

Abstract
A-to-I RNA editing, the deamination of adenosine (A) to inosine (I) that occurs in regions of RNA with double-stranded character, is catalyzed by a family of Adenosine Deaminases Acting on RNA (ADARs). In mammals there are three ADAR genes. Two encode proteins that possess demonstrated deaminase activity: ADAR1, which is interferon-inducible, and ADAR2 which is constitutively expressed. ADAR3, by contrast, has not yet been shown to bean active enzyme. The specificity of the ADAR1 and ADAR2 deaminases ranges from highly site-selective to non-selective, dependent on the duplex structure of the substrate RNA. A-to-I editing is a form of nucleotide substitution editing, because I is decoded as guanosine (G) instead of A by ribosomes during translation and by polymerases during RNA-dependent RNA replication. Additionally, A-to-I editing can alter RNA structure stability as I:U mismatches are less stable than A:U base pairs. Both viral and cellular RNAs are edited by ADARs. A-to-I editing is of broad physiologic significance. Among the outcomes of A-to-I editing are biochemical changes that affect how viruses interact with their hosts, changes that can lead to either enhanced or reduced virus growth and persistence dependent upon the specific virus.
The Three ADARs: ADAR1, ADAR2, and ADAR3
ADARs, RNA adenosine deaminases, catalyze the hydrolytic C6 deamination of adenosine to produce inosine in RNA substrates with double-stranded (ds) character (Bass, 2002; Nishikura, 2010; Toth et al., 2006). This reaction is commonly referred to as A-to-I RNA editing (Fig. 1). ADAR activity was first described during antisense RNA studies as a dsRNA duplex unwinding activity in Xenopus oocytes (Bass and Weintraub, 1987; Rebagliati and Melton, 1987). But instead of unwinding dsRNA, the deamination of adenosine in duplexes leads to destabilization of the dsRNA structure because I:U mismatch base pairs are less stable than A:U pairs (Bass and Weintraub, 1988; Strobel et al., 1994; Wagner et al., 1989). A-to-I editing is of two general types: the editing can be highly site-selective with the adenosine deamination occurring at one or very few specific A’s in an RNA; alternatively, multiple A’s may be deaminated in RNA substrates with near perfect duplex structure (Bass, 2002; Gott and Emeson, 2000; Nishikura, 2010; Toth et al., 2006).
Figure 1. A-to-I Editing by ADARs.
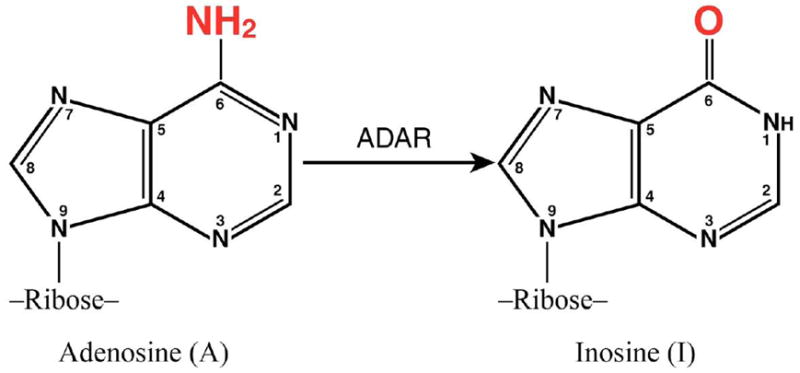
Adenosine deaminases acting on RNA (ADARs) catalyze the hydrolytic C-6 deamination of adenosine (A) to yield inosine (I) in RNA with double-stranded character.
There are three members of the mammalian ADAR gene family, ADAR1, ADAR2 and ADAR3(Bass et al., 1997; Bass, 2002; George et al., 2010; Maas et al., 2003; Nishikura, 2010; Toth et al., 2006). While the three ADAR gene family members share extensive similarity, significant differences also exist among the ADARs in terms of their regulation, their protein domain structure, their subcellular localization and their biological functions. Among the domains present in all of the ADAR proteins are repeated copies of a double-stranded RNA binding domain, designated dsRBD or R, that are found in the central region of the ADAR proteins. The C-terminal region of ADARs has homology to the catalytic domain characteristic of nucleotide deaminases (Fig. 2).
Figure 2. Domain Organization of Human ADAR s.
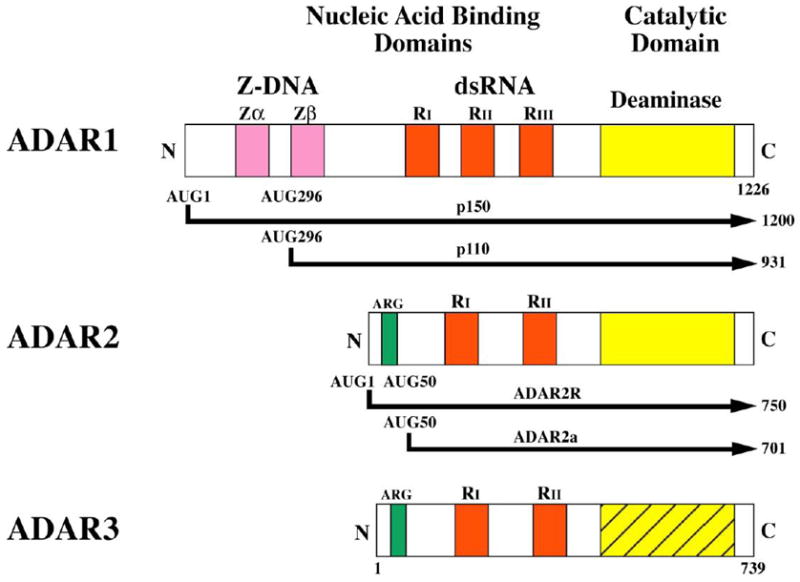
Three ADAR gene family members are known. ADAR1 and ADAR2 are active deaminase enzymes whereas ADAR3 is not known to possess deaminase activity. Alternative promoters and alternative splicing give rise to the different forms of ADAR1 and ADAR2. The p150 isoform of ADAR1 is IFN-inducible. The dsRNA binding domains (RI, RII, RIII), three present in ADAR1 p110 and p150 and two each in ADAR2 and ADAR3, are depicted in red. The C-terminal region deaminase catalytic domain is shown in yellow for ADAR1 and ADAR2, and the homologous region in stipled yellow for ADAR3 that is not yet demonstrated to be an active enzyme. The N-terminal region of the p150 form of ADAR1 possesses two Z-DNA binding domains (Zα and Zβ shown in pink, and the ADAR2R and ADAR3 proteins possess an arginine-rich domain (ARG) shown in green.
ADAR1
The ADAR1 gene is present on human chromosome 1q21 (Wang et al, 1995; Weier et al, 1995) and mouse chromosome 3F2 (Weier et al., 2000). There are two protein size forms of ADAR1 (Patterson and Samuel, 1995), known commonly as p150 and p110 but sometimes referred to as ADAR1-L or large form and ADAR1-S or short form, respectively. ADAR1 also has been known variously as dsRAD, DRADA and K88 in the earlier literature (Bass et al., 1997; Patterson and Samuel, 1995).
ADAR1 p150 is IFN inducible, whereas p110 is constitutively and ubiquitously expressed (George et al, 2005; Patterson et al., 1995; Patterson and Samuel, 1995; Shtrichman et al., 2002). The consensus long open reading frame of the human ADAR1 cDNA is 1226 amino acids in length (Kim et al., 1994b; O’Connell et al., 1995; Patterson and Samuel, 1995). Alternative promoters, only one of which is IFN inducible, drive the expression of the human and mouse ADAR1 genes (George and Samuel, 1999a, 1999b; George et al., 2005; Kawakubo and Samuel, 2000). The IFN inducible ADAR1 promoter possesses a consensus Interferon-stimulated Response Element (ISRE) characteristic of type I IFN regulated genes and an upstream Kinase-consensus Sequence (KCS-like) element similar to that found in the IFN inducible PKR promoter, but the inducible promoter lacks a TATA box (George and Samuel, 1999a,b; George et al., 2005, 2008; Kawakubo and Samuel, 2000; Kuhen and Samuel, 1997; Markle et al., 2003). Because of alternative splicing involving exons 1 and 7, unique transcripts are produced that encode the IFN-inducible p150 protein that is 1200 (human) or 1152 (mouse) amino acids as deduced from the cDNA sequence and the constitutively expressed p110 protein that is 931 (human) or 903 (mouse) amino acids (Fig. 2).
The IFN inducible ADAR1 p150 protein is an N-terminal extension of the constitutively expressed p110 protein. p150 and p110 both possess three copies of the prototypical dsRNA-binding domain, RI, RII and RIII, first characterized in the IFN inducible RNA-dependent protein kinase PKR (Fierro-Monti and Mathews, 2000; Liu and Samuel, 1996; McCormack et al., 1992; Samuel, 1979). The same C-terminal deaminase catalytic domain is present in both p110 and p150 (Kim et al., 1994; O’Connell et al., 1995; Patterson and Samuel, 1995). However, the two ADAR1 proteins differ in their N-terminal region: two copies of a Z-DNA binding domain, Zα and Zβ, are present in p150 but only Zβ is present in p110. The Z-DNA binding domains of ADAR1 have homology to the poxvirus E3L protein N-terminal region that also possesses a Z-DNA binding domain (Herbert et al., 1997, 2001; Patterson and Samuel, 1995; Schwartz et al., 1999).
Mutational analyses have verified the activities assigned to the functional domains as exemplified by the following site-specific amino acid substitutions. Mutation of His910 and Glu912 within the C-terminal region corresponding to the deaminase catalytic core CHAE motif abolished deaminase enzymatic activity of both p110 and p150 (Lai et al., 1995; Liu and Samuel, 1996). The addition of the zinc ion chelator, O-phenanthroline, to deaminase reaction mixtures also inhibits ADAR1 enzyme activity (Kim et al., 1994a). Substitution mutations of a key lysine residue present in the core region of the dsRNA binding domains of ADAR1 (K554, K665, and K777 in RI, RII and RIII, respectively) revealed that RIII was functionally the most important of the three RNA binding domains, and RII the least important, for ADAR1 deaminase activity (Lai et al., 1995; Liu and Samuel, 1996; Liu et al., 2000). The functional importance of the Z-DNA binding domains found in ADAR1, but not ADAR2 or 3, is not yet clear. However, the related Z-DNA binding domain present in the N-terminal region of the poxvirus E3L protein appears to play roles in viral pathogenesis and innate immune signal transduction processes (Kim et al., 2003; Valentine and Smith, 2010). Because the A-form of dsRNA with purine-pyrimidine repeats can be transformed to a left-handed Z-RNA helix and because Zα of ADAR1 can bind Z-RNA (Placido et al, 2007), conceivably a function of the so-called Z-DNA binding domain actually relates to the binding of Z-RNA, but this remains to be established in a physiologically relevant context.
The ADAR1 p150 protein is found in both the cytoplasm and the nucleus, whereas the p110 protein localizes predominantly if not exclusively to the nucleus based on immunofluorescence microscopy of whole cells and biochemical fractionation of cell extracts (Patterson and Samuel, 1995). Nuclear localization and nuclear export signal sequences have been characterized in ADAR1 and the p150 protein has been demonstrated to shuttle between the cytoplasm and nucleus of cells (Eckmann et al., 2001; Fritz et al., 2009; Poulsen et al., 2001; Strehblow et al., 2002). Thus, A-to-I RNA editing catalyzed by ADAR1 p150 could occur either in the nucleus or cytoplasm, whereas editing by ADAR1 p110 would be a nuclear event. RNA editing by ADAR1 can be highly site-selective or the editing can occur at multiple sites. Examples of RNAs edited by ADAR1 with high positional selectivity include cellular transcripts in the nervous system that encode neurotransmitter receptors for glutamate (GluR-B) and serotonin (5-HT2cR) (Burns et al., 1997; Higuchi et al, 1993; Liu et al., 1999; Liu and Samuel, 1999; Niswander et al., 1999) and viral RNAs including hepatitis delta virus antigenome RNA (Taylor, 2003; Casey, 2006) and the kaposin K12 transcript of human herpes virus 8 (Gandy et al., 2007). A-to-I editing of GluR-B and 5-HT2cR transcripts takes place prior to splicing and is specified by imperfect duplex RNA structures formed between adjacent exon and intron sequences; hence, these edits are nuclear events (Seeburg and Hartner, 2003; Toth et al., 2006; Valente and Nishikura, 2005). Beginning with the discovery of A-to-G hyper-editing of measles virus RNA (Cattaneo et al., 1988), several examples of viral RNAs have been identified that possess A-to-G transitions at multiple sites, sequence changes that are presumed to reflect editing by an ADAR. These examples will be discussed later for respective specific viruses, although in most instances it has not yet been established whether ADAR1 or ADAR2 is responsible for the observed sequence changes. Finally, two viral gene products, the vaccinia virus E3L IFN resistance protein (Liu et al., 2001) and the adenovirus VAI RNA (Lei et al., 1998; Taylor et al., 2005) have been demonstrated to antagonize ADAR1 enzymatic activity. It is also conceivable that virus-induced post-translational modifications alter ADAR activity, although this has not yet been demonstrated. However, sumoylation of ADAR1 at Lys418 does alter A-to-I editing activity (Desterro et al., 2005).
ADAR2
The ADAR2 gene is found on human chromosome 21q22 (Mittaz et al., 1997) and mouse chromosome 10C1 (Maas and Gommans, 2009a; Slavov and Gardiner, 2002). ADAR2 also has been known variously as RED1 and DRADA2 in the earlier literature (Bass et al., 1997).
ADAR2, similar to ADAR1, is ubiquitously expressed in most tissues but is most abundant in the brain (Melcher et al., 1996a, 1996b; Gan et al., 2006; Jacobs et al., 2009). ADAR2, like ADAR1, also undergoes alternative splicing events and the diversity of transcripts seen include alternative forms of exons 2, 5, 9 and 10, all of which suggests the existence of different ADAR2 protein isoforms with potentially different activities (Gerber et al., 1997; Maas and Gommans, 2009a; Mittaz et al., 1997; Slavov and Gardiner, 2002). The consensus open reading frame of the human ADAR2 cDNA is 701 amino acids and this form is now referred to as ADAR2a (Bass et al., 1997; Lai et al, 1997; Maas and Gommans, 2009; Mecher et al, 1996b; Mittaz et al., 1997; O’Connell et al., 1997). A second isoform of 750 amino acids designated ADAR2R with an N-terminal extension of the open reading frame by 49 amino acids has been identified (Maas and Gommans, 2009a). The unique N-terminal region of ADAR2R that is not present in ADAR2a includes an arginine-rich domain (ARG, or R) similar in sequence, length and relative position to the ARG or R-domain found in ADAR3 (Maas and Gommans, 2009a; Melcher et al., 1996b). ADAR2a and ADAR2R both possess two copies of the dsRNA-binding domain (RI, RII) instead of three copies as found in ADAR1; the deaminase catalytic domain is present at the C-terminus of ADAR2 similar to the organization for ADAR1 (Fig. 2).
Atomic level structural insights have been gained from the study of ADAR2 domains (Macbeth et al., 2005; Stefl et al., 2006). The NMR solution structure has been solved for the dsRNA-binding domain region of rat ADAR2 (Stefl et al., 2006). The crystal structure of the catalytic domain region of human ADAR2 also has been solved. The structure revealed that the catalytic domain of ADAR2 contains one zinc ion and one molecule of inositol hexakisphosphate. The inositol hexakisphosphate was shown to be a required cofactor for ADAR2 enzymatic activity but not for ADAR1 activity (Macbeth et al., 2005).
ADAR2 localizes predominantly if not exclusively to the nucleus (Desterro et al., 2003; Sansam et al, 2003). ADAR2 displays some overlap with ADAR1 in specificity and substrates that can be edited (Lehmann and Bass, 2000), but also has some uniqueness among substrates. For example, combined biochemical and genetic analyses have revealed that the Glu-R Q/R site is edited by ADAR2, whereas both ADAR1 and ADAR2 can edit the R/G site (Hartner et al., 2004; Higuchi et al., 2000; Liu and Samuel, 1999; Wang et al., 2004). Both ADAR1 and ADAR2 are necessary for complete site-selective editing of the 5-HT2cR substrate RNA (Burns et al., 1997; Hartner et al., 2004; Liu et al., 1999). Editing of transcripts encoding the GluR and 5-HT2cR receptors results in selective amino acid substitutions that affect the functional activities of these neurotransmitter receptor proteins and hence the ensuing neurophysiology. In the case of GluR Q/R editing the calcium permeability of the channel is reduced, whereas the R/G site editing alters kinetic properties of the GluR-B receptor (Seeburg and Hartner, 2003). In the case of 5-HT2cR, full editing reduces G-protein mediated signaling by the receptor (Burns et al. 1997; Niswender et al., 1999). Whether viral infections, for example with neurotropic viruses, leads to alterations in GluR or 5-HT2cR receptor function as a result of changes in the ADAR editing pattern remains to be explored.
ADAR3
The third ADAR, ADAR3, displays significant similarity in size, sequence and domain organization to that of ADAR2R, but otherwise differs from both ADAR1 and 2 in two important ways: ADAR3 lacks enzymatic activity; and, ADAR3 shows restricted tissue expression. While ADAR3 possesses a C-terminal region homologous to the deaminase catalytic core of all known ADARs (Fig. 2), ADAR3 does not display demonstrable deaminase catalytic activity with known A-to-I RNA editing substrates including the GluR-B Q/R, R/G and intronic hot spot sites, the 5-HT2cR sites, or with synthetic dsRNA substrates (Melcher et al, 1996b; Chen et al., 2000). Although ADAR3 lacks editing activity, ADAR3 has been implicated as an inhibitor of both ADAR1 and ADAR2 enzymes (Chen et al., 2000; Cho et al., 2003; Melcher et al., 1996b). Both ADAR1 and ADAR2 function as dimers and are subject to dominant negative effects (Chilibeck et al., 2006; Valente and Nishikura, 2007). Unlike ADAR1 and 2 which are ubiquitously expressed, expression of ADAR3 is restricted to brain regions including amygdala and thalamus (Chen et al., 2000; Melcher et al., 1996b). The N-terminal region of ADAR3, like that of ADAR2R, includes an arginine-rich ARG or R domain (Fig. 2). This ARG/R domain has been shown to possess single-stranded RNA binding activity (Chen et al., 2000) and also specifically interacts with importin-α1 and functions as a nuclear localization sequence (Maas and Gommans, 2009b).
Effects of ADARs and A-to-I Editing on Virus-Host Interactions
ADARs play important roles during viral infections. They can have either a proviral or an antiviral consequence, dependent upon the virus-host combination. A-to-I editing of viral RNAs by ADARs first was implicated from sequence analyses that showed multiple A-to-I (G) substitutions, an occurrence now termed hyper-editing. A-to-I (G) mutations attributed to the action of ADARs during lytic and persistent infections have been described for several different RNA viruses, initially with measles virus (Cattaneo et al., 1988) and then with other RNA viruses including human parainfluenza virus (Murphy et al., 1991), respiratory syncytial virus (Martinez et al., 1997), influenza virus (tenOever et al., 2007), lymphocytic choriomeningitis (LCM) virus (Zhan et al., 2007), Rift Valley fever virus (Suspène et al, 2008), mumps virus (Chambers et al., 2009) and hepatitis C virus (Taylor et al., 2005), and a DNA virus, mouse polyoma virus (Kumar and Carmichael, 1997). Among the RNA viruses implicated as editing targets of ADARs, most are negative-stranded or ambisense in genome organization and coding strategy (Knipe et al., 2007). These examples of viral RNA editing characterized by multiple sequence changes, and others that are decidedly more site-selective including hepatitis delta virus (Jayan and Casey, 2002b; Luo et al., 1990; Wong and Lazinski, 2002), Kaposi’s sarcoma-associated herpesvirus (HHV8) (Gandy et al., 2007) and Epstein-Barr virus (Iizasa et al., 2010), will be considered in further detail. For some of these viruses, the observed A-to-I (G) sequence changes are known to be catalyzed by a specific ADAR and the editing has a subsequent functional consequence that affects the outcome of the virus-host interaction. For viruses that replicate in the cytoplasm of the infected host cell and for which ADAR-mediated A-to-I editing of viral RNA is implicated, based on subcellular localization considerations, it is reasonable to presume that the p150 isoform of ADAR1 is most likely the protein responsible because p150 is the sole ADAR protein found in the cytoplasm. For viruses that replicate in the nucleus, either ADAR1 p110 or p150 or ADAR2 presumably could be responsible for the viral editing events as all of these ADAR proteins are active deaminases and all are found in the nucleus (Bass, 2002; George et al., 2010; Nishikura, 2010; Toth et al., 2006).
The fundamental importance of A-to-I editing to normal physiology of uninfected cells and animals is illustrated by the striking phenotypes seen in the mouse model following either genetic disruption of Adar genes or overexpression of ADAR proteins. Knockouts have been described for Adar1 that disrupt expression of both p110 and p150 (Hartner et al., 2004, 2009; Wang et al., 2004; XuFeng et al. 2009) or only p150 (Ward et al., 2010). The knockout of mouse Adar1 results in embryonic lethality; Adar1−/− embryos die between E11.5 and 12.5 and are characterized by apoptosis in many tissues, liver disintegration and defects in hematopoiesis. By contrast, the Adar2 gene disruption does not result in embryonic lethality, but rather Adar2−/− mice instead display behavioral abnormalities including epileptic seizures (Higuchi et al., 2000). The behavioral dysfunction of Adar2−/− mice interestingly is rescued by the knock-in of GluR-B with an edited (CGG) codon in place of the unedited CAG codon (Higuchi et al., 2000). Finally, transgenic overexpression of either wild-type or deaminase-deficient ADAR2 protein in mice leads to metabolic alterations characterized by hyperphagia and adult-onset obesity (Singh et al., 2007). Adargene targeting studies in the mouse have provided mouse embryo fibroblast cell lines genetically deficient in either ADAR1 or ADAR2. Studies with these cells established unequivocally, in some instances, the site selective roles of ADAR1 and ADAR2 deaminases for editing of viral and cellular RNAs (George et al, 2010; Nishikura, 2010). A-to-I editing has been demonstrated to occur during virus growth and persistence for members of several virus families. Surprisingly, in some instances the ADAR-mediated A-to-I editing has a proviral effect, and in other instances an antiviral effect, as illustrated by the following examples.
Measles Virus and additional Paramyxoviruses
A-to-I (G) substitution editing by in situ C-6 deamination of adenosine has a long history with viruses of the Paramyxoviridae family (Cattaneo and Billeter, 1992), and this form of nucleotide substitution RNA editing should not be confused with the co-transcriptional pseudo-templated G nucleotide insertion editing that causes a frameshift in the transcript that encodes the V protein product of the P/V/C gene (Knipe et al., 2007). Measles virus, a member of the Morbillivirus genus of the family Paramyxoviridae, is an enveloped virus with a nonsegmented negative-stranded ~16-kb RNA genome that specifies five monocistronic genes and the polycistronic P/V/C gene. Measles virus causes a typically acute childhood infection spread by the respiratory route (Moss and Griffin, 2006). However, a rare complication is a persistent infection of the central nervous system (CNS) resulting in a fatal degenerative neurological disease, subacute sclerosing panencephalitis (SSPE), and it is in this context where A-to-G transitions of MV RNA became apparent (Oldstone, 2009).
When MV RNAs from brain autopsies of SSPE patients were characterized by cloning and sequence analysis, numerous differences were found between the sequences of viruses that cause acute infection and those from SSPE patients that included extensive hypermutations in the matrix M gene as well as other viral genes (Baczko et al., 1993; Cattaneo et al., 1986, 1988; Schmid et al., 1992; Wong et al., 1991). The MV sequence changes found in the diseased human brains, U-to-C transitions (or A-to-G dependent upon the strand sequenced), are characteristic of A-to-I (G) editing by an ADAR deaminase (Cattaneo and Billeter, 1992). Reduction of viral M protein synthesis leads to decreased production of infectious virions, and defective M protein production is associated with SSPE (Hall et al., 1979; Liebert et al., 1986). Biologic evidence that the hypermutated matrix M protein of SSPE virus contributes to the persistent infection and progressive CNS disease was provided by transgenic CD46 mouse model studies with MV, including the Edmonston strain virus and recombinant virus engineered to replace the Edmonstron M gene with the M gene of Biken strain SSPE virus (Oldstone et al., 1999; Patterson et al., 2001).
A PCR-based strategy for analysis of viral RNAs has provided additional evidence for A-to-G sequence changes characteristic of ADAR editing events ( Suspène et al., 2008). By inversing the natural hydrogen bonding it was possible to selectively amplify GC-rich viral genomes. The recovery of hyper-edited genomes of Schwarz strain measles virus from IFN sensitive MRC5 cells included multiple A-to-G transitions, whereas genomes amplified from MV infected Vero cells showed typical quasispecies variation of an RNA virus (Suspène et al., 2008). While one study suggested that modification of MV RNAs by ADAR must occur at a very low level if at all based on levels of ADAR activity in cells (Horikami and Moyer, 1995), direct evidence consistent with a functional role of the IFN inducible ADAR1 deaminase as a restriction factor for controlling measles virus infection was provided by the study of mouse embryo fibroblasts (MEFs) stably expressing the SLAM receptor for MV (Ward et al., 2010). MEFs genetically null for the p150 form of ADAR1 displayed extensive syncytium formation and virus-induced cytotoxicity following infection with Edmonston MV strain, and produced infectious yields ~3 to 4 log10 higher at early times after infection, compared to wild-type MEFs (Ward et al., 2010). Studies with human HeLa cells made stably deficient in both ADAR1 p110 and p150 through a short hairpin RNA-mediated knockdown strategy likewise revealed that ADAR1 suppresses MV-induced cytotoxicity. Furthermore, the growth of Moraten vaccine strain MV mutants lacking expression of the accessory proteins V or C was decreased in ADAR1-deficient HeLa cells compared to ADAR1-sufficient cells (Toth et al., 2009). Enhanced apoptosis and virus-induced cytotoxicity in ADAR1-deficient cells correlated with enhanced activation of PKR kinase and interferon regulatory factor IRF3 (Toth et al., 2009). Interestingly, induction of IFN by MV is enhanced by PKR (McAllister et al., 2010) and synthetic dsRNAs containing multiple I:U pairs are sufficient to suppress IFN induction and apoptosis in transfected cells in a manner that correlates with the inhibition of activation of IRF3 (Vitali and Scadden, 2010). Presumably I:U mismatched dsRNA formed under physiologic conditions by the deaminase action of ADARs could act in a similar manner.
Human respiratory syncytial virus (RSV), a member of the Pneumovirus genus of the Paramyxoviridae family, is an important human pathogen. The G glycoprotein of RSV is the virus attachment protein. Antibodies specific for the G protein of RSV have been used to study the antigenic structure of the glycoprotein and to select antibody escape mutants. Among the mutants obtained were hypermutated viruses with multiple A-to-G nucleotide substitution transitions that caused a loss of epitopes of the G glycoprotein (Martínez et al., 1997; Rueda et al., 1994). Such biased sequence changes found in the glycoprotein G gene are consistent with the editing action of an ADAR, and appeared not to be scattered generally throughout the RSV genome as no A-to-G sequence differences were found in the glycoprotein F gene in three G gene mutants (Martínez et al., 1997; Martínez and Melero, 2002). A model has been presented for the generation of the multiple A-to-G transitions in the RSV genome based on the predicted RNA secondary structures that serve as substrates for editing by ADARs (Martínez and Melero, 2002).
Human parainfluenza virus 3 (HPIV3), a member of the Respirovirus genus of the Paramyxoviridae, also has been shown to undergo hyper-mutation consistent with A-to-I editing by an ADAR (Murphy et al., 1991). The 3′-end of HPIV3 RNA recovered from cultured cells persistently infected for 29 months was highly mutated with A-to-G and U-to-C transitions. Deamination catalyzed by ADAR has been implicated as a contributing factor in the sequence changes seen in the live-attenuated Jeryl Lynn vaccine against mumps virus, a member of the Rubulavirus genus of the Paramyxoviridae family (Amexis et al., 2002; Chambers et al., 2009). The Jeryl Lynn vaccine against mumps virus contains two components, JL5 and JL2, that differ by more than 400 nucleotides in sequence (Amexis et al., 2002) with significant sequence variation of the JL2 component (Chambers et al., 2009). Finally, similar to the results obtained with the Edmonston strain MV, ADAR1 p150 protected MEFs from virus-induced cytopathic effects following infection with additional members of the Paramyxoviridae including Newcastle disease virus (NDV, genus Rubulavirus), Sendai virus (genus Respirovirus) and canine distemper virus (CDV, genus Morbillivirus) (Ward et al., 2010).
Vesicular stomatitis virus
In contrast to the results seen with NDV, Sendai and CDV viruses, no significant difference was found between wild-type and ADAR1 p150−/− MEF cells following infection with vesicular stomatitis virus (VSV). The single-cycle yields of VSV were comparable in wild-type and p150−/− mutant MEFs (Ward et al., 2010). The stable over-expression of ADARs in HEK 293 cells also did not affect VSV growth (Li et al., 2010). The single-cycle yield of Indiana serotype VSV was comparable in human 293 cells in which either ADAR1 p150 or ADAR2 was overexpressed compared to parental 293 cells. Furthermore, passage of VSV for up to ten rounds of growth showed similar cycling yields of infectious VSV progeny by 293 parental cells and 293-ADAR1 and 293-ADAR2 overexpressing lines (Li et al., 2010), a cycling phenomenon earlier described for VSV (Palma and Huang, 1974). The stable knockdown of both p110 and p150 ADAR1 in HeLa cells to less than ~10 to 15% of the parental cell levels of p110 and p150 also had no significant effect on VSV growth in the absence of IFN treatment. However, inhibition of VSV growth in IFN-treated cells was ~1 log10 further reduced in the ADAR1 knockdown cells compared to parental cells, and the reduced VSV yield correlated with an increased phosphorylation and activation of the IFN-induced protein kinase PKR (Li et al., 2010).
Prior to the discovery of ADARs, it was found that VSV Indiana defective interfering (DI) particles, when characterized at intervals from a series of undiluted lytic passages of BHK cells, exhibited extensive sequence rearrangements and base substitutions (O’Hara et al., 1984). In one case, the DI particle exhibited a clustering of A-to-G (and U-to-C) transitions in a short but intrinsically A-rich region, sequence changes that would be consistent with editing by an ADAR. Alternatively, the sequence changes might reflect an error-prone VSV polymerase complex rather than A-to-I editing given the lack of an effect of either ADAR1 p150 knockout, ADAR1 p110 and p150 knockdown, or ADAR1 or ADAR2 overexpression on VSV Indiana replication (Li et al., 2010; Ward et al., 2010). Sequence evidence for an ADAR-mediated hypermutation of the PP3 protein of Drosophila sigma virus has been described (Carpenter et al., 2009). Sigma virus is a negative-stranded RNA virus and, like VSV, is a member of the Rhabdoviridae family. Both the biased nature and the nucleotide context of the A-to-G sequence changes seen in sigma virus are consistent with A-to-I editing, although the frequency of their detection was low; the sequence changes were found in only 2 of 104 viral isolates examined. Nonetheless, the observations raise the possibility that ADAR deaminases may plan a role in antiviral immunity in insects (Carpenter et al., 2009) that are known to possess some immune defense strategies similar to those found in mammals (Ding, 2010).
A proviral effect of ADAR1 that enhances VSV replication has been described (Nie et al., 2007). ADAR1 was found to interact with PKR and inhibit PKR activation and eIF-2α phosphorylation, and transfected ADAR1 increased host susceptibility to VSV in wild-type (PKR+) MEFs but not in PKR−/− mutant MEFs (Nie et al., 2007). Both the VSV proviral effect of ADAR1 in transfected MEFs (Nie et al., 2007) as well as the enhancement of plasmid-based ectopic gene expression by ADAR1 seen at the translational level by decreasing PKR-dependent eIF-2α phosphorylation (Wang and Samuel, 2009) required the N-terminal dsRNA binding domain but not the C-terminal deaminase domain of ADAR1.
Influenza virus
Influenza A viruses, enveloped viruses with segmented, negative-stranded RNA genomes consisting of eight different ssRNAs, are members of the Orthomyxoviridae family of viruses. A-to-G transitions are observed in influenza virus RNA isolated from infected mice that possess an intact innate immune signaling system (tenOever et al., 2007). However, mice lacking the IKKε kinase are hyper-susceptible to influenza virus infection, likely because of a defect in IFN signaling in which a subset of ISGs including ADAR1 are poorly expressed. The frequency of A-to-G transitions, the sequence change characteristic of A-to-I editing by an ADAR, was reduced in mice null for IKKε following infection with the WSN strain of influenza virus. When influenza virus RNA isolated from infected lung tissue was analysed, >30% of the clones corresponding to a stem-loop structure present in the viral matrix M1 mRNA showed one or more A-to-G changes in control mice with normal ADAR1 induction, whereas <5% of the clones from IKKε null mice with poor ADAR1 induction possessed an A-to-G transition (tenOever et al., 2007). Whether the editing of influenza virus RNAs in addition to M matrix RNA is altered in the absence of IKKε is not yet known. The p150 isoform of ADAR1 protects against influenza virus-induced cytopathic effects (Ward et al., 2010), but the mechanism of protection is not yet known. Infection of p150−/− MEFs with the mouse adapted WSN strain of influenza A virus resulted in enhanced virus-induced cytopathic effect (CPE) compared to minimal CPE seen with wild-type infected MEFs (Ward et al., 2010). These results taken together are consistent with an antiviral role of ADAR1 against influenza virus.
The influenza virus NS1 non-structural protein is a known antagonist of the interferon response (Randall and Goodbourn, 2008; Samuel, 2001). Among the cellular proteins identified in a yeast two-hybrid screen as an interacting partner with NS1 is ADAR1. The NS1 RNA binding domain and ADAR1 interacted in yeast and co-localized in MDCK cells infected with an H5N1 strain of influenza A (Ngamurulert et al., 2009). Whether the interaction of ADAR1 with NS1 reflects a direct protein: protein interaction or an indirect interaction through an RNA bridge is not yet established. Finally, quantitative analysis of the nucleolar proteome following infection with H1NI PR8 or H3N2 Udorn influenza A strains revealed an increase in nucleolar ADAR1, both p150 and p110, in infected cells (Emmott et al., 2010). The functional consequence of the enhanced amount of ADAR1 present in the nucleolus following influenza infection remains to be established.
Lymphocytic choriomeningitis virus and Rift Valley fever virus
Lymphocytic choriomeningitis virus (LCMV) and Rift Valley fever virus (RVFV) are both enveloped single-stranded RNA viruses with segmented genomes. LCMV is a member of the Arenaviridae family of viruses and has two single-stranded genomic RNA segments, S and L, that display an ambisense coding strategy. An increased frequency of A-to-G and U-to-C mutations has been observed in LCMV infections (Grande-Perez et al., 2002, 2005; Zahn et al., 2007). Again, while the observed LCMV sequence changes are consistent with A-to-I editing, they have not yet been unequivocally established to be caused by the editing action of a specific ADAR isoform.
Infection with LCMV leads to increased expression of the p150 form of ADAR1, both in L929 mouse fibroblasts and in the spleens of C57BL/6 mice (Zahn et al., 2007). The induction of p150 correlates with induced IFN levels in the serum of mice. Although the mutation rate of genomic clones of LCMV in L929 cells did not show any specific pattern at 2 days after infection, after seven days a distinct A-to-G (and U-to-C) bias emerged when the sequence analysis was focused on the glycoprotein GP region within the S segment. Furthermore, infrequent clones showed a hyper-mutation pattern in a region of GP in which 38% of the A’s and 14% of the U’s were mutated. The spleen is a site of high LCMV virus replication in the mouse, and a clear preference for A-to-G and U-to-C transitions was found at four days after infection when mouse spleen tissue was analyzed. Up to 50% of the mutations found in GP in cell culture and mice would be expected to cause mutations at the GP protein, and in some instances the mutations altered the functionality of the viral GP (Zahn et al., 2007). These results suggest that ADAR1, likely p150, is antiviral in the context of the WE strain of LCMV virus in cell culture and the mouse model, with the effects most pronounced after late times after infection. However, no significant differences were seen in the yields of the clone 13 strain of LCMV between wild-type and ADAR1 p150−/− mutant MEFs (Ward et al., 2010).
Rift Valley fever virus is a member of the Bunyaviridae family of viruses and possesses three negative-stranded RNA genomic segments, S, M and L. The RVFV NSs protein product of the S segment is an antagonist of the IFN response, by blocking IFN production and by mediating the degradation of the PKR kinase (Billecocq et al., 2004; Habjan et al., 2009). When a region of the L gene was isolated from RVFV-infected cells by the 3DI-PCR-based strategy that amplifies GC-rich ADAR-edited RNAs, characterization by cloning and sequencing of the PCR products revealed extensive A-to-G hyper-editing of the viral RNA isolated from infected IFN competent MRC5 cells but only quasispecies variation of the RNA from infected Vero cells (Suspene et al., 2008).
Hepatitis C virus
HCV is a member of the Flaviviridae family of viruses and possesses a ~9.6-kb positive-stranded RNA genome contained within an enveloped virion. Chronic liver disease caused by HCV infection, including liver cirrhosis and heptaceullar carcinoma, are estimated to affect ~180 million individuals globally. The present therapy for chronic HCV infection is a combination therapy consisting of pegylated IFNα and ribavirin. While the therapeutic responsiveness of HCV genotypes 2 and 3 approaches 80%, a sustained virologic response as measured by HCV RNA load reduction is much lower for genotype 1 HCV-infected individuals (Fried et al., 2002). The analysis of polymorphisms in several genes of the IFNα system not unexpectedly revealed that host genetic factors affect HCV responsiveness to IFN therapy, in addition to factors including HCV virus genotype, patient age and cirrhosis status (Welzel et al., 2009). Among these genes identified is ADAR. When European American patients in the Hepatitis C Antiviral Long-term Treatment against Cirrhosis (HALT-C) trial were analyzed for relationships between polymorphisms of 13 IFN system pathway genes and sustained response, the benchmark of undetectable HCV RNA at week 72 of therapy was associated with ADAR Ex9 +14A in addition to polymorphisms in IFNAR1and IFNAR2 and JAK1 (Welzel et al., 2009). These latter genes encode receptor subunits for IFN and a tyrosine kinase involved in IFN signal transduction leading to induction of IFN stimulated genes, including Adar1 (George et al., 2008; Samuel, 2001)
While the analysis of polymorphisms in genes such as ADAR1 suggests that ADAR1 may play an important role in the host response to HCV infection (Welzel09), only limited mechanistic studies have been described that directly test this possibility. The HCV replicon system has provided a valuable experimental strategy to study HCV RNA replication and innate antiviral immunity (Appel et al., 2006). Evidence consistent with an antiviral role of ADAR1 emerged when it was found that IFNα treatment of HCV RNA replicon-containing cells inhibited HCV viral RNA production, and that the siRNA-mediated knockdown of ADAR1 or the inhibition of ADAR (and PKR) activity by adenovirus VAI RNA stimulated HCV viral RNA production and reduced inosine-containing viral RNA (Taylor et al, 2005).
The availability of the JFH-1HCV virus and Huh7 hepatoma cell system for production of infectious HCV in cell culture now permits mechanistic analyses of innate antiviral responses to HCV in virus-infected cells (Lemon, 2010). It will be interesting to learn from the study of HCV-infected cells whether ADAR1 is an important contributor to the outcome of the HCV virus-host interaction in the infectious virus cell culture system. However, it is known already that ADAR1 can modulate PKR activation status in cells infected with negative-stranded RNA viruses (Li et al., 2010; Nie et al., 2007; Toth et al., 2009); PKR activation and eIF-2 phosphorylation is suppressed by ADAR1. It also is known that PKR activation in HCV-infected cells results in the inhibition of IFN-stimulated cellular protein synthesis that initiates translation through a 5′-cap dependent mechanism, but not HCV IRES-dependent viral protein synthesis (Garaigoita and Chisari, 2009). Furthermore, HCV also controls IFN production through modulation of PKR activation; PKR-mediated inhibition of IFN induction occurs in JFH1 HCV-infected Huh cells at the level of eIF2-mediated inhibition of translation (Arnaud et al., 2010). Thus, an interesting possibility is that ADAR1 is antiviral in the context of HCV infection through a combination of mechanisms that includes A-to-I editing of HCV RNA that reduces HCV protein expression directly through mutation of viral coding sequences, and editing that inhibits HCV indirectly through suppression of PKR activation because of the A-to-I destablization of viral RNAs that otherwise would activate PKR and prevent expression of both IFN and ISGs that do block HCV replication.
Hepatitis D virus
HDV provides an excellent example of site-selective editing of a viral RNA by ADAR in which the effect of A-to-I editing is proviral (Casey, 2006). HDV is a viroid-like infectious agent that occurs as a subviral satellite of hepatitis B virus (HBV) and is dependent upon HBV for replication to provide the HBV surface antigen as the capsid coat of HDV. The ~1.7 kb HDV genome is a circular RNA that forms a rod-like imperfect double-stranded RNA duplex in which ~70% of the nucleotides are based-paired. HDV encodes two forms of virus-coded delta antigen protein (HDAg), a shorter form (HDAg-S) that is necessary for viral RNA replication and a longer form (HDAg-L) that mediates packaging of the viral genome and is necessary for HDV particle formation (Casey, 2006; Taylor, 2003). Early during replication HDAg-S is produced, but at later times HDAg-L is synthesized. A-to-I editing by ADAR at an “amber/W” site is necessary for production of the HDAg-L protein.
The ADAR editing of HDV antigenome RNA changes the amber (UAG) translation termination codon that stops HDAg-S synthesis to a tryptophan (UIG) codon that permits translation elongation and production of the HDAg-L protein. Without A-to-I editing to generate the U(I=G)G Trp codon, HDAg-L synthesis and subsequent genome packaging into HDV particles does not occur. HDV RNA editing is highly specific for the amber/W site (Polson et al., 1998; Sato et al., 2001). The constitutively expressed p110 isoform of ADAR1 is the ADAR deaminase primarily responsible for editing of the amber/tryptophan site under normal conditions (Jayan and Casey 2002b; Wong and Lazinski, 2002). An important determinant of the amount of editing of the amber/W site that occurs during viral RNA replication is the metastable secondary structure of the HDV RNA substrate that may differ among HDV genotypes and isolates (Jayan and Casey, 2005; Linnstaedt et al., 2010).
While under normal physiologic conditions ADAR1 p110 editing of HDV RNA is proviral, enhanced levels of editing by either ADAR1 or ADAR2 can result in an antiviral response (Jayan and Casey 2002a; Hartwig et al., 2004). Inhibition of HDV replication occurs under conditions of increased HDV RNA editing in Huh cells achieved by ectopic overexpression of either ADAR1 or ADAR2, by IFN treatment to induce increased expression of the endogenous p150 isoform of ADAR1, or by generation of a replication competent HDV that produces increased amounts of HDAg-L due to increased editing (Jayan and Casey 2002a; Hartwig et al, 2004; Sato et al., 2004). Whether inhibition of HDV occurs in natural infections because of enhanced editing by ADAR is unkown. However, when pegylated IFNα is utilized as a therapy for treatment of HBV, if the level of ADAR1 p150 is increased in patients treated with IFN, it is not unreasonable to presume that the elevated p150 might display antiviral activity against HDV if also present as a satellite of HBV in the patient.
Retroviruses including human immunodeficiency virus
Human immunodeficiency virus type 1 (HIV-1), a member of the Lentivirus genus of the family Retroviridae, is an enveloped virus with a positive-stranded RNA genome that is reverse transcribed into a dsDNA provirus that subsequently is integrated into the host’s genome. Several host cell machineries and factors are utilized during HIV-1 replication including those for viral RNA transcription and processing (Brass et al., 2008; Bushman et al., 2009; Knipe et al., 2007). ADAR1 generally has been found to have a proviral role, enhancing HIV-1 expression (Clerzius et al., 2009; Doria et al., 2009; Phuphuakrat et al., 2008). Thus, the action of ADAR1 is in contrast to the mutator activity of APOBEC3G and related cytidine deaminases which fucntion as potent host restriction factors to impair retrovirus growth, including HIV-1, through dC-to-dU editing of retroviral DNA (Chiu and Green, 2008; Malim, 2009).
The expression of ADAR1 but not ADAR2 was increased following PHA-activation of CD4+ T lymphocytes (Phuphuakrat et al., 2008). Overexpression of wild-type ADAR1 but not a catalytic domain HAE to QAA 910912 substitution mutant of ADAR1 lacking deaminase activity upregulated HIV p24 protein expression. While ADAR overexpression increased HIV-1 production, siRNA-mediated knockdown of ADAR1 decreased production of the Gag precursor p55 and the capsid p24 proteins (Phuphuakrat et al., 2008). HIV-1 RNA was cloned and sequenced corresponding to two known regions of structured viral RNA, the transactivation responsive element TAR and the Rev responsive element RRE. The sequences around TAR did not show any repetitive editing, whereas A-to-G changes were found around the RRE region at positions 8164–8166 in the ADAR1 overexpressing cells corresponding to sites within the env gene. Mutant HIV-1 with the A-to-G edited sequence was expressed more efficiently that wild-type HIV-1 (Phuphuakrat et al., 2008). The TAR stem-loop structure of HIV was earlier shown to be a substrate for A-to-I editing by ADARs when injected into Xenopus oocytes (Sharmeen et al., 1991), although the significance of TAR editing in a physiologically relevant context of HIV-1 infected T cells has not yet been clearly established (Phuphuakrat et al., 2008).
Subsequent studies showed that over-expression of ADAR1 in HIV-1 producer cells increased viral replication by both editing-dependent and editing-independent mechanisms. HIV-1 protein expression was enhanced by ADAR1 p150 overexpression in a manner that required the dsRNA binding domains of ADAR but not ADAR1 deaminase activity, and the increased HIV-1 expression correlated with a reduction in PKR activation and eIF-2 phosphorylation (Clerzius et al., 2009; Doria et al., 2009). However, the release of progeny HIV-1 virions was increased by overexpression of wild-type but not a catalytic mutant ADAR1, and these virions displayed enhanced infectivity. ADAR1-mediated editing was detected in an A-rich hairpin region region of the the 5′-UTR shared by all HIV-1 transcripts as well as at sites in the Rev and Tat coding sequences (Doria et al., 2009). Thus, HIV-1 not only takes advantage of ADAR1 to impair antiviral pathways including PKR, but HIV-1 also seemingly employs mechanisms to avoid potentially deleterious A-to-I editing of its genome RNA transcripts in a manner that would impair virus production.
Finally, sequence changes presumed to be caused by A-to-I editing catalyzed by ADARs have been described for Rous-associated virus (RAV-1) (Hajjar and Linial, 1995) and avian leucosis virus (ALV) (Felder et al, 1994). A c-mil/c-raf transducing retrovirus, designated IC4, was isolated that contained a highly mutated U3 long terminal repeat sequence in which 48% of the A was converted to G, a substitution consistent with A-to-I editing (Felder et al., 1994). An avian recombinant provirus generated during in vitro passage was described that contained a ~150 nucleotide repeat region with A-to-G hypermutations, likely arising from editing by an ADAR activity (Hajjar and Linial, 1995).
Human herpesviruses 4 (EBV) and 8 (KSHV)
Epstein-Barr virus (EBV) also known as human herpesvirus 4, and Kaposi’s sarcoma-associated herpesvirus (KSHV) also known as human herpesvirus 8, are both members of Herpesviridae family of dsDNA viruses. Both of these herpesviruses encode RNA transcripts that undergo site-selective A-to-I editing (Gandy et al., 2007; Iizasa et al., 2010). In one case, a viral transcript is edited in a manner that affects both a protein coding sequence and a microRNA (miRNA); in the other case, a virus-derived miRNA is edited.
KSHV is associated with two AIDS-associated malignancies, Kaposi’s sarcoma (KS) and primary effusion lymphoma (PEL). The K12 RNA transcript of KSHV produces multiple kaposin protein variants (A,B,C) and also a miRNA. K12 is abundantly expressed in latent KS-associated KSHV infection and also is induced in lytic infection. The region of the K12 transcript that encodes the miR-K10 microRNA also includes the kaposin A ORF, and has tumorigenic potential (Damania 2004; Muralidhar et al., 1998; Pfeffer et al., 2005). However, selective A-to-I RNA editing corresponding to genome position 117990 within the K12 transcript eliminates transforming activity and is increased during lytic KSHV infection (Gandy et al., 2007). ADAR1 transcripts are ~10 to 20 times more abundant than ADAR2 transcripts in PEL cells, and purified ADAR1 p110 was able to edit the K12 RNA at adenosine 117990 in a highly selective manner (Gandy et al., 2007). Thus, in the case of KSHV, A-to-I editing appears to contribute to the control of the kaposin A and miR-K10 transcript expression and subsequent biology: an unedited A at 117990 is associated with tumorigenicty; the presence of a G (I) at 117990 resulting from A-to-I editing is associated lytic KSHV replication.
EBV is associated with infectious mononucleosis and a variety of lymphomas. Among virus-derived small regulatory RNAs that function as miRNAs, the first was identified in EBV-infected human B cells (Pfeffer et al., 2004). More than 20 EBV miRNA genes are now known and they include BART6. EBV miRNAs are implicated in the transition from lytic to latent infection and the attenuation of antiviral responses (Berkhout and Jeang, 2007; Skalsky and Cullen, 2010). EBV BART6 miRNA primary transcripts (pri-miR BART6) have been shown to undergo A-to-I editing (Iizasa et al., 2010). BART6 miRNA, which targets the Dicer nuclease that cleaves pre-miRNAs into miRNA duplexes, affects the latent state of EBV infection. Editing of the BART6 miRNA inhibits its activity and thus is potentially an important determinant in the regulation of EBV replication and latency (Iizasa et al., 2010). In the context of the known human tropism of EBV, it is interesting that the sites in the 3′-UTR of human Dicer mRNA targeted by the EBV BART6 miRNA are not conserved in either mouse or chimp Dicer mRNAs. In contrast to the editing of EBV BART6 miRNA (Iizasa et al., 2010), the full significance of KSHV K12 miRNA editing is not yet established (Gandy et al., 2007). It is not yet known which of the ADARs is responsible for editing either the EBV or the KSHV miRNAs.
Mouse polyoma virus
Mouse polyoma virus (PyV), a member of the Polyomaviridae, is a small DNA virus that possesses a ~5-kb double-stranded circular DNA genome. In mouse cells permissive for productive PyV infection, viral transcription occurs in the nucleus whereby early and late promoters drive expression from opposite strands of the viral genome. Spliced early RNA transcripts encode the three T antigens including large T important for DNA replication, and the late viral transcripts encode the three capsid structural proteins. At late times in PyV multiplication cycle the early mRNA is down-regulated by nuclear antisense late-strand RNA (Liu et al., 1994). It has been convincingly demonstrated by sequence analysis that the overlap between early and late transcripts and their 3′-polyadenylation signals leads to extensive A-to-G substitutions (Gu et al., 2009; Kumar and Carmichael, 1997). Early-strand RNAs present in the nucleus at late times after infection possess extensive A to G (I) sequence changes consistent with hyper-deamination by an ADAR activity (Kumar and Carmichael, 1997). After knocking down ADAR1 by only about 4-fold in NIH3T3 cells by siRNA transfection, a defect in the early-to-late switch was reported (Gu et al., 2009). MEFs genetically null in either Adar1 or Adar2, both of which are nuclear enzymes, would provide an approach to establish which of the ADARs is responsible for the biased hyper-editing of PyV RNA.
Biochemical Mechanisms by which ADARs and A-to-I Editing Mediate Biologic Changes that Affect Virus-Host Interactions
A-to-I editing by the ADAR deaminases is of immense physiologic importance, with demonstrated profound consequences both in virus-infected cells and in uninfected cells in culture and intact animals (George et al., 2010; Hundley and Bass, 2010; Nishikura, 2010; Seeburg and Hartner, 2003; St. Laurent et al., 2009). Because an edited RNA possesses a different sequence than its unedited counterpart (A-to-I, C-to-U) whose sequence corresponds to the genome specified sequence, the edited RNAs may have different functional activities. Established and potential biochemical mechanisms by which A-to-I RNA editing by ADARs may affect gene expression and function and alter the outcome of virus-host interactions are summarized in Figure 3.
Figure 3. Mechanisms by which A-to-I RNA Editing may affect Gene Expression and Function.
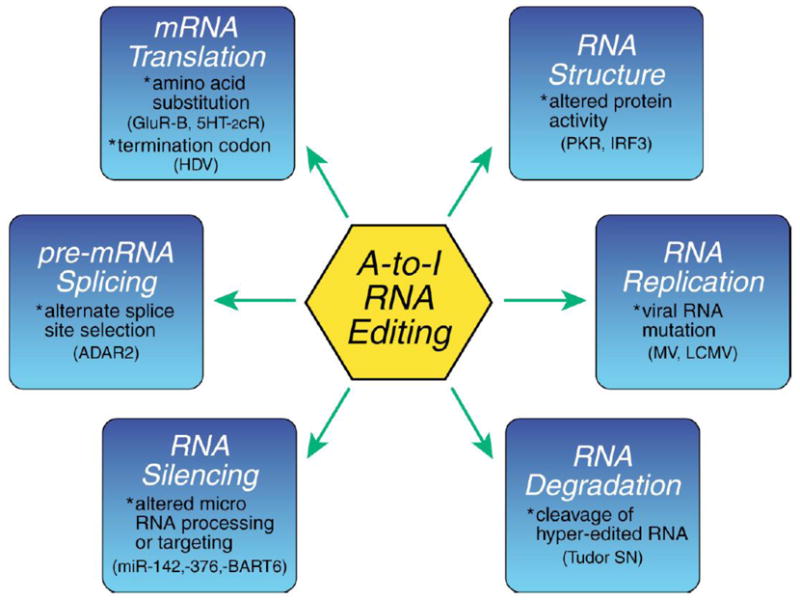
Deamination of adenosine (A) to produce in osine (I) in duplex RNA structures catalyzed by ADARs leads to the nucleotide substitution of an I for an A in an RNA sequence. Because I base pairs as G instead of A, A-to-I editing may effect gene expression and function in virus-infected cells by a number of ways that includes: mRNA translation, by changing codons and hence the amino acid sequence of synthesized proteins; pre-mRNA splicing, by changing a conserved A in splice site recognition sequences; RNA stability by altering sequences and structures involved in nuclease recognition; viral genome genetic stability, by changing template and thus product sequences by deamination leading to A-to-G (U-to-C) transitions during viral RNA synthesis; and RNA structure-dependent activities including microRNA production or targeting or dsRNA protein-RNA interactions involved in innate immune responses. [Adapted from Samuel 2003 with permission])
Translation
A-to-I editing may alter the process of mRNA translation (Fig. 3). The purine inosine base pairs as guanosine with cytidine instead of adenosine with uridine. Hence, I in an mRNA triplet codon is read as G instead of A by ribosomes during translation. The genetic recoding that happens as a result of editing, if occurring in a highly site-selective manner, can lead to amino acid substitutions and hence protein products with altered activity. Among the best characterized examples of site-selective editing is the HDV viral RNA in which a stop codon becomes a tryptophan codon permitting the synthesis of large delta antigen (Casey, 2006; Taylor, 2003). Two well characterized examples of site-selective editing of cellular mRNAs leading to amino acid substitutions are the transcripts in the nervous system for the glutamate receptor GluR-B and the serotonin receptor 5-HT2CR (Jepson and Reenan, 2008; Seeburg and Hartner, 2003). In these cases, the receptors produced following transcript editing by ADARs display altered kinetic properties and calcium permeability (GluR) and altered G-protein mediated signaling (5-HT2CR). Conceivably neurotropic viruses, or for that matter viral infections generally that lead to induction of IFN and hence increased levels of ADAR1 deaminase, might under some pathological conditions lead to changes in editing of neurotransmitter receptor transcripts and hence altered neurophysiology, although this has yet to be established. Editing of viral transcripts can occur at multiple sites, thereby generating altered reading frames with multiple amino acid substitutions if not premature termination codons. These changes to ORFs altogether may result in aberrant or reduced viral protein production as exemplified by matrix and envelope glycoprotein products of MV, RSV and LCMV (Cattaneo et al., 1988; Martinez et al., 1997; Schmid et al., 1992; Zahn et al., 2007).
RNA structure
A-to-I editing also can cause a change in RNA structure (Fig. 3), as more stable Watson-Crick A:U base pairs became less stable I:U wobble base pairs (Bass, 2002; George et al., 2010; Serra et al., 2004; Toth et al, 2006; Valente and Nishikura, 2005). Indeed, the reduced stability of a duplex RNA following ADAR-catalyzed A-to-I editing of dsRNA is the basis of discovery of ADARs in Xenopus oocytes during the course of antisense RNA studies (Rebagliati and Melton 1987; Bass and Weintraub 1987). A quantitation of the reduced duplex stability observed upon conversion of a A:U pair to a I:U base pair, as would be seen following adenosine deamination in a dsRNA by an ADAR, has been described (Strobel et al., 1994). An internal I:U pair is less stable (approximately 1 kcal per mol) than an internal G:U pair, and substantially less stable (approximately 2 kcal per mol) than an internal A:U base pair. ADAR-mediated suppression of proapoptotic and dsRNA-dependent activities exemplified by PKR and IRF3 have been described in virus-infected cells (Li et al., 2010; Nie et al., 2007; Toth et al., 2009). Furthermore, synthetic dsRNAs containing multiple I:U base pairs, as would be formed by the action of ADAR, suppress the IFN response and apoptosis. I:U dsRNA inhibits the activation of IRF3, possibly the result of impairment of IPS-adaptor dependent cytosolic RNA sensors RIG-I and MDA5 (Vitali and Scadden, 2010).
Pre-mRNA Splicing
A-to-I editing may alter pre-mRNA splicing patterns (Fig. 3). As exemplified by ADAR2 transcripts, editing can change a splice site-recognition sequence. Among the cellular RNA transcripts edited in the nucleus by ADAR2 is its own pre-mRNA transcript. ADAR2 autoregulation by autoediting of ADAR2 pre-mRNA creates a 3′-splice site (from AA to AI) for a subsequent alternative splicing event that leads to the insertion of 47 nucleotides from intron I into the mature transcript (Rueter et al., 1999; Feng et al., 2006). While not yet shown to occur for a viral mRNAs, it nevertheless is possible that an ADAR could affect alternative splicing of a nuclear viral transcript in a manner conceptually similar to that established for the editing of pre-mRNA by ADAR2.
RNA replication
A-to-I editing may conceivably contribute to the generation of viral genome mutations in the case of viruses that undergo RNA-dependent RNA replication (Fig. 3). Again, because the purine I base pairs as G instead of A, editing by ADAR could generate A-to-G or U-to-C transitions. If a secondary structure in the template RNA was edited by an ADAR-mediated adenosine deamination, this would be expected to lead to a complementary change in the product strand following RNA-dependent RNA replication by viral polymerases in virus-infected cells. For single-stranded RNA viruses, editing of either the genome or antigenome RNA strand is theoretically possible (Casey, 2006; Cattaneo and Billeter, 1992; Toth et al., 2006).
RNA silencing
A-to-I editing has been demonstrated to alter microRNA (miR) and siRNA silencing processes (Fig. 3). Editing of both cellular and viral microRNAs by ADARs has been demonstrated, exemplified by miR-142, miR-151 and miR-376 (cellular) and miR-BART6 (EBV). The known effects of ADARs on RNA silencing include altered processing of miR precursors and altered targeting of miRs and siRNAs (Habig and others 2007; Heale et al., 2009; Luciano et al., 2004; Nishikura, 2010).
Drosha and Dicer endonucleases function together with dsRNA binding proteins to process pri-miRNA transcripts to produce mature miRs (Berkhout and Jeang, 2007; Filipowicz et al. 2008). It is estimated that ~20% of the human pri-miRs are subject to A-to-I editing by ADAR1 and ADAR2 (Kawahara et al., 2008). A-to-I editing of the pri-miR-142 leads to an inhibition of processing of the precursor transcript in the nucleus by the Drosha-DGCR8 complex, consequently reducing the level of mature miR-142 produced (Yang et al., 2006). A-to-I editing of pri-miR-151 inhibits cleavage by Dicer-TRBP in the cytoplasm (Kawahara et al, 2007a). ADAR2-mediated A-to-I editing within the seed sequence of miR-376 alters the targeting of the miR to silence different genes than the unedited miR-376 (Kawahara et al, 2007b). A-to-I editing of the viral pri-miR-BART6 in latently EBV-infected cells inhibits its expression or loading onto functionally active RISC complexes (Iizasa et al., 2010). Finally, ADAR proteins were found in some instances to modulate gene silencing independent of deaminase catalytic activity (Heale et al., 2009; Yang et al., 2005). Both ADAR1 and ADAR2 bind to siRNAs, and ADARs can impair the knockdown efficiency of synthetic siRNAs (Peacock et al., 2010; Scadden and Smith, 2001a; Yang et al., 2005). ADAR2 also can modulate the processing of miRNA-376a independent of catalytic activity (Heale and others 2009). Finally, modulation of cellular miRs by IFN has been described as an antiviral defense mechanism in the case of HCV (Pedersen et al., 2007).
RNA Degradation
Finally, A-to-I editing may target RNAs for degradation (Fig. 3). A ribonuclease was identified that specifically targeted hyper-edited duplex RNAs, with efficient cleavage within a region of multiple I:U base pairs (Scadden and Smith, 2001b). ADAR1 and ADAR2 generated the cleavage site when hyper-editing long dsRNA (Scadden and O’Connell, 2005). Tudor staphylococcal nuclease (Tudor-SN), a component of the RISC complex in RNAi, was then identified as a nuclease that bound and cleaved I-containing dsRNA (Scadden, 2005).
Bioinformatic analyses of the human transcriptome identified several candidate A-to-I editing sites. Curiously most of these sites were found in non-coding regions, introns and 3′-UTRs that contain inverted repeats exemplified by Alu repeats (Athanasiadis et al., 2004; Kawahara et al., 2008; Kim et al., 2004; Levanon et al., 2004, 2005; Sakurai et al., 2010) in addition to miRs (Kawahara et al., 2008). Among the functions of the noncoding intronic editing of Alu sequences mediated by ADAR1 is to prevent aberrant exonization of Alu sequences into mature processed mRNA (Sakurai et al., 2010). Another function of the A-to-I editing appears to target hyper-edited I-containing RNAs for degradation (Osenberg et al., 2009; Scadden, 2005; Scadden and O’Connell, 2005). Possibly additional factors are needed in some instances to achieve selectivity (Agranat et al, 2008), but the consistent levels of A-to-I editing seen including in Alu repeats (Greenberger et al., 2010)suggests a tightly regulated process. Because ADAR1 p150 is IFN inducible (Patterson and Samuel, 1995; George et al., 1999a,b; 2005) and because editing of specific transcripts can become increased in human cells treated with IFN (Hartwig et al., 2004; Yeo et al., 2010), it is tempting to speculate that the RNase selective for I-containing duplex RNA could play an antiviral role if the targeted RNA was viral or even cellular perhaps encoding a component required by the virus for replication (Scadden and Smith, 1997; 2001b).
Summary and Future Prospectives
Do we need editors in virology? Dependent upon the circumstances, the answer is an emphatic and unequivocal “yes”, whether the editors are ADARs editing RNA or APOBECs editing DNA. In the case of the former, the subject of this review, the outcome of altered ADAR protein expression and changes in A-to-I editing during viral infection can be either antiviral or proviral, dependent upon the virus-host combination. Considerable progress has been made toward understanding the biochemistry and molecular biology of the ADAR family of proteins and the physiological consequences of their expression and actions.
We now know two mammalian genes that encode an enzymatically active RNA adenosine deaminase, Adar1and Adar2. Furthermore, through a gene organization strategy that includes alternative promoters, one of which for Adar1 is interferon inducible, together with several different alternative splice variants, multiple different protein variants of ADAR1 and ADAR2 potentially can be expressed that potentially possess different cellular localizations and different biochemical activities. But with few exceptions, such differences have yet to be established. One precedent, though, is the expression of two size isoforms of ADAR1, p150 and p110, through the use of altnerative promoters and splicing. The large protein size isoforms of ADAR1, p150, is expressed from a promoter that is interferon and pathogen inducible, and the subcellular localization and some biochemical activities of ADAR1 p150 differ from those of the other known active ADAR deaminases, ADAR1 p110 and ADAR2.
Studies of the regulation of expression of ADARs and identification of sequence changes characterized by A-to-G and U-to-C nucleotide substitutions observed during persistent infections initially established a link between the interferon system and a possible role of ADARs in the host response to viral infection. Knockouts of ADARs in mice and knockdowns in cultured cells further established a relationship between the innate immune interferon response and ADAR1, and between stress-induced apoptotic responses and ADAR1. While insight has been gained regarding how the ADAR1 deaminase and A-to-I editing may affect IFN induction and action and virus-induced cytotoxicity, including through modulation of PKR and IRF3 activation and RIG-I/MDA5 cytosolic RNA sensing, the precise molecular mechanisms remain to be elucidated.
Most effects attributed to ADARs in virus-infected cells are dependent upon ADAR catalytic activity and A-to-I editing, but unexpectedly some catalytic-independent effects also have been found for ADAR1 and ADAR2, both in infected and uninfected cells and mice. These editing independent effects possibly reflect the consequence of ADAR protein interactions with other cellular or viral proteins, or alternatively they may arise either because of sequestration or steric effects following RNA binding by ADAR. Whether these editing independent changes occur during viral infections and under physiologically relevant conditions of ADAR expression remain to be fully elucidated.
There remains much to be learned about ADARs and their roles in virus-infected cells. The details of the molecular mechanisms by which ADARs modulate virus-host interactions, in some cases observed as an antiviral effect and in other cases as a proviral effect, with a few exceptions largely remain unresolved. For some viruses, the functional effect of ADAR is clearly a direct consequence of enzymatic editing of the viral RNA, as exemplified by HDV. But in other situations the effect that an ADAR may exert on the host response to infection may be indirect, for example through the editing of a cellular RNA that encodes a membrane channel or neurotransmitter receptor that subsequently alters the virus-induced cytopathogenic response. The discovery that noncoding RNAs are frequent targets of ADARs, including microRNAs and Alu elements in UTRs, has expanded the possible role of ADARs beyond the mechanistic changes attributed to alternations of coding potential by virtue of editing within open reading frames. Both viral and cellular microRNA transcripts have been identified that are edited by ADARs. Conceivably virus infection together with IFN treatment leads to changes in the profile of viral and cellular microRNAs as a consequence of A-to-I editing, thereby altering the virus-host interaction.
With few examples, it is not yet fully clear what determines the balance between an antiviral and a proviral effect of ADAR in a viral infection. From insights gained so far, the type of virus examined, the kind of cell infected, and whether the cell is interferon treated or not, altogether contribute to determining the outcome of the virus-host interaction and the contributing roles of ADARs. But whether the rate-limiting action of an ADAR in a specific viral infection always requires A-to-I editing activity, or in some instances may be independent of ADAR enzymatic activity, and whether the targeting of a viral or alternatively a cellular RNA represents the rate-limiting step attributed to ADAR action that contributes to determining the outcome of a given infective process, in most instances remains to be established. Precedents have been established for both editing dependent and editing independent ADAR responses, and for ADAR actions in virus-infected cells that are exerted toward both viral and cellular targets.
Acknowledgments
I thank the members of our laboratory for their helpful comments and the many investigators within the RNA editing and virology fields for their scholarly contributions that taken together made this review possible. Work from the author’s laboratory was supported in part by the National Institute of Allergy and Infectious Diseases, NIH (Research Grants AI-12520 and AI-20611).
Abbreviations
- ADAR
adenosine deaminase acting on RNA
- bp
base pair
- dsRNA
double-stranded RNA
- IFN
interferon
- ISRE
interferon-stimulated response element
- nt
nucleotide
- PKR
protein kinase regulated by RNA
- ssRNA
single-stranded RNA
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agranat L, Raitskin O, Sperling J, Sperling R. The editing enzyme ADAR1 and the mRNA surveillance protein hUpf1 interact in the cell nucleus. Proc Natl Acad Sci USA. 2008;105:5028–5033. doi: 10.1073/pnas.0710576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amexis G, Rubin S, Chizhikov V, Pelloquin F, Carbone K, Chumakov K. Sequence diversity of Jeryl Lynn strain of mumps virus: quantitative mutant analysis for vaccine quality control. Virology. 2002;300(2):171–179. doi: 10.1006/viro.2002.1499. [DOI] [PubMed] [Google Scholar]
- Appel N, Schaller T, Penin F, Bartenschlager R. From structure to function: new insights into hepatitis C virus RNA replication. J Biol Chem. 2006;281:9833–9836. doi: 10.1074/jbc.R500026200. [DOI] [PubMed] [Google Scholar]
- Arnaud N, Dabo S, Maillard P, Budkowska A, Kalliampakou KI, Mavromara P, Garcin D, Hugon J, Gatignol A, Akazawa D, Wakita T, Meurs EF. Hepatitis C virus controls interferon production through PKR activation. PLoS ONE. 2010;5:e10575. doi: 10.1371/journal.pone.0010575. doi: 10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2(12):e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baczko K, Lampe J, Liebert UG, Brinckmann U, ter Meulen V, Pardowitz I, Budka H, Cosby SL, Isserte S, Rima BK. Clonal expansion of hypermutated measles virus in a SSPE brain. Virology. 1993;197:188–195. doi: 10.1006/viro.1993.1579. [DOI] [PubMed] [Google Scholar]
- Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Nishikura K, Keller W, Seeburg PH, Emeson RB, O’Connell MA, Samuel CE, Herbert A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA. 1997;3:947–949. [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. A developmentally regulated activity that unwinds RNA duplexes. Cell. 1987;48:607–613. doi: 10.1016/0092-8674(87)90239-x. [DOI] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–1098. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- Berkhout B, Jeang KT. RISCy business: MicroRNAs, pathogenesis, and viruses. J Biol Chem. 2007;282:26641–26645. doi: 10.1074/jbc.R700023200. [DOI] [PubMed] [Google Scholar]
- Billecocq A, Spiegel M, Vialat P, Kohl A, Weber F, Bouloy M, Haller O. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J Virol. 2004;78:9798–9806. doi: 10.1128/JVI.78.18.9798-9806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- Bushman FD, Malani N, Fernandes J, D’Orso I, Cagney G, Diamond TL, Zhou H, Hazuda DJ, Espeseth AS, König R, Bandyopadhyay S, Ideker T, Goff SP, Krogan NJ, Frankel AD, Young JA, Chanda SK. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter JA, Keegan LP, Wilfert L, O’Connell MA, Jiggins FM. Evidence for ADAR-induced hypermutation of the Drosophila sigma virus (Rhabdoviridae) BMC Genet. 2009;10:75. doi: 10.1186/1471-2156-10-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JL. RNA editing in hepatitis delta virus. Curr Top Microbiol Immunol. 2006;307:67–89. doi: 10.1007/3-540-29802-9_4. [DOI] [PubMed] [Google Scholar]
- Cattaneo R, Billeter MA. Mutations and A/I hypermutations in measles virus persistent infections. Curr Top Microbiol Immunol. 1992;176:63–74. doi: 10.1007/978-3-642-77011-1_5. [DOI] [PubMed] [Google Scholar]
- Cattaneo R, Schmid A, Eschle D, Baczko L, ter Meulen V, Billeter MA. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;55:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo R, Schmid A, Rebmann G, Baczko K, Ter Meulen V, Bellini WJ, Rozenblatt S, Billeter MA. Accumulated measles virus mutations in a case of subacute sclerosing panencephalitis: interrupted matrix protein reading frame and transcription alteration. Virology. 1986;154:97–107. doi: 10.1016/0042-6822(86)90433-2. [DOI] [PubMed] [Google Scholar]
- Chambers P, Rima BK, Duprex WP. Molecular differences between two Jeryl Lynn mumps virus vaccine component strains, JL5 and JL2. J Gen Virol. 2009;90:2973–2981. doi: 10.1099/vir.0.013946-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6:755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilibeck KA, Wu T, Liang C, Schellenberg MJ, Gesner EM, Lynch JM, MacMillan AM. FRET analysis of in vivo dimerization by RNA-editing enzymes. J Biol Chem. 2006;281:16530–16535. doi: 10.1074/jbc.M511831200. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J Biol Chem. 2003;278:17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- Clerzius G, Gélinas JF, Daher A, Bonnet M, Meurs EF, Gatignol A. ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J Virol. 2009;83:10119–10128. doi: 10.1128/JVI.02457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damania B. Oncogenic gamma-herpesviruses: comparison of viral proteins involved in tumorigenesis. Nat Rev Microbiol. 2004;2:656–668. doi: 10.1038/nrmicro958. [DOI] [PubMed] [Google Scholar]
- Desterro JM, Keegan LP, Lafarga M, Berciano MT, O’Connell M, Carmo-Fonseca M. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci. 2003;116:1805–1818. doi: 10.1242/jcs.00371. [DOI] [PubMed] [Google Scholar]
- Desterro JMP, Keegan LP, Jaffray E, Hay RT, O’Connell MA, Carmo-Fonseca M. SUMO-1 modification alters ADAR1 editing activity. Mol Biol Cell. 2005;16:5115–5126. doi: 10.1091/mbc.E05-06-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SW. RNA-based antiviral immunity. Nat Rev Immunol. 2010;10:632–644. doi: 10.1038/nri2824. [DOI] [PubMed] [Google Scholar]
- Doria M, Neri F, Gallo A, Farace MG, Michienzi A. Editing of HIV-1 RNA by the double-stranded RNA deaminase ADAR1 stimulates viral infection. Nucleic Acids Res. 2009;37:5848–5858. doi: 10.1093/nar/gkp604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann CR, Neunteufl A, Pfaffstetter L, Jantsch MF. The human but not the xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol Biol Cell. 2001;12:1911–1924. doi: 10.1091/mbc.12.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmott E, Wise H, Loucaides EM, Matthews DA, Digard P, Hiscox JA. Quantitative proteomics using SILAC coupled to LC-MS/MS reveals changes in the nucleolar proteome in influenza A virus-infected cells. J Proteome Res. 2010;9:5335–5345. doi: 10.1021/pr100593g. [DOI] [PubMed] [Google Scholar]
- Felder MP, Laugier D, Yatsula B, Dezélée P, Calothy G, Marx M. Functional and biological properties of an avian variant long terminal repeat containing multiple A to G conversions in the U3 sequence. J Virol. 1994;68:4759–4767. doi: 10.1128/jvi.68.8.4759-4767.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Sansam CL, Singh M, Emeson RB. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol Cell Biol. 2006;26:480–488. doi: 10.1128/MCB.26.2.480-488.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro-Monti I, Mathews MB. Proteins binding to duplexed RNA: One motif, multiple functions. Trends Biochem Sci. 2000;25:241–246. doi: 10.1016/s0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Jr, Häussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- Fritz J, Strehblow A, Taschner A, Schopoff S, Pasierbek P, Jantsch MF. RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol Cell Biol. 2009;29:1487–1497. doi: 10.1128/MCB.01519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy SZ, Linnstaedt SD, Muralidhar S, Cashman KA, Rosenthal LJ, Casey JL. RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. J Virol. 2007;81:13544–13551. doi: 10.1128/JVI.01521-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6:513–522. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Das S, Samuel CE. Organization of the mouse RNA-specific adenosine deaminase Adar1 gene 5′-region and demonstration of STAT1-independent, STAT2-dependent transcriptional activation by interferon. Virology. 2008;380:338–343. doi: 10.1016/j.virol.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Gan Z, Liu Y, Samuel CE. Adenosine deaminases acting on RNA (ADARs), RNA editing and interferon action. J Interferon Cytokine Res. 2010 doi: 10.1089/jir.2010.0097. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci U S A. 1999a;96:4621–4626. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Samuel CE. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene. 1999b;229:203–213. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- George CX, Wagner MV, Samuel CE. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem. 2005;280:15020–15028. doi: 10.1074/jbc.M500476200. [DOI] [PubMed] [Google Scholar]
- Gerber A, O’Connell MA, Keller W. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA. 1997;3:453–463. [PMC free article] [PubMed] [Google Scholar]
- Gott JM, Emeson RB. Functions and mechanisms of RNA editing. Annu Rev Genet. 2000;34:499–531. doi: 10.1146/annurev.genet.34.1.499. [DOI] [PubMed] [Google Scholar]
- Grande-Pérez A, Lázaro E, Lowenstein P, Domingo E, Manrubia SC. Suppression of viral infectivity through lethal defection. Proc Natl Acad Sci USA. 2005;102:4448–4452. doi: 10.1073/pnas.0408871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande-Pérez A, Sierra S, Castro MG, Domingo E, Lowenstein PR. Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc Natl Acad Sci U S A. 2002 Oct 1;200299(20):12938–43. doi: 10.1073/pnas.182426999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberger S, Levanon EY, Paz-Yaacov N, Barzilai A, Safran M, Osenberg S, Amariglio N, Rechavi G, Eisenberg E. Consistent levels of A-to-I RNA editing across individuals in coding sequences and non-conserved Alu repeats. BMC Genomics. 2010;11:608. doi: 10.1186/1471-2164-11-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu R, Zhang Z, Decerbo JN, Carmichael GG. Gene regulation by sense-antisense overlap of polyadenylation signals. RNA. 2009;15:1154–1163. doi: 10.1261/rna.1608909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habjan M, Pichlmair A, Elliott RM, Overby AK, Glatter T, Gstaiger M, Superti-Furga G, Unge H, Weber F. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J Virol. 2009;83:4365–4375. doi: 10.1128/JVI.02148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall WW, Lamb RA, Choppin PW. Measles and subacute sclerosing panencephalitis virus proteins: lack of antibodies to the M protein in patients with subacute sclerosing panencephalitis. Proc Natl Acad Sci USA. 1979;76:2047–2051. doi: 10.1073/pnas.76.4.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar AM, Linial ML. Modification of retroviral RNA by double-stranded RNA adenosine deaminase. J Virol. 1995;69:5878–5882. doi: 10.1128/jvi.69.9.5878-5882.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig D, Schoeneich L, Greeve J, Schutte C, Dorn I, Kirchner H, Hennig H. Interferon-alpha stimulation of liver cells enhances hepatitis delta virus RNA editing in early infection. J Hepatol. 2004;41:667–672. doi: 10.1016/j.jhep.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O’Connell MA. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009;28:3145–3156. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert A, Alfken J, Kim YG, Mian IS, Nishikura K, Rich A. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc Natl Acad Sci U S A. 1997;94:8421–8426. doi: 10.1073/pnas.94.16.8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert A, Rich A. The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc Natl Acad Sci U S A. 2001;98:12132–12137. doi: 10.1073/pnas.211419898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Stefan M, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an ampa receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- Horikami SM, Moyer SA. Double-stranded RNA adenosine deaminase activity during measles virus infection. Virus Res. 1995;36:87–96. doi: 10.1016/0168-1702(94)00103-j. [DOI] [PubMed] [Google Scholar]
- Hundley HA, Bass BL. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci. 2010;35:377–383. doi: 10.1016/j.tibs.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizasa H, Wulff BE, Alla NR, Maragkakis M, Megraw M, Hatzigeorgiou A, Iwakiri D, Takada K, Wiedmer A, Showe L, Lieberman P, Nishikura K. Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J Biol Chem. 2010;285:33358–33370. doi: 10.1074/jbc.M110.138362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MM, Fogg RL, Emeson RB, Stanwood GD. ADAR1 and ADAR2 expression and editing activity during forebrain development. Dev Neurosci. 2009;31:223–237. doi: 10.1159/000210185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayan GC, Casey JL. Increased RNA editing and inhibition of hepatitis delta virus replication by high-level expression of ADAR1 and ADAR2. J Virol. 2002a;76:3819–3827. doi: 10.1128/JVI.76.8.3819-3827.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayan GC, Casey JL. Inhibition of hepatitis delta virus RNA editing by short inhibitory RNA-mediated knockdown of ADAR1 but not ADAR2 expression. J Virol. 2002b;76:12399–12404. doi: 10.1128/JVI.76.23.12399-12404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayan GC, Casey JL. Effects of conserved RNA secondary structures on hepatitis delta virus genotype I RNA editing, replication, and virus production. J Virol. 2005;79(17):11187–11193. doi: 10.1128/JVI.79.17.11187-11193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Reenan RA. RNA editing in regulating gene expression in the brain. Biochim Biophys Acta. 2008;1779:459–470. doi: 10.1016/j.bbagrm.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36:5270–5280. doi: 10.1093/nar/gkn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007a;8:763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007b;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakubo K, Samuel CE. Human RNA-specific adenosine deaminase (ADAR1) gene specifies transcripts that initiate from a constitutively active alternative promoter. Gene. 2000;258:165–172. doi: 10.1016/s0378-1119(00)00368-1. [DOI] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res. 2004;14:1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, Garner TL, Sanford T, Speicher D, Murray JM, Nishikuraaa K. Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts. J Biol Chem. 1994b;269:13480–13489. [PubMed] [Google Scholar]
- Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Molecular cloning of c DNA for double-stranded-RNA adenosine-deaminase, a candidate enzyme for nuclear-RNA editing. Proc Natl Acad Sci U S A. 1994a;91:11457–11461. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, Rich A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci U S A. 2003;100:6974–6979. doi: 10.1073/pnas.0431131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe D, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. Fields Virology. 5. Lippincott Williams and Wilkins; Philadelphia: 2007. [Google Scholar]
- Kuhen KL, Samuel CE. Isolation of the interferon-inducible RNA-dependent protein kinase PKR promoter and identification of a novel DNA element within the 5′-flanking region of human and mouse Pkr genes. Virology. 1997;227:119–130. doi: 10.1006/viro.1996.8306. [DOI] [PubMed] [Google Scholar]
- Kumar M, Carmichael GG. Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts. Proc Natl Acad Sci U S A. 1997;94:3542–3547. doi: 10.1073/pnas.94.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Chen CX, Carter KC, Nishikura K. Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol Cell Biol. 1997;17:2413–2424. doi: 10.1128/mcb.17.5.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Drakas R, Nishikura K. Mutagenic analysis of double-stranded-RNA adenosine-deaminase, a candidate enzyme for RNA editing of glutamate-gated ion-channel transcripts. J Biol Chem. 1995;270:17098–17105. doi: 10.1074/jbc.270.29.17098. [DOI] [PubMed] [Google Scholar]
- Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- Lei M, Liu Y, Samuel CE. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology. 1998;245:188–196. doi: 10.1006/viro.1998.9162. [DOI] [PubMed] [Google Scholar]
- Lemon SM. Induction and evasion of innate antiviral responses by hepatitis C virus. J Biol Chem. 2010;285:22741–22747. doi: 10.1074/jbc.R109.099556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, Rechavi G, Jantsch MF, Eisenberg E. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res. 2005;33:1162–1168. doi: 10.1093/nar/gki239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wolff KC, Samuel CE. RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology. 2010;396:316–322. doi: 10.1016/j.virol.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebert UG, Baczko K, Budka H, ter Meulen V. Restricted expression of measles virus proteins in brains from cases of subacute sclerosing panencephalitis. J genl Virol. 1986;67:2435–2444. doi: 10.1099/0022-1317-67-11-2435. [DOI] [PubMed] [Google Scholar]
- Linnstaedt SD, Kasprzak WK, Shapiro BA, Casey JL. The fraction of RNA that folds into the correct branched secondary structure determines hepatitis delta virus type 3 RNA editing levels. RNA. 2009;15:1177–1187. doi: 10.1261/rna.1504009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Emeson RB, Samuel CE. Serotonin-2c receptor pre-mRNA editing in rat brain and in vitro by splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J Biol Chem. 1999;274:18351–18358. doi: 10.1074/jbc.274.26.18351. [DOI] [PubMed] [Google Scholar]
- Liu Y, George CX, Patterson JB, Samuel CE. Functionally distinct double-stranded RNA-binding domains associated with alternative splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase. J Biol Chem. 1997;272:419–4428. doi: 10.1074/jbc.272.7.4419. [DOI] [PubMed] [Google Scholar]
- Liu Y, Herbert A, Rich A, Samuel CE. Double-stranded RNA-specific adenosine deaminase: nucleic acid binding properties. Methods. 1998;15:199–205. doi: 10.1006/meth.1998.0624. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lei M, Samuel CE. Chimeric double-stranded RNA-specific adenosine deaminase ADAR1 proteins reveal functional selectivity of double-stranded RNA- binding domains from ADAR1 and protein kinase PKR. Proc Natl Acad Sci U S A. 2000;97:12541–12546. doi: 10.1073/pnas.97.23.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel CE. Mechanism of interferon action: Functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA- specific adenosine deaminase. J Virol. 1996;70:1961–1968. doi: 10.1128/jvi.70.3.1961-1968.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel CE. Editing of glutamate receptor subunit B pre-mRNA by splice-site variants of interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J Biol Chem. 1999;274:5070–5077. doi: 10.1074/jbc.274.8.5070. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wolff KC, Jacobs BL, Samuel CE. Vaccinia virus E3L interferon resistance protein inhibits the interferon-induced adenosine deaminase A-to-I editing activity. Virology. 2001;289:378–387. doi: 10.1006/viro.2001.1154. [DOI] [PubMed] [Google Scholar]
- Liu Z, Batt DB, Carmichael GG. Targeted nuclear antisense RNA mimics natural antisense-induced degradation of polyoma virus early RNA. Proc Natl Acad Sci USA. 1994;91:4258–4262. doi: 10.1073/pnas.91.10.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–1177. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo GX, Chao M, Hsieh SY, Sureau C, Nishikura K, Taylor J. A specific base transition occurs on replicating hepatitis delta virus-RNA. J Virol. 1990;64:1021–1027. doi: 10.1128/jvi.64.3.1021-1027.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Gommans WM. Novel exon of mammalian ADAR2 extends open reading frame. PLoS One. 2009a;4:e4225. doi: 10.1371/journal.pone.0004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Gommans WM. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res. 2009b;37:5822–5829. doi: 10.1093/nar/gkp599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Rich A, Nishikura K. A-to-I RNA Editing: Recent News and Residual Mysteries. J Biol Chem. 2003;278:1391–1394. doi: 10.1074/jbc.R200025200. [DOI] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009;364:675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez I, Melero JA. A model for the generation of multiple A to G transitions in the human respiratory syncytial virus genome: predicted RNA secondary structures as substrates for adenosine deaminases that act on RNA. J Gen Virol. 2002 Jun;83(Pt 6):1445–55. doi: 10.1099/0022-1317-83-6-1445. [DOI] [PubMed] [Google Scholar]
- Martínez I, Dopazo J, Melero JA. Antigenic structure of the human respiratory syncytial virus G glycoprotein and relevance of hypermutation events for the generation of antigenic variants. J Gen Virol. 1997;78:2419–22429. doi: 10.1099/0022-1317-78-10-2419. [DOI] [PubMed] [Google Scholar]
- Markle D, Das S, Ward SV, Samuel CE. Functional analysis of the KCS-like element of the interferon-inducible RNA-specific adenosine deaminase ADAR1 promoter. Gene. 2003;304:143–149. doi: 10.1016/s0378-1119(02)01200-3. [DOI] [PubMed] [Google Scholar]
- McAllister CS, Toth AM, Zhang P, Devaux P, Cattaneo R, Samuel CE. Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J Virol. 2010;84:380–386. doi: 10.1128/JVI.02630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack SJ, Thomis DC, Samuel CE. Mechanism of interferon action: identification of a RNA binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2 alpha protein kinase. Virology. 1992;188:47–56. doi: 10.1016/0042-6822(92)90733-6. [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996b;271:31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996a;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- Mittaz L, Scott HS, Rossier C, Seeburg PH, Higuchi M, Antonarakis SE. Cloning of a human RNA editing deaminase (ADARB1) of glutamate receptors that maps to chromosome 21q22.3. Genomics. 1997;41:210–217. doi: 10.1006/geno.1997.4655. [DOI] [PubMed] [Google Scholar]
- Moss WJ, Griffin DE. Global measles elimination. Nat Rev Microbiol. 2006;4:900–908. doi: 10.1038/nrmicro1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DF, Dimock K, Kang CY. Numerous transitions in human parainfluenza virus 3 RNA recovered from persistently infected cells. Virology. 1991;181:760–763. doi: 10.1016/0042-6822(91)90913-v. [DOI] [PubMed] [Google Scholar]
- Muralidhar S, Pumfery AM, Hassani M, Sadaie MR, Kishishita M, Brady JN, Doniger J, Medveczky P, Rosenthal LJ. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J Virol. 1998;72:4980–4988. doi: 10.1128/jvi.72.6.4980-4988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DG, Dimock K, Kang CY. Numerous transitions in human parainfluenza virus 3 RNA recovered from persistently infected cells. Virology. 1991;181:760–763. doi: 10.1016/0042-6822(91)90913-v. [DOI] [PubMed] [Google Scholar]
- Nie Y, Hammond GL, Yang JH. Double-stranded RNA deaminase ADAR1 increases host susceptibility to virus infection. J Virol. 2007;81:917–923. doi: 10.1128/JVI.01527-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Copeland SC, Herrick-Davis K, Emeson RB, Sanders-Bush E. RNA editing of the human serotonin 5-hydroxytryptamine 2c receptor silences constitutive activity. J Biol Chem. 1999;274:9472–9478. doi: 10.1074/jbc.274.14.9472. [DOI] [PubMed] [Google Scholar]
- Ngamurulert S, Limjindaporn T, Auewaraku P. Identification of cellular partners of Influenza A virus (H5N1) non-structural protein NS1 by yeast two-hybrid system. Acta Virol. 2009;53:153–159. doi: 10.4149/av_2009_03_153. [DOI] [PubMed] [Google Scholar]
- Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, Jenny A, Keller W. Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol Cell Biol. 1995;15:1389–1397. doi: 10.1128/mcb.15.3.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MA, Gerber A, Keller W. Purification of human double-stranded RNA-specific editase 1 (hRED1) involved in editing of brain glutamate receptor B pre-mRNA. J Biol Chem. 1997;272:473–478. doi: 10.1074/jbc.272.1.473. [DOI] [PubMed] [Google Scholar]
- O’Hara P, Nichol S, Horodyski F, Holland J. Vesicular stomatitis virus defective interfering particles can contain extensive genomic sequence rearrangements and base substitutions. Cell. 1984;36:915–924. doi: 10.1016/0092-8674(84)90041-2. [DOI] [PubMed] [Google Scholar]
- Oldstone MB. Modeling subacute sclerosing panencephalitis in a transgenic mouse system: uncoding pathogenesis of disease and illuminating components of immune control. Curr Top Microbiol Immunol. 2009;330:31–54. doi: 10.1007/978-3-540-70617-5_2. [DOI] [PubMed] [Google Scholar]
- Oldstone MB, Lewicki H, Thomas D, Tishon A, Dales S, Patterson J, Manchester M, Homann D, Naniche D, Holz A. Measles virus infection in a transgenic model: virus-induced immunosuppression and central nervous system disease. Cell. 1999;98:629–640. doi: 10.1016/s0092-8674(00)80050-1. [DOI] [PubMed] [Google Scholar]
- Osenberg S, Dominissini D, Rechavi G, Eisenberg E. Widespread cleavage of A-to-I hyperediting substrates. RNA. 2009;15:1632–1639. doi: 10.1261/rna.1581809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma EL, Huang A. Cyclic production of vesicular stomatitis virus caused by defective interfering particles. J Infect Dis. 1974;129:402–410. doi: 10.1093/infdis/129.4.402. [DOI] [PubMed] [Google Scholar]
- Patterson JB, Cornu TI, Redwine J, Dales S, Lewicki H, Holz A, Thomas D, Billeter MA, Oldstone MB. Evidence that the hypermutated M protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virology. 2001;291:215–225. doi: 10.1006/viro.2001.1182. [DOI] [PubMed] [Google Scholar]
- Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol Cell Biol. 1995;15:5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JB, Thomis DC, Hans SL, Samuel CE. Mechanism of interferon action - double-stranded RNA-specific adenosine-deaminase from human-cells is inducible by alpha-interferon and gamma-interferon. Virology. 1995;210:508–511. doi: 10.1006/viro.1995.1370. [DOI] [PubMed] [Google Scholar]
- Peacock H, Fostvedt E, Beal PA. Minor-Groove-Modulating Adenosine Replacements Control Protein Binding and RNAi Activity in siRNAs. ACS Chem Biol. 2010 doi: 10.1021/cb100245u. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–822. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grässer FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269–776. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Zavolan M, Grässer FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, Tuschl T. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- Phuphuakrat A, Kraiwong R, Boonarkart C, Lauhakirti D, Lee TH, Auewarakul P. Double-stranded RNA adenosine deaminases enhance expression of human immunodeficiency virus type 1 proteins. J Virol. 2008;82:10864–10872. doi: 10.1128/JVI.00238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placido D, Brown BA, Lowenhaupt K, Rich A, Athanasiadis A. A left-handed RNA double helix bound by the Z alpha domain of the RNA-editing enzyme ADAR1. Structure. 2007;15:395–404. doi: 10.1016/j.str.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson AG, Ley HL, 3rd, Bass BL, Casey JL. Hepatitis delta virus RNA editing is highly specific for the amber/W site and is suppressed by hepatitis delta antigen. Mol Cell Biol. 1998;18:1919–1926. doi: 10.1128/mcb.18.4.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J. Crm1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol Cell Biol. 2001;21:7862–7871. doi: 10.1128/MCB.21.22.7862-7871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Rebagliati MR, Melton DA. Antisense RNA injections in fertilized frog eggs reveal an RNA duplex unwinding activity. Cell. 1987;48:599–605. doi: 10.1016/0092-8674(87)90238-8. [DOI] [PubMed] [Google Scholar]
- Rueda P, García-Barreno B, Melero JA. Loss of conserved cysteine residues in the attachment (G) glycoprotein of two human respiratory syncytial virus escape mutants that contain multiple A-G substitutions (hypermutations) Virology. 1994;198:653–662. doi: 10.1006/viro.1994.1077. [DOI] [PubMed] [Google Scholar]
- Rueter SM, Dawson RT, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Yano T, Kawabata H, Ueda H, Suzuki T. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol. 2010;6:733–740. doi: 10.1038/nchembio.434. [DOI] [PubMed] [Google Scholar]
- Samuel CE. Mechanism of interferon action: Phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc Natl Acad Sci U S A. 1979;76:600–604. doi: 10.1073/pnas.76.2.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. RNA editing minireview series. J Biol Chem. 2003;278:1389–1390. doi: 10.1074/jbc.R200032200. [DOI] [PubMed] [Google Scholar]
- Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci USA. 2003;100:14018–14023. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Cornillez-Ty C, Lazinski DW. By inhibiting replication, the large hepatitis delta antigen can indirectly regulate amber/W editing and its own expression. J Virol. 2004;78:8120–8134. doi: 10.1128/JVI.78.15.8120-8134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Wong SK, Lazinski DW. Hepatitis delta virus minimal substrates competent for editing by ADAR1 and ADAR2. J Virol. 2001;75:8547–55. doi: 10.1128/JVI.75.18.8547-8555.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol. 2005;12:489–496. doi: 10.1038/nsmb936. [DOI] [PubMed] [Google Scholar]
- Scadden AD. Inosine-containing dsRNA binds a stress-granule-like complex and downregulates gene expression in trans. Mol Cell. 2007;28:491–500. doi: 10.1016/j.molcel.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD, O’Connell MA. Cleavage of dsRNAs hyper-edited by ADARs occurs at preferred editing sites. Nucleic Acids Res. 2005;33:5954–5964. doi: 10.1093/nar/gki909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD, Smith CW. A ribonuclease specific for inosine-containing RNA: a potential role in antiviral defence? EMBO J. 1997;16(8):2140–2149. doi: 10.1093/emboj/16.8.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD, Smith CW. RNAi is antagonized by A-->I hyper-editing. EMBO Rep. 2001a;(12):1107–1111. doi: 10.1093/embo-reports/kve244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD, Smith CW. Specific cleavage of hyper-edited dsRNAs. EMBO J. 2001b;20:4243–4252. doi: 10.1093/emboj/20.15.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Spielhofer P, Cattaneo R, Baczko K, ter Meulen V, Billeter MA. Subacute sclerosing panencephalitis is typically characterized by alterations in the fusion protein cytoplasmic domain of the persisting measles virus. Virology. 1992;188:910–915. doi: 10.1016/0042-6822(92)90552-z. [DOI] [PubMed] [Google Scholar]
- Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A. Crystal structure of the Z domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science. 1999;284:1841–1845. doi: 10.1126/science.284.5421.1841. [DOI] [PubMed] [Google Scholar]
- Seeburg PH, Hartner J. Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr Opin Neurobiol. 2003;13(3):279–283. doi: 10.1016/s0959-4388(03)00062-x. [DOI] [PubMed] [Google Scholar]
- Serra MJ, Smolter PE, Westhof E. Pronounced instability of tandem IU base pairs in RNA. Nucleic Acids Res. 2004;32:1824–1828. doi: 10.1093/nar/gkh501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharmeen L, Bass B, Sonenberg N, Weintraub H, Groudine M. Tat-dependent adenosine-to-inosine modification of wild-type transactivation response RNA. Proc Natl Acad Sci USA. 1991;88:8096–8100. doi: 10.1073/pnas.88.18.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman R, Heithoff DM, Mahan MJ, Samuel CE. Tissue selectivity of interferon-stimulated gene expression in mice infected with dam(+) versus dam(−) Salmonella enterica Serovar typhimurium strains. Infect Immun. 2002;70:5579–5588. doi: 10.1128/IAI.70.10.5579-5588.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Kesterson RA, Jacobs MM, Joers JM, Gore JC, Emeson RB. Hyperphagia-mediated obesity in transgenic mice misexpressing the RNA-editing enzyme ADAR2. J Biol Chem. 2007;282:22448–22459. doi: 10.1074/jbc.M700265200. [DOI] [PubMed] [Google Scholar]
- Skalsky RL, Cullen BR. Viruses, microRNAs, and host interactions. Annu Rev Microbiol. 2010;64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov D, Gardiner K. Phylogenetic comparison of the pre-mRNA adenosine deaminase ADAR2 genes and transcripts: Conservation and diversity in editing site sequence and alternative splicing patterns. Gene. 2002;299:83–94. doi: 10.1016/s0378-1119(02)01016-8. [DOI] [PubMed] [Google Scholar]
- St Laurent G, 3rd, Savva YA, Reenan R. Enhancing non-coding RNA information content with ADAR editing. Neurosci Lett. 2009;466:89–98. doi: 10.1016/j.neulet.2009.09.009. [DOI] [PubMed] [Google Scholar]
- Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Strehblow A, Hallegger M, Jantsch MF. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol Biol Cell. 2002;13:3822–3835. doi: 10.1091/mbc.E02-03-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel SA, Cech TR, Usman N, Beigelman L. The 2,6-diaminopurine riboside. 5-methylisocytidine wobble base pair: an isoenergetic substitution for the study of G.U pairs in RNA. Biochemistry. 1994;33:13824–13835. doi: 10.1021/bi00250a037. [DOI] [PubMed] [Google Scholar]
- Suspène R, Renard M, Henry M, Guétard D, Puyraimond-Zemmour D, Billecocq A, Bouloy M, Tangy F, Vartanian JP, Wain-Hobson S. Inversing the natural hydrogen bonding rule to selectively amplify GC-rich ADAR-edited RNAs. Nucleic Acids Res. 2008;36(12):e72. doi: 10.1093/nar/gkn295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Puig M, Darnell MER, Mihalik K, Feinstone SM. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J Virol. 2005;79:6291–6298. doi: 10.1128/JVI.79.10.6291-6298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM. Replication of human hepatitis delta virus: Recent developments. Trends Microbiol. 2003;11:185–190. doi: 10.1016/s0966-842x(03)00045-3. [DOI] [PubMed] [Google Scholar]
- tenOever BR, Ng SL, Chua MA, McWhirter SM, García-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- Toth AM, Li Z, Cattaneo R, Samuel CE. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem. 2009;284:29350–29356. doi: 10.1074/jbc.M109.045146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AM, Zhang P, Das S, George CX, Samuel CE. Interferon action and the double-stranded RNA-dependent enzymes ADAR1 adenosine deaminase and PKR protein kinase. Prog Nucleic Acid Res Mol Biol. 2006;81:369–434. doi: 10.1016/S0079-6603(06)81010-X. [DOI] [PubMed] [Google Scholar]
- Valente L, Nishikura K. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J Biol Chem. 2007;282:16054–16061. doi: 10.1074/jbc.M611392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente L, Nishikura K. ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog Nucleic Acid Res Mol Biol. 2005;79:299–338. doi: 10.1016/S0079-6603(04)79006-6. [DOI] [PubMed] [Google Scholar]
- Valentine R, Smith GL. Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J Gen Virol. 2010;91:2221–2229. doi: 10.1099/vir.0.021998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali P, Scadden AD. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol. 2010;17:1043–1050. doi: 10.1038/nsmb.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RW, Smith JE, Cooperman BS, Nishikura K. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc Natl Acad Sci U S A. 1989;86:2647–2651. doi: 10.1073/pnas.86.8.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of Adar1 RNA editing deaminase gene. J Biol Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Samuel CE. Adenosine deaminase ADAR1 increases gene expression at the translational level by decreasing protein kinase PKR-dependent eIF-2alpha phosphorylation. J Mol Biol. 2009;393:777–787. doi: 10.1016/j.jmb.2009.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zeng Y, Murray JM, Nishikura K. Genomic organization and chromosomal location of the human dsRNA adenosine deaminase gene: The enzyme for glutamate-activated ion channel RNA editing. J Mol Biol. 1995;254:184–195. doi: 10.1006/jmbi.1995.0610. [DOI] [PubMed] [Google Scholar]
- Ward SV, George CX, Welch JJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MBO. The RNA editing enzyme ADAR1 is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.1017241108. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weier HUG, George CX, Greulich KM, Samuel CE. The interferon-inducible, double-stranded RNA-specific adenosine deaminase gene (DSRAD) maps to human chromosome 1q21.1–21.2. Genomics. 1995;30:372–375. doi: 10.1006/geno.1995.0034. [DOI] [PubMed] [Google Scholar]
- Weier HUG, George CX, Lersch RA, Breitweser S, Cheng JF, Samuel CE. Assignment of the RNA-specific adenosine deaminase gene (Adar) to mouse chromosome 3f2 by in situ hybridization. Cytogenet Cell Genet. 2000;89:214–215. doi: 10.1159/000015615. [DOI] [PubMed] [Google Scholar]
- Welzel TM, Morgan TR, Bonkovsky HL, Naishadham D, Pfeiffer RM, Wright EC, Hutchinson AA, Crenshaw AT, Bashirova A, Carrington M, Dotrang M, Sterling RK, Lindsay KL, Fontana RJ, Lee WM, Di Bisceglie AM, Ghany MG, Gretch DR, Chanock SJ, Chung RT, O’Brien TR HALT-C Trial Group. Variants in interferon-alpha pathway genes and response to pegylated interferon-Alpha2a plus ribavirin for treatment of chronic hepatitis C virus infection in the hepatitis C antiviral long-term treatment against cirrhosis trial. Hepatology. 2009;49(6):1847–1858. doi: 10.1002/hep.22877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SK, Lazinski DW. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc Natl Acad Sci U S A. 2002;99:15118–15123. doi: 10.1073/pnas.232416799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TC, Ayata M, Ueda S, Hirano A. Role of biased hypermutation in evolution of subacute sclerosing panencephalitis virus from progenitor acute measles virus. J Virol. 1991;65:2191–2199. doi: 10.1128/jvi.65.5.2191-2199.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, Yang Q, Wang Q, Cheng T. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci U S A. 2009;106:17763–17768. doi: 10.1073/pnas.0903324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WD, Wang QD, Howell KL, Lee JT, Cho DSC, Murray JM, Nishikura K. ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J Biol Chem. 2005;280:3946–3953. doi: 10.1074/jbc.M407876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.1009231107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn RC, Schelp I, Utermöhlen O, von Laer D. A-to-G hypermutation in the genome of lymphocytic choriomeningitis virus. J Virol. 2007;81:457–464. doi: 10.1128/JVI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]