoietic stress. (A-B) Recovery of Hb values in wt and Hfe-KO mice after phenylhydrazine-induced hemolytic anemia and phlebotomy, respectively. (B) In the phlebotomy studies, the results obtained in wt mice fed a low-iron (Low Fe) or high-iron (High Fe) diet, and Hfe-KO mice fed the low-iron diet are also shown. Serum iron (C) and Tf saturation (D) over the course of the first 2 days of the phlebotomy regimen are shown for wt and Hfe-KO mice fed a regular or low-iron diet. (E) Hamp expression in wt and Hfe-KO mice at steady state and after erythropoietic stimulation by phlebotomy or Epo administration. The graph is shown in logarithmic scale. Data are mean ± SEM of 5 to 20 age- and sex-matched mice. *P ≤ .05 relative to wt controls. **P ≤ .01 relative to wt controls. ***P ≤ .001 relative to wt controls.
The role of macrophages in erythropoiesis and iron metabolism has long been recognized.32 It has been suggested that interaction of macrophages with erythroblasts supports the maturation of the latter cells through a yet unknown mechanism. In addition, macrophages are responsible for the clearance of senescent RBCs, exporting recycled iron via Fpn1.32,33 It has been shown that iron export from macrophages is enhanced under conditions of low hepcidin activity, such as in Hfe-related hemochromatosis.34 Therefore, we evaluated the ability of macrophages to cause the increased Hb production seen in Hfe-KO mice. We analyzed erythropoiesis after macrophage depletion by liposome-encapsulated clodronate, a technique commonly used to specifically target and eliminate macrophages in vivo.28 Our data showed that almost complete macrophage depletion (supplemental Figure 3) was associated with milder hematologic alterations and anemia in Hfe-KO mice compared with wt mice (Figure 2). Although macrophages are important in maintaining a normal erythropoietic rate in both Hfe-KO and wt mice, in their absence Hfe-KO mice still exhibited increased Hb levels, associated with an increased cellular Hb content, suggesting that macrophages are not the primary factor responsible for the differences observed in these parameters.
Figure 2.

Hematologic values are less affected in Hfe-KO than wt mice after macrophage depletion. Macrophages were eliminated by weekly injections of clodronate-containing liposomes. Mice injected with PBS-containing liposomes served as controls. The effect of macrophage depletion on Hb (A), RBC (B), hematocrit (C), and MCH (D) values during the course of the treatment is shown. Data are mean ± SEM of 5 age- and sex-matched mice. *P ≤ .05 relative to PBS controls. **P ≤ .01 relative to PBS controls. ***P ≤ .001 relative to PBS controls.
Hfe-KO mice regulate hepcidin normally in response to erythropoietic stimuli
Hepcidin is strongly modulated in response to erythropoietic stimuli,1–3,7 but the regulatory mechanism(s) has not yet been established. We analyzed the response of hepcidin to phlebotomy and Epo administration in Hfe-KO mice to determine whether this could be responsible for the differences observed. Our data show that Hfe-KO mice down-regulate hepcidin expression to the same extent as wt mice in response to both phlebotomy and Epo injections. Yet, the response of hepcidin to erythropoietic stimulation is increased in Hfe-KO compared with iron overloaded wt mice (Figure 1E). These data indicate that Hfe is not directly involved in regulation of hepcidin response to erythropoietic stimulation. However, under conditions of enhanced erythropoietic activity, hepcidin expression is inappropriately low in Hfe-KO mice, considering their iron load. This further corroborates our hypothesis that Hfe-KO mice exhibit increased iron mobilization for erythropoiesis explaining, at least in part, the increased response seen in these animals.
Combination of the hemochromatosis and thalassemic phenotypes leads to increased liver iron content and Hb levels
Our data showed that lack of Hfe is advantageous under conditions of stress. Therefore, we determined how the absence of Hfe affects a pathologic condition associated with anemia and ineffective erythropoiesis, such as β-thalassemia. We generated 2 groups of experimental animals using mice affected by β-thalassemia intermedia (th3/+): th3 → wt mice (wt recipients engrafted with th3/+ hematopoietic stem cells [HSCs]) and th3 → Hfe-KO mice (Hfe-KO recipients engrafted with th3/+ HSCs). Using these mice, we assessed how lack of Hfe in the liver affected iron distribution and hematologic parameters in β-thalassemia. Compared with wt → wt mice, th3 → wt mice exhibited 4- and 4.5-fold increases in the iron content of their livers and spleens, respectively, whereas those in the serum were only slightly increased (Table 2). In th3 → Hfe-KO animals, hepatic iron overload increased dramatically (10-fold compared with wt → wt controls), whereas splenic iron levels were similar to those of th3/+ animals (Table 2). In addition, serum iron parameters were increased compared with both wt → wt and th3 → wt mice. We also compared the hematologic values in th3 → wt and th3 → Hfe-KO mice, observing that the latter group exhibited increased Hb levels, RBC counts, and MCH (Table 2). This supports the observation by Ginzburg et al that administration of additional iron ameliorated the anemia in mice affected by β-thalassemia intermedia35 and confirms that lack of Hfe provides additional protection under conditions of anemia.
Table 2.
Hematologic parameters in bone marrow transplanted animals
| Hb, g/dL | RBC, ×106/μL | Hct, % | MCV, fL | MCH, pg | Retic, ×109/L | LIC, μg/mg dry | SIC, μg/mg dry | SI, μg/dL | Tf Sat, % | |
|---|---|---|---|---|---|---|---|---|---|---|
| Wt → wt | 14.3 ± 0.2 | 10.2 ± 0.2 | 46.6 ± 0.7 | 45.7 ± 0.2 | 14.1 ± 0.1 | 276 ± 12 | 0.29 ± 0.02 | 1.97 ± 0.18 | 117 ± 10 | 56 ± 3 |
| Hfe → Hfe | 15.3 ± 0.2† | 9.7 ± 0.1* | 47.8 ± 0.2 | 49.4 ± 0.8‡ | 15.8 ± 0.1‡ | 238 ± 26 | 0.98 ± 0.08‡ | 2.47 ± 0.75 | 225 ± 13‡ | 88 ± 2† |
| th3/+ → wt | 7.8 ± 0.2 | 8.0 ± 0.2 | 29.2 ± 1.0 | 36.5 ± 1.5 | 9.7 ± 0.1 | 1587 ± 76 | 1.29 ± 0.27 | 8.22 ± 1.12 | 162 ± 11 | 49 ± 4 |
| th3/Hfe → wt | 8.1 ± 0.3 | 8.3 ± 0.2 | 30.8 ± 1.6 | 36.7 ± 1.0 | 9.7 ± 0.2 | 1807 ± 149 | 1.08 ± 0.05 | 10.31 ± 0.74 | 152 ± 12 | 51 ± 3 |
| th3/+ → Hfe | 9.4 ± 0.1‡ | 8.9 ± 0.2† | 36.9 ± 0.3† | 41.6 ± 0.7* | 10.6 ± 0.2† | 1958 ± 102* | 3.09 ± 0.34‡ | 8.29 ± 1.3 | 232 ± 7* | 82 ± 4† |
Values are mean ± SEM of 5 to 20 female mice.
Hct indicates hematocrit; MCV, mean corpuscular volume; Retic, reticulocyte count; LIC, liver iron concentration; SIC, splenic iron concentration; SI, serum iron; and Tf Sat, transferrin saturation.
P ≤ .05 relative to controls (wt → wt for Hfe → Hfe and th3 → wt for th3/+ → Hfe).
P ≤ .01 relative to controls (wt → wt for Hfe → Hfe and th3 → wt for th3/+ → Hfe).
P ≤ .001 relative to controls (wt → wt for Hfe → Hfe and th3 → wt for th3/+ → Hfe).
We also generated th3/+Hfe-KO mice. To determine how specific ablation of Hfe in the hematopoietic compartment affected the thalassemic phenotype, we transplanted wt mice with th3/+Hfe-KO HSCs (th3/Hfe → wt). Although these mice showed no differences in the aforementioned parameters compared with th3 → wt mice (Table 2), they did exhibit a decreased level of Tfrc on erythroid plasma membranes (not shown), similar to what we observed in Hfe-KO mice (supplemental Figure 2). These data suggest, that even though thalassemic erythroid cells lacking Hfe present lower levels of membrane Tfrc during their development, this does not alter their ability to produce Hb, supporting our notion that Hfe interferes with erythroid iron metabolism.
Hfe modulates hepcidin in response to iron overload in thalassemic mice
To evaluate the role of Hfe in modulating the response of hepcidin to iron overload in thalassemic mice, we looked at the levels of Hamp, Bmp6 (a strong modulator of hepcidin in response to iron),36 and Id1 (another target of the Bmp/Smad pathway).37 All of these genes are up-regulated by iron overload in wt mice.38 Our data show that th3 → Hfe-KO mice have similar levels of hepcidin and Id1 expression compared with th3 → wt and wt → wt mice, despite increased iron overload. The levels of Bmp6 were elevated in both thalassemic groups compared with wt mice. However, when the levels of hepcidin, Bmp6, and Id-1 were normalized to liver iron content, we observed that these levels were markedly reduced in th3 → wt mice compared with wt → wt animals (Figure 3). After normalization, these levels were further decreased in th3 → Hfe-KO mice. Collectively, these observations indicate that iron overload can partially counteract the repressive effect of ineffective erythropoiesis on hepcidin expression in β-thalassemia. Moreover, lack of Hfe further aggravates iron overload in thalassemic mice, indicating that Hfe plays a positive role in the regulation of hepcidin in this disorder.
Figure 3.

Hfe is important for the control of hepcidin in response to iron in β-thalassemic mice. Expression of Hamp (A), Bmp6 (C), and Id1 (E) in the liver of wt and thalassemic mice at 5 months of age. Values for wt → wt (wt mice engrafted with wt HSCs), th3 → wt (wt mice engrafted with th3/+ HSCs), and th3 → Hfe-KO (Hfe-KO mice engrafted with th3/+ HSCs) mice are shown. Expression levels of Hamp, Bmp6, and Id1 were normalized to liver iron concentrations and are shown in panels B, D, and F, respectively. All of these genes are abnormally low in th3 → wt mice considering their iron load but are further down-regulated in th3 → Hfe-KO mice. Data are mean ± SEM of 5 to 8 mice. *P ≤ .05. **P ≤ .01.
Hfe is expressed in early erythroid cells and interferes with Tf bound iron uptake
Our data show altered RBC parameters and an increased response to erythropoietic stress in Hfe-KO mice. Prior studies indicated that there was no Hfe expression in erythroid cells. To resolve this issue, we analyzed the expression of Hfe in erythroid cells selected from the spleens of phlebotomized wt mice. As a control, we also analyzed expression of other genes known to be associated with iron metabolism in erythroid cells, such as Fpn139 and Tfr2.40 Our results revealed expression of Hfe, Tfr2 and Fpn1 in freshly isolated splenocytes, highly enriched in erythroid progenitors (Figure 4C). Moreover, we corroborated this finding by demonstrating the expression of Hfe in erythroid colonies isolated from specific erythroid colony-forming assays (Figure 4D). We also looked for Hamp, Hjv, and BMPs, but they were not expressed. These results argue strongly against Hfe being a sensor for hepcidin in erythroid cells, although they do not exclude completely this possibility.
Figure 4.

Hfe is expressed in erythroid progenitors. (A) Fluorescence-activated cell sorter profiles of wt splenic erythroid cells after selection. (Top panel) Tfrc and Ter119 costaining. (Bottom panel) CD44 versus forward scatter (FSC) as previously described by Chen et al.31 (B) Reverse-transcribed polymerase chain reaction showing expression of Hfe, Tfr2, Fpn1, and β-actin in erythroid cells harvested from the spleens of phlebotomized wt mice and (C) in erythroid colonies derived from BM cells.
To determine whether Hfe interacts with Tfrc in erythroid cells, possibly interfering with iron uptake, an Hfe-GFP fusion protein was expressed in MEL cells using a lentiviral vector. We observed a high degree of colocalization of Hfe-GFP and Tfrc by immunofluorescence (Figure 5A) and confirmed their interaction by coimmunoprecipitation (Figure 5B). To determine whether Hfe affects iron uptake, we isolated wt and Hfe-KO erythroid cells and measured iron uptake using 59Fe-saturated Tf. Our data showed no statistically significant difference in iron uptake between Hfe-KO and wt cells (Figure 5C). Interestingly, the level of Tfrc expression in Hfe-KO cells was 80% of that seen in wt cells, as measured by flow cytometry (Figure 5D). This was not associated with altered Tfrc recycling but instead seemed to reflect an overall decrease in Tfrc on the plasma membrane of erythroid cells (not shown). When iron uptake was normalized to the Tfrc levels, it was significantly increased in Hfe-KO compared with wt cells (Figure 5E), showing that Hfe-KO cells take up more iron per Tfrc. These data suggest that Hfe interferes with erythroid transferring-bound iron uptake.
Figure 5.

Hfe interacts with Tfrc in erythroid cells and impairs Tf-bound iron uptake. (A) Immunofluorescence of MEL cells showing colocalization (yellow) of Hfe-GFP fusion protein (green) and Tfrc (red). Cells were prepared as described in supplemental Methods. Images were captured on a Nikon TE2000-U inverted microscope equipped with DIC (Nikon), a CoolSnap cf digital camera (Photometrics), and a Plan Apo objective 60×/1.40 oil (Nikon) and acquired using IP Lab Version 3.6.5a software (Scanalytics). Brightness/contrast and color balance were adjusted using Adobe Photoshop Version 7.0.1 (Adobe Systems). (B) Western blot for Tfrc after immunoprecipitation of GFP in protein lysates from MEL cells transduced with lentivirus expressing GFP or Hfe-GFP fusion protein. After stripping, the membranes were blotted for GFP, showing the expected size for GFP and Hfe-GFP in the corresponding lanes. (C) Uptake of 59Fe by wt and Hfe-KO erythroid cells from 59Fe-saturated Tf. Cells were harvested from phlebotomized mice and erythroid cells selected as previously described.30 Counts per minute (cpm) were normalized to the percentage of Tfrc+ cells as detected by fluorescence-activated cell sorter (usually > 85%). Five independent experiments were performed. (D) Tfrc mean fluorescence intensity in isolated erythroid cells shows down-regulation of Tfrc in Hfe-KO cells. Twenty independent measures were made. (E) Iron uptake measured in panel C was normalized to the Tfrc mean fluorescence intensity ratio between wt and Hfe-KO cells for each experiment to control for the differences observed in Tfrc expression. Data are mean ± SEM. *P ≤ .05. ***P ≤ .001.
Direct hepatic injection of lentiviral vectors carrying Hfe reduces the iron burden in Hfe-KO mice with little impact on hematologic parameters
We hypothesized that expression of Hfe solely in the liver might allow us to discriminate between the role of Hfe in the hematopoietic compartment and that of iron overload in erythropoiesis. For this purpose, we developed a lentiviral vector to transduce the Hfe gene directly into the liver of Hfe-KO mice. Hepatic injection of lentiviral vectors into adult animals is quite cumbersome and not very efficient.41 Therefore, we modified this technique by directly injecting the liver of 3-day-old pups. To validate this approach, we used the TGW vector (Figure 6A), which harbors the GFP gene under the control of the synthetic TTR hepatocyte-specific promoter. Injection of lentiviral vectors into the liver of pups presents several advantages: (1) the liver is small and visible through the skin, thereby requiring only small amounts of vector and making injection extremely simple; (2) the liver cells are proliferating and thus more permissive for lentiviral infection than adult cells42; and (3) at this stage of development, the immune system is expected to better tolerate the protein encoded by the transgene. Up to 3 months after injection, livers transduced with TGW exhibited high levels of viral integration and expression of the transgene (not shown), as well as widespread synthesis of GFP (Figure 6B). Using this approach, we injected Hfe-KO mice with lentiviral vectors carrying GFP (TGW) or Hfe (THW), these mice being designated Hfe-KO-Ctr and Hfe-KO-THW, respectively. Two months after injection, Hfe-KO-THW mice exhibited elevated expression of Hfe in the liver (Figure 6C). In addition, hepcidin expression was significantly increased compared with the levels in Hfe-KO-Ctr mice (Figure 6C). This was reflected in a reduction in serum iron, Tf saturation, and liver iron content to levels that were almost normal (Figures 6D-F). However, this marked decrease in iron values did not produce major changes in their erythroid parameters compared with those seen in Hfe-KO-Ctr mice (Table 3), suggesting an additional role for Hfe in the hematopoietic compartment.
Figure 6.

Hepatic injection of a lentivirus carrying the Hfe gene is effective in preventing HH. (A) Schematic representations of the lentiviral vectors THW and TGW. The various components shown are the long terminal repeat (LTR), self-inactivating long terminal repeat (SIN-LTR), the HIV rev-response and central polypurine tract elements (RRE, cPPT), the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE), the liver-specific TTR promoter (TTR), the GFP, and the mouse Hfe gene (Hfe). (B) Representative fluorescence analyses of liver sections from animals 2 months after being transduced with PBS or TGW. Liver was fixed in 4% paraformaldehyde and frozen in OCT embedding medium. Images were captured on a Nikon TE2000-U inverted microscope equipped with DIC, a CoolSnap cf digital camera (Photometrics), and a Plan fluor objective 20×/0.45 (Nikon), and acquired using IP Lab Version 3.6.5a software (Scanalytics). Brightness/contrast and color balance were adjusted using Adobe Photoshop Version 7.0.1 (Adobe Systems). (C) Expression of Hfe and Hamp in the livers of mice injected with lentivirus. (D) Serum iron, (E) Tf saturation, and (F) organ iron content of mice injected with THW together with the appropriate controls. Data are mean ± SEM of 5 or 6 individual age- and sex-matched mice. *P ≤ .05. **P ≤ .01. ***P ≤ .001.
Table 3.
Hematologic parameters in liver injected animals
| Hb, g/dL | RBC, ×106/μL | Hct, % | MCV, fL | MCH, pg | Retic, ×109/L | |
|---|---|---|---|---|---|---|
| Wt-Ctr | 15.6 ± 0.3 | 10.2 ± 0.1 | 50.7 ± 0.6 | 49.7 ± 0.2 | 15.3 ± 0.1 | 281 ± 186 |
| Hfe-KO-Ctr | 16.7 ± 0.3 | 10.4 ± 0.1 | 52.1 ± 0.5 | 50.1 ± 0.9 | 16.0 ± 0.2* | 231 ± 13 |
| Hfe-KO-THW | 16.3 ± 0.2 | 10.4 ± 0.1 | 51.0 ± 0.7 | 49.0 ± 0.9 | 15.6 ± 0.1* | 351 ± 18*† |
Values are mean ± SEM of 5 to 20 female mice.
Hct indicates hematocrit; MCV, mean corpuscular volume; and Retic, reticulocyte count.
P ≤ .05 relative to wt-Ctr.
P ≤ .05 relative to Hfe-KO-Ctr.
Wt animals transplanted with Hfe-KO BM recover faster from erythropoietic stress than wt animals transplanted with wt BM
To further differentiate between the function of Hfe in the liver and that in the hematopoietic compartment, we performed a series of additional BM transplantation experiments. The first 2 groups of animals were wt → wt (wt recipients engrafted with wt HSCs) and Hfe-KO → wt (wt recipients engrafted with Hfe-KO HSCs). These mice do not develop iron overload (supplemental Table 2), allowing us to assess how the absence of Hfe in the hematopoietic compartment affects erythropoiesis in the absence of iron overload. As a further control for evaluating erythropoiesis in mice developing iron overload, we also generated wt → Hfe-KO (Hfe-KO recipients engrafted with wt HSCs) and Hfe-KO → Hfe-KO (Hfe-KO recipients engrafted with Hfe-KO HSCs). When erythropoiesis was challenged by phlebotomy, we observed that wt → Hfe-KO and Hfe-KO → Hfe-KO animals recovered faster than Hfe-KO → wt and wt → wt mice (Figure 7A-B), supporting the hypothesis that lack of Hfe in the liver accelerates recovery when iron overload exists. Surprisingly, however, Hfe-KO → wt mice still had an increased response to anemia compared with wt → wt mice (Figure 7A). This was associated with an increased MCH and occurred, even though these mice do not develop iron overload (supplemental Table 2). These data suggest that lack of Hfe in hematopoietic organs increases iron uptake by erythroid cells. Although this seems to be a mild effect, it is sufficient to accelerate recovery from profound anemia under conditions of normal iron homeostasis.
Figure 7.
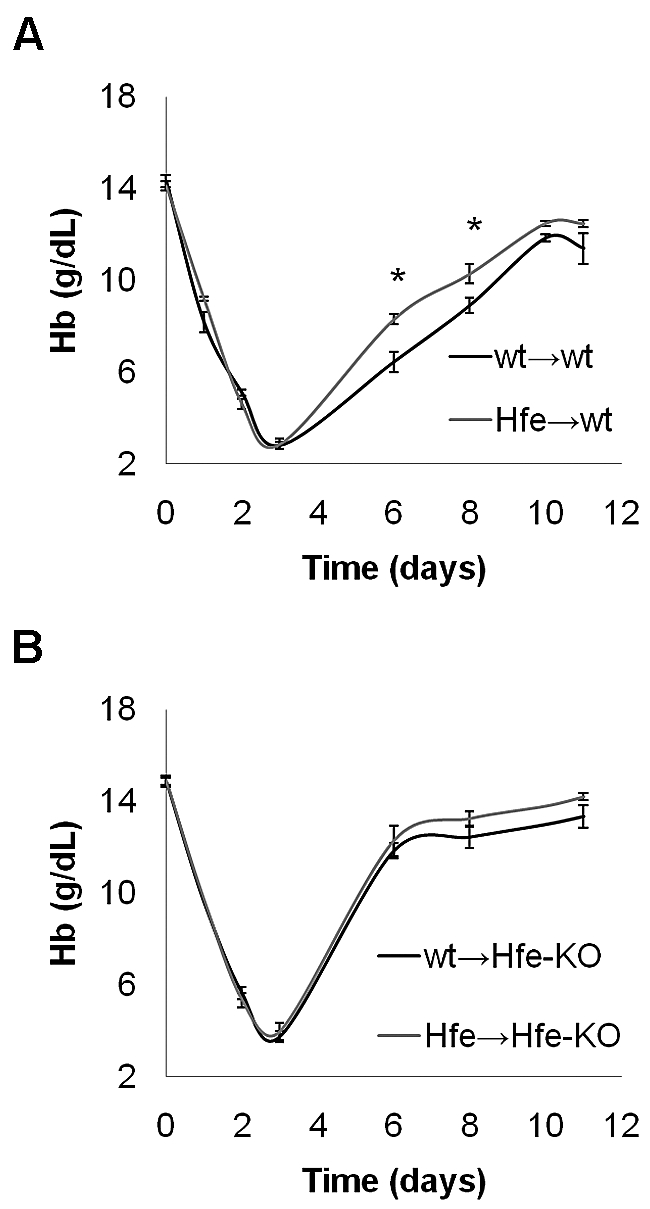
Wt mice transplanted with Hfe-KO BM respond better to erythropoietic stress. Recovery of Hb values in wt → wt versus Hfe → wt (A) and wt → Hfe-KO versus Hfe → Hfe-KO (B) mice after phlebotomy, as in Figure 1. Phlebotomies were performed 2-3 months after BM transplantation. Data are mean ± SEM of 5-10 mice. *P ≤ .05 relative to controls.
Discussion
HH is a disease mainly associated with iron overload, its phenotype being primarily linked with expression of Hfe in the liver because of its role in the regulation of hepcidin.10,11,14,24 However, several lines of evidence point to additional hematologic functions for Hfe: (1) macrophages from HH patients and Hfe-KO mice seem to be iron deficient43,44; (2) reconstitution of Hfe-KO mice with BM from wt animals leads to organ iron redistribution45; (3) numerous erythropoietic abnormalities have been reported in HH patients23,25,26; and (4) analysis of the Hfe promoter revealed the presence of several GATA1-responsive elements.27 In addition, the main partner of Hfe (Tfrc) is highly expressed in erythroid progenitor cells.22 Accordingly, we decided to investigate the role of Hfe in erythropoiesis, the goal being to understand how lack of this gene in the liver or the hematopoietic compartment affected the erythropoietic response.
From our data, we can conclude that erythropoiesis is altered in Hfe-KO mice at steady state. Differences in several hematologic parameters could be seen between age-matched Hfe-KO and wt mice. We also demonstrate that Hfe-KO mice are able to respond better to stress erythropoiesis induced by phenylhydrazine, phlebotomy, or macrophage depletion. The response to phlebotomy was increased in Hfe-KO mice, even compared with iron-overloaded wt mice. Our data suggest that this is associated, at least in part, with increased down-regulation of hepcidin in response to erythropoietic stimulation in Hfe-KO mice compared with iron-loaded wt mice. Hepcidin regulation requires integration of different signals for a proper response.7 In Hfe-KO mice, the iron-regulatory pathway of hepcidin is not fully functional, and erythropoiesis-induced hepcidin down-regulation is stronger than what is expected under conditions of iron overload. This leads to increased mobilization of iron for erythropoiesis, possibly conferring an advantage under conditions of stress erythropoiesis. In addition, iron absorption from the diet seems to be crucial for recovery from anemia, being a major source of iron in wt and Hfe-KO mice as demonstrated by a decreased response when mice were kept on a low-iron diet. Phlebotomy is the main treatment for iron overload in HH patients. However, our data suggest that this treatment leads to both mobilization of iron from stores and increased intestinal iron absorption. These observations indicate that patients might benefit from a controlled iron diet or treatment with hepcidin/hepcidin agonists so as to limit iron absorption and thereby increase the benefit of the phlebotomy. We also determined that lack of Hfe expression in the liver under pathologic conditions prevents proper regulation of hepcidin in response to iron. We showed that th3 → Hfe-KO mice had hepcidin levels similar to those of th3 → wt mice despite increased iron overload, presenting a phenotype consistent with HH. This resulted in increased Hb production, with little effect on ineffective erythropoiesis (not shown). All these data indicate that lack of an appropriate hepcidin response to iron overload is beneficial under conditions of stress erythropoiesis, where fast recovery from anemia is required. In β-thalassemia, lack of Hfe is associated with increased Hb levels. This, however, also leads to increased iron overload that, over time, probably aggravates the overall phenotype in this disorder.
We also present evidences that some of the hematologic differences could not be attributed exclusively to iron overload. Thus: (1) age- and sex-matched Hamp-KO mice had a milder increase in Hb and MCH than Hfe-KO mice; (2) prevention of iron overload in Hfe-KO mice by injection of lentiviral vectors carrying Hfe did not result in complete normalization of the hematologic changes seen in the Hfe-KO controls; and (3) faster recovery from anemia was observed in Hfe-KO → wt mice compared with wt → wt mice. In accordance with a role for Hfe in erythropoiesis, we detected Hfe expression in erythroid progenitors. Early studies showed that Hfe is tightly bound to Tfrc and modulates cellular iron uptake.16–21 However, the physiologic relevance of this process is still unclear. When we overexpressed an Hfe-GFP fusion protein in an erythroid cell line, we observed a strong interaction with Tfrc. To our knowledge, this is the first time that such an interaction has been shown in erythroid cells. In addition, we present evidence that Hfe actively participates in modulation of iron uptake in erythroid progenitors. Because iron is essential during erythropoiesis, coordination of iron uptake, heme biosynthesis, and Hb production is highly regulated. The availability of heme was shown to positively modulate protein synthesis by the heme-regulated inhibitor kinase.46 Iron status was demonstrated to control heme biosynthesis, in particular by modulating the activity of 5-aminolevulinic acid synthetase.47 In addition, a heme export protein has been identified in human erythroid progenitors, possibly functioning to protect against cytotoxicity associated with heme accumulation.48 It has been suggested that a number of other proteins involved in iron metabolism regulate the content and distribution of iron within erythroid cells, including Tfrc, mitoferrin, and Fpn1.22,39,49 Of particular relevance was the discovery of an alternative form of Fpn1 in erythroid precursors and intestinal epithelium.39 This form is insensitive to regulation by intracellular iron, being controlled instead by systemic iron levels, probably through hepcidin-induced degradation. Interestingly, Tfrc regulation in erythroid cells is distinct from that in other cell types, as it too seems to be less responsive to iron. Indeed, Tfrc was shown to be strongly modulated through Irp2 in response to Epo.50 It is reasonable to assume that Tfrc activity in early erythroid progenitors, like that of Fpn1, might be modulated post-translationally, with Hfe being an obvious candidate to serve in this capacity. Hfe might be expressed in erythroid cells to modulate Tf-bound iron uptake. This is supported by our data showing an elevated iron uptake in erythroid cells, when normalized to Tfrc levels on the cell surface. Alternatively, Hfe might modulate erythroid iron metabolism by interfering with the association between Tfr2 and EpoR, in light of the fact that Tfr2 increases the sensitivity of EpoR to Epo.40 The synthesis of many iron-related proteins, including Tfr1, is modulated by Irp2, a target of the Epo/EpoR/Jak2/Sta5 pathway in erythroid cells.50 Therefore, it is possible that Hfe might also modulate the association between Tfr2 and EpoR, further controlling the expression of Tfr1 or other Irp2 targets involved in iron storage or its availability for heme synthesis. Nevertheless, increased Hb concentration seen in Hfe-KO erythroid cells is probably a consequence of combining 2 favorable factors: mild increase in erythroid iron uptake and the presence of iron overload. Our findings support the hypothesis that Hfe helps maintain iron homeostasis in erythroid cells by 2 distinct means. On the one hand, expression of Hfe in the liver indirectly influences erythropoiesis by modulating the response of hepcidin to iron load, whereas on the other, Hfe seemingly has a direct effect in erythroid cells, influencing iron uptake. This dual role for Hfe could be physiologically relevant as a means to avoid toxicity associated with iron overload under conditions of high iron availability or to limit erythroid iron intake under conditions of iron deficiency. Although it could serve to prioritize the various physiologic demands for iron, it would not be advantageous after erythropoietic stress. Erythroid iron homeostasis must be strictly controlled and coordinated with iron metabolism. Our results point to Hfe as a key player in regulating that control.
Supplementary Material
Acknowledgments
The authors thank the members of the Pasta and Red Cells Society of New York for technical support and helpful discussions. Hamp-KO mice were a kind gift from Drs Seth Rivera and Tom Ganz.
This work was supported by the Carlo and Mico'l Schejola Foundation and the Children's Cancer and Blood Foundation (S.R.), R01DK55463 (R.W.G.), and the American Portuguese Biomedical Research Fund (Inova grant; M.d.S., P.R.). P.R is a fellow from Fundacao para a Ciencia e Tecnologia, Portugal (SFRH/BD/24813/2005).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: P.R. collected and analyzed the data, designed the experiments, and wrote the manuscript; E.G. and C.C. maintained the mouse colony and performed animal experiments; N.C. and S.G cloned and produced lentiviral vectors; C.C.P. performed immunoprecipitation and reviewed the manuscript; N.V.R. provided clodronate liposomes; A.F. contributed vital reagents and reviewed the manuscript; R.W.G. performed atomic absorption analysis and reviewed the manuscript; M.d.S. analyzed data and reviewed the manuscript; and S.R. designed the experiments, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stefano Rivella, 515 East 71st St, Rm S-702, New York, NY 10021; e-mail: str2010@med.cornell.edu.
References
- 1.Vokurka M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res. 2006;55(6):667–674. doi: 10.33549/physiolres.930841. [DOI] [PubMed] [Google Scholar]
- 2.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110(7):1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108(12):3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276(11):7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 5.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98(15):8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 7.Huang H, Constante M, Layoun A, Santos MM. Contribution of STAT3 and SMAD4 pathways to the regulation of hepcidin by opposing stimuli. Blood. 2009;113(15):3593–3599. doi: 10.1182/blood-2008-08-173641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feder JN, Tsuchihashi Z, Irrinki A, et al. The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J Biol Chem. 1997;272(22):14025–14028. doi: 10.1074/jbc.272.22.14025. [DOI] [PubMed] [Google Scholar]
- 9.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7(3):205–214. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361(9358):669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 12.Nicolas G, Viatte L, Lou DQ, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34(1):97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 13.Muckenthaler M, Roy CN, Custodio AO, et al. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet. 2003;34(1):102–107. doi: 10.1038/ng1152. [DOI] [PubMed] [Google Scholar]
- 14.Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9(3):217–227. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace DF, Summerville L, Crampton EM, Frazer DM, Anderson GJ, Subramaniam VN. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology. 2009;50(6):1992–2000. doi: 10.1002/hep.23198. [DOI] [PubMed] [Google Scholar]
- 16.Chua AC, Herbison CE, Drake SF, Graham RM, Olynyk JK, Trinder D. The role of Hfe in transferrin-bound iron uptake by hepatocytes. Hepatology. 2008;47(5):1737–1744. doi: 10.1002/hep.22180. [DOI] [PubMed] [Google Scholar]
- 17.Waheed A, Grubb JH, Zhou XY, et al. Regulation of transferrin-mediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc Natl Acad Sci U S A. 2002;99(5):3117–3122. doi: 10.1073/pnas.042701499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lebron JA, West AP, Jr, Bjorkman PJ. The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor. J Mol Biol. 1999;294(1):239–245. doi: 10.1006/jmbi.1999.3252. [DOI] [PubMed] [Google Scholar]
- 19.Feder JN, Penny DM, Irrinki A, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci U S A. 1998;95(4):1472–1477. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gross CN, Irrinki A, Feder JN, Enns CA. Cotrafficking of HFE, a nonclassical major histocompatibility complex class I protein, with the transferrin receptor implies a role in intracellular iron regulation. J Biol Chem. 1998;273(34):22068–22074. doi: 10.1074/jbc.273.34.22068. [DOI] [PubMed] [Google Scholar]
- 21.Lebron JA, Bennett MJ, Vaughn DE, et al. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93(1):111–123. doi: 10.1016/s0092-8674(00)81151-4. [DOI] [PubMed] [Google Scholar]
- 22.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet. 1999;21(4):396–399. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- 23.Feeney GP, Carter K, Masters GS, Jackson HA, Cavil I, Worwood M. Changes in erythropoiesis in hereditary hemochromatosis are not mediated by HFE expression in nucleated red cells. Haematologica. 2005;90(2):180–187. [PubMed] [Google Scholar]
- 24.Vujic Spasic M, Kiss J, Herrmann T, et al. Hfe acts in hepatocytes to prevent hemochromatosis. Cell Metab. 2008;7(2):173–178. doi: 10.1016/j.cmet.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Barton JC, Bertoli LF, Rothenberg BE. Peripheral blood erythrocyte parameters in hemochromatosis: evidence for increased erythrocyte hemoglobin content. J Lab Clin Med. 2000;135(1):96–104. doi: 10.1016/s0022-2143(00)70026-6. [DOI] [PubMed] [Google Scholar]
- 26.McLaren CE, Barton JC, Gordeuk VR, et al. Determinants and characteristics of mean corpuscular volume and hemoglobin concentration in white HFE C282Y homozygotes in the hemochromatosis and iron overload screening study. Am J Hematol. 2007;82(10):898–905. doi: 10.1002/ajh.20937. [DOI] [PubMed] [Google Scholar]
- 27.Mura C, Le Gac G, Jacolot S, Ferec C. Transcriptional regulation of the human HFE gene indicates high liver expression and erythropoiesis coregulation. FASEB J. 2004;18(15):1922–1924. doi: 10.1096/fj.04-2520fje. [DOI] [PubMed] [Google Scholar]
- 28.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174(1):83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 29.May C, Rivella S, Callegari J, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406(6791):82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 30.Libani IV, Guy EC, Melchiori L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875–885. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci U S A. 2009;106(41):17413–17418. doi: 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood. 2008;112(3):470–478. doi: 10.1182/blood-2008-03-077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Ahmad KA, Ahmann JR, Migas MC, et al. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol Dis. 2002;29(3):361–366. doi: 10.1006/bcmd.2002.0575. [DOI] [PubMed] [Google Scholar]
- 35.Ginzburg YZ, Rybicki AC, Suzuka SM, et al. Exogenous iron increases hemoglobin in beta-thalassemic mice. Exp Hematol. 2009;37(2):172–183. doi: 10.1016/j.exphem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 36.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531–539. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 37.Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277(7):4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- 38.Kautz L, Meynard D, Monnier A, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503–1509. doi: 10.1182/blood-2008-03-143354. [DOI] [PubMed] [Google Scholar]
- 39.Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009;9(5):461–473. doi: 10.1016/j.cmet.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forejtnikova H, Vieillevoye M, Zermati Y, et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood. 2010;116(24):5357–5367. doi: 10.1182/blood-2010-04-281360. [DOI] [PubMed] [Google Scholar]
- 41.Follenzi A, Battaglia M, Lombardo A, Annoni A, Roncarolo MG, Naldini L. Targeting lentiviral vector expression to hepatocytes limits transgene-specific immune response and establishes long-term expression of human antihemophilic factor IX in mice. Blood. 2004;103(10):3700–3709. doi: 10.1182/blood-2003-09-3217. [DOI] [PubMed] [Google Scholar]
- 42.Park F, Ohashi K, Chiu W, Naldini L, Kay MA. Efficient lentiviral transduction of liver requires cell cycling in vivo. Nat Genet. 2000;24(1):49–52. doi: 10.1038/71673. [DOI] [PubMed] [Google Scholar]
- 43.Moura E, Noordermeer MA, Verhoeven N, Verheul AF, Marx JJ. Iron release from human monocytes after erythrophagocytosis in vitro: an investigation in normal subjects and hereditary hemochromatosis patients. Blood. 1998;92(7):2511–2519. [PubMed] [Google Scholar]
- 44.Drakesmith H, Sweetland E, Schimanski L, et al. The hemochromatosis protein HFE inhibits iron export from macrophages. Proc Natl Acad Sci U S A. 2002;99(24):15602–15607. doi: 10.1073/pnas.242614699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makui H, Soares RJ, Jiang W, Constante M, Santos MM. Contribution of Hfe expression in macrophages to the regulation of hepatic hepcidin levels and iron loading. Blood. 2005;106(6):2189–2195. doi: 10.1182/blood-2005-02-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109(7):2693–2699. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ponka P. Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood. 1997;89(1):1–25. [PubMed] [Google Scholar]
- 48.Quigley JG, Yang Z, Worthington MT, et al. Identification of a human heme exporter that is essential for erythropoiesis. Cell. 2004;118(6):757–766. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 49.Shaw GC, Cope JJ, Li L, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440(7080):96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 50.Kerenyi MA, Grebien F, Gehart H, et al. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood. 2008;112(9):3878–3888. doi: 10.1182/blood-2008-02-138339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.