

Abstract
BACKGROUND AND PURPOSE
Paulownia tomentosa is a rich source of geranylated flavanones, some of which we have previously shown to have cytotoxic activity. To identify members of this class of compounds with cytostatic effects, we assessed the effects of the geranylated flavanone tomentodiplacone B (TOM B) on cell cycle progression and cell cycle regulatory pathways of THP-1 human monocytic leukaemia cells.
EXPERIMENTAL APPROACH
Cell viability was measured by dye exclusion and proliferation by WST-1 assays; cell cycle was monitored by flow cytometry. Regulatory proteins were assessed by immunoprecipitation and kinase assays, and Western blotting.
KEY RESULTS
Tomentodiplacone B had no effect during the first 24 h of cell growth at concentrations between 1 and 2.5 µM, but inhibited cell growth in a dose-dependent manner at concentrations of 5 µM or higher. Growth inhibition during the first 24 h of exposure to TOM B was not accompanied by cytotoxicity as cells were accumulated in G1 phase dose-dependently. This G1 phase accumulation was associated with down-regulation of cyclin-dependent kinase 2 activity and also protein levels of cyclins E1 and A2. However, key stress-related molecules (γ-H2AX, p53 and p21) were not induced, suggesting that TOM B acts by directly inhibiting the cyclin-dependent kinase 2 signalling pathway rather than initiating DNA damage or cellular stress.
CONCLUSIONS AND IMPLICATIONS
Our study provides the first evidence that TOM B directly inhibits proliferation of human monocytic leukaemia cells, and thus is a potential anticancer agent, preventing leukaemia cells from progressing from G1 phase into DNA synthesis.
Keywords: flavonoids, antiproliferative effect, CDK2, cyclins, cell cycle regulators
Introduction
Mammalian G1 cyclins and their associated cyclin-dependent kinases (CDKs) regulate cell division by communicating extracellular and intracellular mitogenic signals to the molecular machinery that drives the cell from G1 to S phase (Sherr and Roberts, 2004). Regulation of the G1 to S phase transition (G1/S checkpoint) is often compromised in malignant cells. Members of the inhibitors of CDK4 and Cip/Kip families of CDK inhibitors (CKI) regulate CDK activity and cell cycle progression. The inhibitors of CDK4 family include proteins p15, p16, p18 and p19, which bind to and inhibit CDK4 and CDK6 specifically during the G1 phase. The Cip/Kip family includes proteins p21, p27 and p57, which halt the cell cycle by binding to and inactivating diverse cyclin/CDK complexes. DNA damage triggers G1 arrest by activating p53, which in turn stimulates the transcription of p21. Thus, targeting CDK and cyclin/CDK complexes is a promising strategy for cancer treatment.
Natural plant products and their derivatives have been found to possess CDK inhibitory activity. For example, flavopiridol, a semisynthetic flavone derivative of rohitukine (an unusual chromane alkaloid from Amoora or Dysoxylum), is the first clinically useful natural inhibitor of CDK activity to enter human clinical trials for cancer treatment and has stimulated interest in CDK inhibitor research (Senderowicz, 1999). Other natural compounds trigger programmed cell death or act as tumour suppressors by inhibiting CDKs, and are called ‘small molecule’ CKIs (Maddika et al., 2007). Flavonoids are widespread natural compounds and their cytotoxicity to different cell types and potential usefulness in cancer therapy has been thoroughly reviewed (López-Lázaro, 2002; Chang et al., 2008). Flavonoids can influence cell cycle progression by various mechanisms. Quercetin inhibits protein kinase C, causes G1 arrest or G2/M arrest, and induces apoptosis (Formica and Regelson, 1995; Zhang et al., 2009). Murakami et al. (2008) showed that quercetin has multiple targets that contribute to its anticancer properties.
Flavanones are a subclass of flavonoids, some of which, such as hesperidin and taxifolin, cause cell cycle arrest (Ramos, 2007). In addition, prenylated flavonoid derivatives have different pharmacological activity in living organisms, when compared with that of their non-prenylated analogues (Wätjen et al., 2007; 2008; Musthapa et al., 2009; Rao et al., 2009). The activity and pharmacological effects of these derivatives is further influenced by structural modifications of the prenyl group, including group substitution, addition of hydroxyl groups, formation of carbonyl bonds, or cyclization. For example, the cell cycle inhibitory activity of the prenylated chalcones found in hops and beer is suppressed by cyclization and hydration of the prenyl groups in chalcones (Miranda et al., 1999).
Paulownia tomentosa is a rich source of geranylated flavanones, a subclass of flavanoids with members that we have previously shown to have cytotoxic activity (Smejkal et al., 2008a). Similar geranyl flavanones, found in Taiwanese propolis, directly induce apoptosis without first inhibiting cell cycle progression (Chen et al., 2003). In our most recent study, we observed that C-geranyl flavanones have strong cytotoxic activity in normal human fibroblasts and five human cancer cell lines (Smejkal et al., 2010). Notably, when compared with several standard anticancer drugs, some C-geranyl flavanones showed similar and/or stronger antiproliferative affects. Thus we have expanded our study to identify prenylated and geranylated flavanone compounds from Moraceae and Scrophulariaceae families with cytostatic properties, and found that tomentodiplacone B (TOM B; 4′,5,7-trihydroxy-3′-methoxy-6-[7-hydroxy-3,7-dimethyl-2,5-octadienyl]flavanone; Figure 1A), which has a hydroxy methoxy modified B-ring and hydroxylated geranyl side chain (Figure 1A), has cytostatic effects. Here we have analysed these cytostatic effects further in terms of progression through the cell cycle, using the THP-1 human monocytic leukaemia cell line as a model system.
Figure 1.
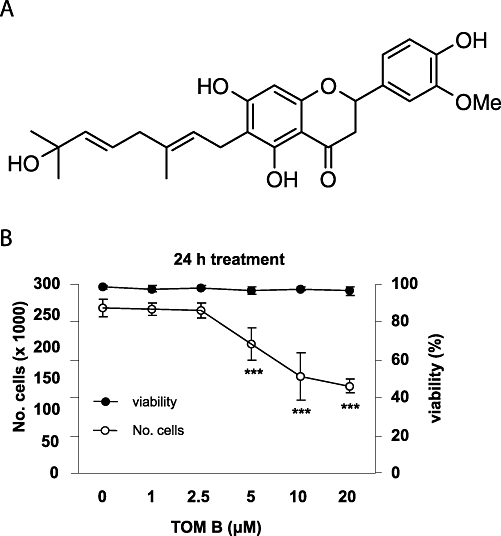
Tomentodiplacone B (TOM B) inhibits proliferation of THP-1 leukaemia cells. (A) Structure of TOM B. (B) THP-1 cells were seeded (2 × 105 cells·mL−1), treated with the indicated concentrations of TOM B for 24 h, cell numbers counted, and viability determined by erythrosin B exclusion. The results shown are expressed as the means ± SD of three independent experiments, with each condition tested in triplicate. *P < 0.05; **P < 0.01; ***P < 0.001, significantly different from control.
Methods
Cell culture
The human monocytic leukaemia THP-1 cell line was purchased from the European Collection of Cell Cultures (Salisbury, UK; Methods of characterization: DNA Fingerprinting (Multilocus probes) and isoenzyme analysis). Cells were cultured in RPMI 1640 medium supplemented with antibiotics (100 U·mL−1 penicillin, 100 mg·mL−1 streptomycin), 10% fetal bovine serum (FBS), and 2 mM L-glutamine. Cultures were kept in an incubator at 37°C in a water-saturated 5% CO2 atmosphere in air. Cells were passaged at approximately 1 week intervals. Cells were routinely tested for the absence of mycoplasma (Hoechst 33258 staining method). Human embryonic stem cells (hESC) and human foreskin fibroblasts (HFF) were included as positive controls in some experiments. HFF (SCRC 1041) were obtained from the American Type Culture Collection (Manassas, VA, USA, http://www.atcc.org). The hESC (cell line CCTL14) (Adewumi et al., 2007) and HFF were cultured as described previously (Eiselleova et al., 2008). Briefly, hESC were maintained on mitotically inactivated mouse embryonic fibroblasts in culture media consisting of DMEM/F12 supplemented with 15% knockout serum replacement, L-glutamine, MEM non-essential amino acids, 0.5% penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA), 100 µM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA) and 10 ng·mL−1 FGF-2 (PeproTech, Rocky Hill, NJ, USA). HFF were maintained in KnockOut DMEM supplemented with 20% FBS, 2 mM L-glutamine, 1× MEM non-essential amino acids solution, 1% penicillin-streptomycin (all media components from Invitrogen), and 100 µM β-mercaptoethanol (Sigma-Aldrich).
Analysis of cell viability and cell proliferation
THP-1 cells (2 × 105 cells·mL−1) were incubated in 2 mL of RPMI 1640 medium in six-well plates at 37°C for 24 h with 1, 2.5, 5, 10 or 20 µM TOM B. Numbers and viabilities of the cells were determined by counting with a haemocytometer after staining with erythrosin B [0.1% erythrosin B (w·v−1) in phosphate-buffered saline (PBS), pH 7.2–7.4]. Unstained cells were considered to be viable. Cell proliferation was determined using a WST-1 assay kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. For WST-1 assays, cells were seeded into 96-well plates (5 × 104 cells·well−1 in 100 µL culture medium) in triplicate in complete RPMI 1640 medium and measurements were taken 24 and 72 h after the treatment with TOM B. All data were evaluated using GraphPad Prism 5.00 software (GraphPad Software, San Diego, CA, USA, http://www.graphpad.com).
Cell cycle analysis
Human monocytic leukaemia cells were incubated with 5, 10 or 20 µM TOM B for 24 h, washed in PBS (pH 7.4) and fixed for 30 min in an ice-cold 70% ethanol. Fixed cells were washed three times in PBS (pH 7.4) and incubated with RNaseA (0.02 mg·mL−1) (Boehringer, Ingelheim, Germany) for 30 min at 37°C. Nuclei were stained with propidium iodide (40 µg·mL−1) and analysed by flow cytometry using a Beckman Coulter Cytomics FC500 flow cytometer (Beckman Coulter, Brea, CA, USA). Cell cycle distribution was analysed using FlowJo software (Tree Star, Ashland, OR, USA; http://www.flowjo.com) and WinMDI (TSRI, La Jolla, CA, USA; http://facs.scripps.edu/).
Immunoprecipitation and kinase assay
Cells were extracted in ice-cold extraction buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA, 8 mM β-glycerophosphate, 50 mM NaF, 1 mM phenylmethylsulphonyl fluoride, 1 mM tosylphenylalanine chloromethane, 1 mM dithiothreitol, 1 µg·mL−1 leupeptin, 1 µg·mL−1 aprotinin, and 10 µg·mL−1 soybean trypsin inhibitor), and the extracts were stored at −80°C until use. Before use, extracts were thawed and cleared by centrifugation at 15 000×g for 5 min at 4°C, and protein concentrations were determined using the DC Protein Assay Kit (Bio-Rad, Hercules, CA, USA). Extracts were incubated with the appropriate antibody for 1 h on ice. Rabbit polyclonal antibodies against CDK1 (PC25) (Calbiochem, San Diego, CA, USA) and CDK2 (sc-163) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used to immunoprecipitate CDK1 and CDK2. Immunoprecipitates were collected on Protein G agarose beads (Sigma-Aldrich) by rotation at 4°C overnight, followed by washing three times with extraction buffer and twice with kinase assay buffer [50 mM HEPES (pH 7.5) 10 mM MgCl2, 10 mM MnCl2, 8 mM β-glycerophosphate, 1 mM dithiothreitol]. Kinase reactions were carried out for 30 min at 37°C in a total volume of 25 µL of kinase assay buffer supplemented with 100 µg·mL−1 histone H1 (Sigma-Aldrich) and 40 µCi·mL−1[32P]ATP. Reactions were terminated by addition of 2× Laemmli sample buffer, and each reaction mix was subjected to SDS-polyacrylamide gel electrophoresis and autoradiography. The intensity of bands was quantified with ImageJ software (Research Services Branch, Bethesda, MD, USA; http://rsbweb.nih.gov/ij/).
Western blotting
Cells were washed three times with PBS (pH 7.4) and lysed in 100 mM Tris-HCl (pH 6.8) containing 20% glycerol and 1% SDS. Protein concentrations were determined using the DC Protein Assay Kit (Bio-Rad, Hercules, CA, USA). Lysates were supplemented with bromophenol blue (0.01%) and β-mercaptoethanol (1%). Equal amounts of total protein (10 µg) were separated by SDS-polyacrylamide gel electrophoresis, electrotransferred onto PVDF membranes (Millipore, Billerica, MA, USA), immunodetected using the appropriate primary and secondary antibodies, and visualized with ECL Plus reagent (Amersham, Aylesbury, UK) according to the manufacturer's instructions. The intensity of bands was quantified with ImageJ software.
Data analysis
Data are presented as means ± SD or SEM, as shown, from at least three independent experiments. Statistical significance was tested using the one-way anova with Dunnett's post test for comparisons between the means, and differences between two conditions were retained for P < 0.05. Statistical significance was determined at levels of P < 0.05, P < 0.01 and P < 0.001.
Materials
Tomentodiplacone B was isolated and supplied by the Department of Natural Drugs, Faculty of Pharmacy, University of Veterinary and Pharmaceutical Sciences Brno, Czech Republic. The identity of TOM B was confirmed by HRMS, 1H and 13C NMR analysis, as reported by Smejkal et al. (2008b). The purity of TOM B exceeded 95% according to the HPLC analysis. The compound is poorly soluble in water; therefore, a fresh 10 mM stock solution in dimethylsulphoxide (Sigma-Aldrich, St. Louis, MO, USA) was prepared 1 day prior to experiments and stored at −20°C. This solution was further diluted in the culture media to the desired final concentrations. RPMI 1640 culture medium and PBS were purchased from Lonza (Verviers, Belgium); FBS was from Sigma-Aldrich. Antibiotics (penicillin and streptomycin) were from Lonza. Mouse monoclonal antibodies against cyclin B1 (MS-868), cyclin E1 (MS-870), cyclin D1 (MS-210), cyclin D2 (MS-221), cyclin D3 (MS-215) and CDK1 (MS-110) were purchased from Neomarkers (Fremont, CA, USA). Mouse monoclonal antibody against cyclin A2 (sc-53228), and rabbit polyclonal antibodies against CDK2 (sc-163) and p21 (sc-397) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal antibodies against p38 MAPK [pT180/Y182] (9215S) and p38 MAPK (9212) were purchased from Cell Signaling Technologies (Beverly, MA, USA). Mouse monoclonal antibody against γ-H2AX [pS139] (05-636) was purchased from Millipore (Billerica, MA, USA). Mouse monoclonal antibodies against pRb (554136) and p27 (610242) were purchased from BD Biosciences (Franklin Lakes, NJ, USA). Rabbit polyclonal antibody against pRb [pT821] (44-582G) was purchased from Biosource (Carlsbad, CA, USA). Mouse monoclonal antibody against p53 (DO-1) was generously provided by Dr Bořivoj Vojtěšek (Masaryk Memorial Cancer Institute, Brno, Czech Republic). All other reagents were from Sigma-Aldrich.
Results
Growth inhibitory effects of TOM B
We initially assessed the effect of TOM B on the viability and growth of human leukaemia cells. THP-1 cells were exposed to increasing concentrations (1, 2.5, 5, 10 and 20 µM) of TOM B for 24 h, stained for viability, and counted. As shown in Figure 1B, although the viability of cells was not affected at any concentration, TOM B concentrations of 5 µM or higher significantly reduced the numbers of cells (P < 0.001). This demonstrates that TOM B has antiproliferative, rather than cytotoxic, effects on this cell line.
This conclusion was further supported by determining cell number using metabolic activity as the assay, following exposure to TOM B for 24 or 72 h. Concentrations of TOM B of 5 µM and higher caused dose-dependent inhibition of THP-1 cell growth, after 24 h (Figure 1B). Treatment with 20 µM TOM B for 72 h caused an almost 70% reduction in cell metabolic activity (data not shown). Based on these studies, concentrations of 5, 10 and 20 µM TOM B were used in all further experiments.
Effects of TOM B on distribution of cells in cell cycle phases
To determine at which stage of the cell cycle the TOM B-induced growth arrest occurred, we performed cell cycle analysis based on DNA content using flow cytometry of THP-1 cells. The data in Figure 2 show that a 24 h treatment with TOM B caused a concentration-dependent accumulation of THP-1 cells with 2C DNA content, representing G1 phase. While the percentage of S phase cells decreased, the percentage of cells with 4C DNA content, representing G2/M phase, was unchanged by TOM B treatment. As expected, the TOM B-treated cells did not have a hypodiploid sub-G1 peak that would be indicative of cell death. Taken together, these data demonstrate that TOM B-treated human leukaemia cells accumulate in G1 phase without causing significant cytotoxicity in the first 24 h of treatment.
Figure 2.
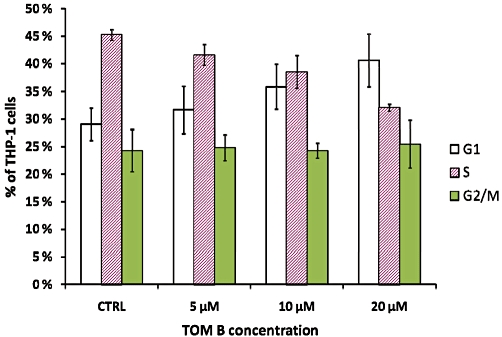
Tomentodiplacone B (TOM B) induces THP-1 cell accumulation in G1 phase. Data from flow cytometric analysis of the DNA content of THP-1 cells treated with TOM B for 24 h. The bars show the distribution of TOM B-treated THP-1 cells in the phases of the cell cycle. Values shown are the means ± SEM of the percentages of cells in individual phases of the cell cycle from three independent experiments. CTRL, control.
Expression and activity of CDKs
Normally, CDK2 activity is low in early G1 phase and upon increase, drives cells into S phase. Here we measured CDK2 kinase activity in THP-1 cells, to determine if it was inhibited by TOM B treatment. As shown in Figure 3A, exposure of cells to TOM B caused a dose-dependent down-regulation of CDK2 activity, reaching a maximal inhibition of about twofold at 20 µM (Figure 3B). In contrast, incubation with TOM B did not inhibit CDK1, a key driver of the G2/M transition, and the levels of this CDK remained unchanged, compared with untreated control cells.
Figure 3.
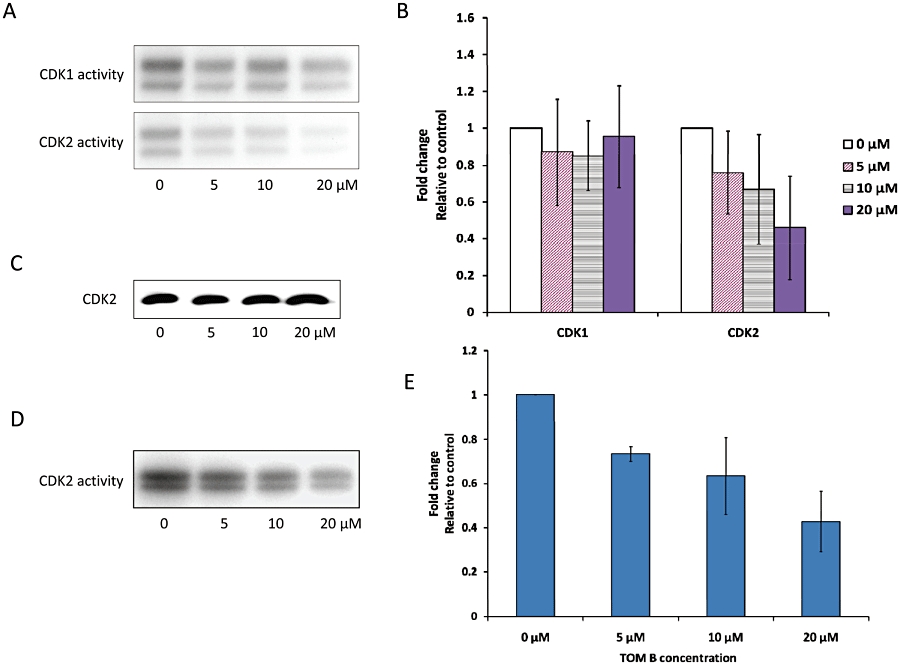
Activity of cyclin-dependent kinase 1 (CDK1) and CDK2 after treatment with tomentodiplacone B (TOM B) for 24 h. (A) The activity of CDK1 and CDK2 in control and TOM B-treated THP-1 cells, determined using histone H1 as a substrate. Data from a typical experiment are shown. (B) Summary data of CDK1 and CDK2 activities after TOM B treatment. Values shown are means ± SD of three independent experiments. (C) Protein levels of CDK2 in THP-1 cells were not affected by TOM B treatment, as determined by Western blot analysis. (D) The activity of CDK2, isolated from control THP-1 cells by immunoprecipitation and incubated with TOM B, was assayed with histone H1. Panel (E) shows summary data from three independent experiments.
To assess whether TOM B was a direct inhibitor of CDK2 catalytic activity, TOM B (5, 10 or 20 µM) was added directly to CDK2 isolated from THP-1 cells by immunoprecipitation and then assayed for kinase activity, using dimethylsulphoxide as vehicle control. As shown in Figure 3D, incubation of isolated CDK2 in the presence of TOM B caused a dose-dependent down-regulation of CDK2 activity, suggesting that TOM B can be a direct inhibitor of CDK2.
Notably, neither CDK2 (Figure 3C) nor CDK1 (not shown) protein levels were altered by TOM B treatment, thus leaving a direct action of TOM B, cyclin down-regulation and a stress response as the three most probable causes of the change to CDK activity in cells.
As the activity of CDK2, but not that of CDK1, was inhibited by TOM B, in agreement with the observed accumulation in G1 phase (Sherr and Roberts, 2004), we focused on identifying proteins affecting the regulation of the G1/S-associated CDK that were altered by exposure to TOM B.
Expression of cell cycle regulators
Several proteins exert their roles in modulation of cell cycle progression via their accumulation and/or degradation so we have assessed their metabolism in THP-1 cells exposed to TOM B. The most relevant proteins included three d-type cyclins (D1, D2 and D3) that, in various combinations, bind to and allosterically regulate one of two CDK subunits, CDK4 and CDK6, as well as cyclin E1 (and its truncated forms), which, in analogous fashion, govern the activity of the catalytic subunit CDK2 (Morgan, 1997) and cyclins B1, which regulate CDK1 activity. Of the five cyclins analysed, only cyclin A2 and E1 levels were altered by TOM B at concentrations that accumulated THP-1 cells in G1 (Figure 4A and B).
Figure 4.
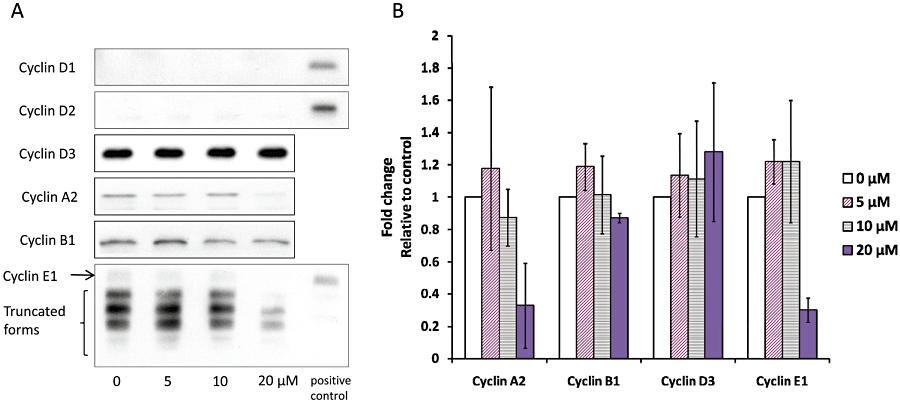
Cyclin and cyclin-dependent kinase (CDK) expression in THP-1 cells after 24 h of tomentodiplacone B (TOM B) treatment. (A) Cyclins D1 and D2 were undetectable in THP-1 cells. The expression of cyclin A2 and cyclin E1 was reduced, and the expression of cyclin D3 and cyclin B1 was not affected, as determined by Western blot analysis. Human foreskin fibroblasts cells were used as a positive control for Western blot analysis of cyclin D1 and cyclin E1 expression and human embryonic stem cells were used as a positive control for cyclin D2 expression. (B) Summary data of cyclin levels in THP-1 cells after 24 h of TOM B treatment. Values shown are means ± SD of three independent experiments.
Consistent with their roles in CDK2 activation, both cyclins A2 and E1 were down-regulated in cells exposed to TOM B, with the most pronounced effects at 20 µM. In many types of cells, both tumour and normal, cyclin E1 is present in full length and truncated forms, which have all been shown to activate CDK2 (Akli and Keyomarsi, 2003). These lower molecular weight forms of cyclin E1 were present also in THP-1 cells and were down-regulated upon exposure to TOM B. Expression of the d-type cyclins was either undetectable (D1 and D2) in untreated THP-1 cells or unchanged (D3) in THP-1 cells after exposure to TOM B (Figure 4A).
These findings demonstrated that TOM B down-regulated cyclin A2 and E1 protein levels, which would inhibiting CDK2 activity. Rb protein is phosphorylated by CDK2 and then performs processes critical for G1/S progression. We therefore examined whether Rb phosphorylation was suppressed in TOM B-treated cells. As shown in Figure 5A, 24 h exposure to TOM B resulted in reduced phosphorylation of the Rb protein. This was detectable as a reduction in the total amount of phosphorylated Rb protein and reduction of the Thr821 phosphorylated form, the known target site of CDK2. The decrease in Rb phosphorylation was maximal at the highest concentration of TOM B, with Thr821 phosphorylation reduced threefold (Figure 5C).
Figure 5.
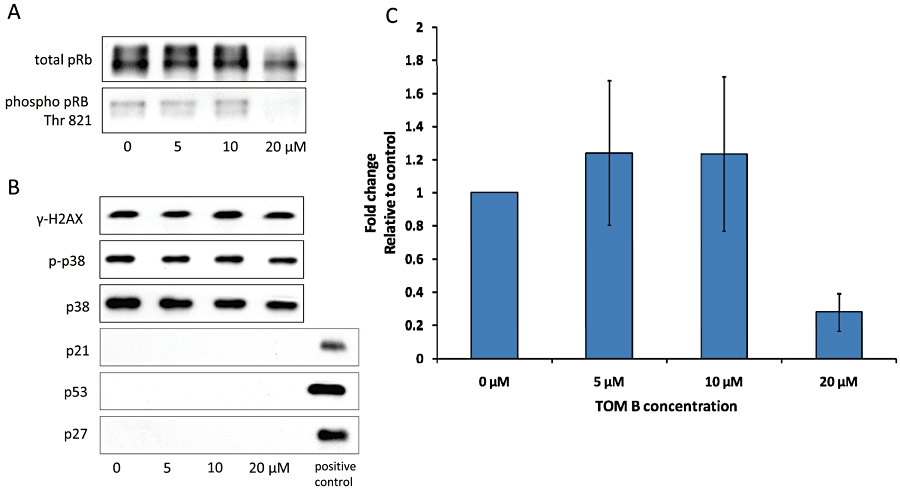
Expression of cell cycle regulators and stress response proteins in THP-1 cells after 24 h of tomentodiplacone B (TOM B) treatment. (A) pRb phosphorylation on Thr 821 was reduced after 24 h of TOM B treatment, as determined by Western blot analysis. Data representative of three other experiments are shown. (C) Summary data (means ± SD) from three independent experiments. (B) Proteins p21 and p27 were undetectable in THP-1 cells. Accumulation of p21, which is typical for stress-associated cell cycle arrest, was not induced by TOM B. Other stress-related protein levels, including histone γ-H2AX, p53 and p38 kinase, were unchanged in TOM B-treated cells as determined by Western blot analysis.
Although no signs of cell death were observed in the THP-1 cell cultures treated with TOM B, it was still possible that stress pathways were activated by TOM B and were mediating the effect of TOM B on the progression through the G1/S border. One known mechanism linking stress signals to the cell cycle regulatory machinery is the transcriptional up-regulation of the CKI, p21. In most cell types, the basal level of p21 protein is very low. In our system, p21 protein was not detectable either in control or TOM B-treated THP-1 cells (Figure 5B). Levels of another CKI, p27, were also undetectable (Figure 5B). Although the absence of p21 induction supported the idea that TOM B did not activate stress signalling pathways, we also tested for the signalling via other stress regulators. As shown in Figure 5B, we did not observe any activation of other stress regulators, such as increased p53 levels, increased p38 phosphorylation or levels, and increased histone γ-H2AX phosphorylation, in TOM B-treated THP-1 cells.
Discussion and conclusions
In the present study, we assessed the effects of the geranylated flavanone TOM B, which was recently isolated and characterized in our laboratory, on cell cycle regulation, because we recently reported that TOM B has potent anticancer activity in cancer cell lines (Smejkal et al., 2010). Using human monocytic leukaemia cells THP-1 as a model system, we found strong antiproliferative effects of TOM B in concentrations between 5 and 20 µM. Although at concentrations of 50 µM and higher, TOM B did cause cell death (data not shown), below 20 µM no cytotoxic effects were detected. The unusually pronounced antiproliferative effect of TOM B, an approximately 70% reduction in cell growth after 72 h (not shown), prompted us to study the underlying molecular mechanisms mediating the effects of TOM B on cell growth.
Cells have two key checkpoints, one at the G1/S transition and one at the G2/M transition, which monitor and regulate progression through the cell cycle. Transition from G1 to S phase is a rate-limiting step in the cell cycle and is referred to as a restriction point (Elledge, 1996). The function of restriction points is often compromised in tumour cells, causing unrestrained cells to freely enter into the next S phase due to the mutation or deletion of tumour suppressor genes like Rb and p53 (Bucher and Britten, 2008). Here we have shown that exposure to TOM B had antiproliferative effect in tumour cells, which most likely mirrored the accumulation of cells in G1 phase of cell cycle and was caused by decreased activity of a key driver of S phase progression, CDK2. Importantly, the down-regulation of CDK2 activity was not due to stimulation of CKI expression, as neither p21 nor p27 levels were increased by TOM B treatment. The p21 protein typically accumulates to arrest cells at the G1/S boundary, in response to various stress stimuli. Thus, its absence in TOM B-treated cells meant that the typical cellular stress responses to cause cell cycle restriction were not activated. This conclusion was further supported by the lack of activation of other stress-related molecular markers including histone γ-H2AX (becomes phosphorylated on damaged DNA), p53 and p38 kinase by TOM B. Indeed, the complete absence of apoptotic cells in TOM B-containing cultures according to DNA laddering assays also supports our conclusion that the cytostatic effects of TOM B are not mediated by stress-response signalling pathways (data not shown).
Two of the characteristic features of tumour cells are that they have developed a block to normal differentiation pathways and have gained unlimited proliferative capacity. However, tumour cells can still respond to cell-cycle regulators with strong differentiation effects. Phosphorylation of Rb by cyclin D/CDK4/6 and subsequently by cyclin E1/CDK2 dissociates the repressor complex from Rb (Ortega et al., 2002). Rb activation permits transcription of key S phase-promoting genes including some that are required for DNA replication (Romani et al., 2009). In contrast, dephosphorylation of Rb slows the progression of cells into S phase, thus enabling cellular differentiation to occur during prolonged G1 phases. In light of these facts, the simultaneous prolonging of G1 phase and inhibition of Rb phosphorylation caused by TOM B could cause THP-1 cells to initiate differentiation programmes. The ability to stimulate tumour cells to accumulate in G1, differentiate and undergo terminal cell division that leads to loss of tumourigenicity is a potentially useful property of TOM B for clinical applications. Determining whether TOM B triggers the differentiation of leukaemia cells is a critical research goal.
The accumulation of THP-1 cells exposed to TOM B at G1 was associated with reduced CDK2 activity, but levels of major CKIs remained unchanged. Instead, levels of activators of CDK2, including cyclins A2 and E1, were reduced by TOM B, and the specificity of this effect in the G1/S transition was supported by our observation that TOM B did not affect levels of cyclin B1, an activator of CDK1 and a driver of the G2/M transition (Dunphy et al., 1988). Cyclin E1 is the predominant regulatory protein that drives progression from G1 to S; thus, its deregulation has profound implications for oncogenesis and tumourigenesis (Pardee, 1989). Interestingly, treatment with TOM B caused selective reduction of cyclin E1 isoform levels, which are the predominant forms of cyclin E1 in THP-1 cells. These truncated forms of cyclin E1 are often differentially expressed in tumour versus normal cells. The expression of these isoforms correlates very strongly with advanced breast cancer stage, and they have also been observed in other tumours, such as colon cancer and haematological malignancies (Akli and Keyomarsi, 2003). The truncated forms have a much higher capacity to induce CDK2 kinase activity than full-length cyclin E1, due to increased affinity for CDK2. Therefore, they can stimulate cells to progress through the cell cycle more effectively than the full-length form (Porter et al., 2001). Although to date, cyclin E1 has not been targeted for cancer therapy, it could represent a target for the design of an antiproliferation adjuvant cancer therapy that is selective for tumour cells over normal proliferating cells, because the truncated forms predominate in tumour cells (Akli and Keyomarsi, 2003). Besides down-regulating the CDK2 activator cyclin E1, when applied to whole cells, TOM B also inhibited CDK2 activity in cell-free assays. As it is not possible to distinguish between these two mechanisms on the basis of the data in this study, we prefer, at present, to consider TOM B as a compound exerting both activities in whole cells.
In conclusion, our study provides the first evidence that TOM B directly inhibits the proliferation of human monocytic leukaemia cells. The TOM B-induced accumulation of cell in G1 phase was found to coincide with down-regulation of the cyclin E1 isoform and cyclin A2 levels, reduced CDK2 activity and reduced Rb phosphorylation. Therefore, TOM B represents a potential anticancer agent capable of preventing leukaemia cells from transitioning from G1 phase into DNA synthesis. Although our results suggest that enhancement of THP-1 differentiation may also be involved in TOM B action, its ability to induce cytodifferentiation of human cancer cells is still unknown. Our data indicate that 20 µM TOM B is sufficient to accumulate cells in G1 phase of the cell cycle and retard the growth of THP-1 cells. This low dose reduces the chances of TOM B side effects in vivo, which requires assessment in animal model systems. Possible modifications of side chains in the TOM B molecule could also significantly lower the effective concentration. The potential of a TOM B-based anticancer single or combination therapy awaits future exploration.
Acknowledgments
This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic (MSM0021622430, LC06077), and the Academy of Sciences of the Czech Republic (AV0Z50390512, AV0Z50390703), and the Masaryk University (MUNI/0118/2009). We thank Darja Urbánková for technical assistance and Dr Enrico Garattini (Mario Negri Institute for Pharmacological Research, Milan, Italy) for critical reading of the manuscript.
Glossary
Abbreviations
- CDK
cyclin-dependent kinase
- CKI
cyclin-dependent kinase inhibitors
- FBS
fetal bovine serum
- hESC
human embryonic stem cells
- HFF
human foreskin fibroblasts
- TOM B
tomentodiplacone B
Conflict of interest
None.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007;25:803–816. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- Akli S, Keyomarsi K. Cyclin E and its low molecular weight forms in human cancer and as targets for cancer therapy. Cancer Biol Ther. 2003;2:38–47. [PubMed] [Google Scholar]
- Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98:523–528. doi: 10.1038/sj.bjc.6604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Mi M, Ling W, Zhu J, Zhang Q, Wei N, et al. Structurally related cytotoxic effects of flavonoids on human cancer cells in vitro. Arch Pharm Res. 2008;31:1137–1144. doi: 10.1007/s12272-001-1280-8. [DOI] [PubMed] [Google Scholar]
- Chen CN, Wu CL, Shy HS, Lin JK. Cytotoxic prenylflavanones from taiwanese propolis. J Nat Prod. 2003;66:503–506. doi: 10.1021/np0203180. [DOI] [PubMed] [Google Scholar]
- Dunphy WG, Brizuela L, Beach D, Newport J. The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell. 1988;54:423–431. doi: 10.1016/0092-8674(88)90205-x. [DOI] [PubMed] [Google Scholar]
- Eiselleova L, Peterkova I, Neradil J, Slaninova I, Hampl A, Dvorak P. Comparative study of mouse and human feeder cells for human embryonic stem cells. Int J Dev Biol. 2008;52:353–363. doi: 10.1387/ijdb.082590le. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Formica JV, Regelson W. Review of the biology of quercetin and related bioflavonoids. Food Chem Toxicol. 1995;33:1061–1080. doi: 10.1016/0278-6915(95)00077-1. [DOI] [PubMed] [Google Scholar]
- López-Lázaro M. Flavonoids as anticancer agents: structure-activity relationship study. Curr Med Chem Anticancer Agents. 2002;2:691–714. doi: 10.2174/1568011023353714. [DOI] [PubMed] [Google Scholar]
- Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007;10:13–29. doi: 10.1016/j.drup.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Miranda CL, Stevens JF, Helmrich A, Henderson MC, Rodriguez RJ, Yang YH, et al. Antiproliferative and cytotoxic effects of prenylated flavonoids from hops (Humulus lupulus) in human cancer cell lines. Food Chem Toxicol. 1999;37:271–281. doi: 10.1016/s0278-6915(99)00019-8. [DOI] [PubMed] [Google Scholar]
- Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- Murakami A, Ashida H, Terao J. Multitargeted cancer prevention by quercetin. Cancer Lett. 2008;269:315–325. doi: 10.1016/j.canlet.2008.03.046. [DOI] [PubMed] [Google Scholar]
- Musthapa I, Juliawaty LD, Syah YM, Hakim EH, Latip J, Ghisalberti EL. An oxepinoflavone from Artocarpus elasticus with cytotoxic activity against P-388 cells. Arch Pharm Res. 2009;32:191–194. doi: 10.1007/s12272-009-1134-0. [DOI] [PubMed] [Google Scholar]
- Ortega S, Malumbres M, Barbacid M. Cyclin d-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- Porter DC, Zhang N, Danes C, McGahren MJ, Harwell RM, Faruki S, et al. Tumor specific proteolytic processing of cyclin E generates hyperactive lower-molecular-weight forms. Mol Cell Biol. 2001;21:6254–6269. doi: 10.1128/MCB.21.18.6254-6269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos S. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J Nutr Biochem. 2007;18:427–442. doi: 10.1016/j.jnutbio.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Rao GV, Swamy BN, Chandregowda V, Reddy GC. Synthesis of (±)-Abyssinone I and related compounds: their anti-oxidant and cytotoxic activities. Eur J Med Chem. 2009;44:2239–2245. doi: 10.1016/j.ejmech.2008.05.032. [DOI] [PubMed] [Google Scholar]
- Romani AA, Desenzani S, Morganti MM, La Monica S, Borghetti AF, Soliani P. Zoledronic acid determines S-phase arrest but fails to induce apoptosis in cholangiocarcinoma cells. Biochem Pharmacol. 2009;78:133–141. doi: 10.1016/j.bcp.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Senderowicz AM. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. 1999;17:313–320. doi: 10.1023/a:1006353008903. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Smejkal K, Babula P, Slapetová T, Brognara E, Dall'Acqua S, Zemlicka M, et al. Cytotoxic activity of C-geranyl compounds from Paulownia tomentosa fruits. Planta Med. 2008a;74:1488–1491. doi: 10.1055/s-2008-1081339. [DOI] [PubMed] [Google Scholar]
- Smejkal K, Chudík S, Kloucek P, Marek R, Cvacka J, Urbanová M, et al. Antibacterial C-geranylflavonoids from Paulownia tomentosa fruits. J Nat Prod. 2008b;71:706–709. doi: 10.1021/np070446u. [DOI] [PubMed] [Google Scholar]
- Smejkal K, Svacinová J, Slapetová T, Schneiderová K, Dall'Acqua S, Innocenti G, et al. Cytotoxic activities of several geranyl-substituted flavanones. J Nat Prod. 2010;73:568–572. doi: 10.1021/np900681y. [DOI] [PubMed] [Google Scholar]
- Wätjen W, Weber N, Lou YJ, Wang ZQ, Chovolou Y, Kampkötter A, et al. Prenylation enhances cytotoxicity of apigenin and liquiritigenin in rat H4IIE hepatoma and C6 glioma cells. Food Chem Toxicol. 2007;45:119–124. doi: 10.1016/j.fct.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Wätjen W, Suckow-Schnitker AK, Rohrig R, Kulawik A, Addae-Kyereme J, Wright CW, et al. Prenylated flavonoid derivatives from the bark of erythrina addisoniae. J Nat Prod. 2008;71:735–738. doi: 10.1021/np070417j. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zhao XH, Wang ZJ. Cytotoxicity of flavones and flavonols to a human esophageal squamous cell carcinoma cell line (KYSE-510) by induction of G2/M arrest and apoptosis. Toxicol In Vitro. 2009;23:797–807. doi: 10.1016/j.tiv.2009.04.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.