

Abstract
BACKGROUND AND PURPOSE
Human ether-a-go-go related gene (HERG) channel inhibitors may be subdivided into compounds that are trapped in the closed channel conformation and others that dissociate at rest. The structural peculiarities promoting resting state dissociation from HERG channels are currently unknown. A small molecule-like propafenone is efficiently trapped in the closed HERG channel conformation. The aim of this study was to identify structural moieties that would promote dissociation of propafenone derivatives.
EXPERIMENTAL APPROACH
Human ether-a-go-go related gene channels were heterologously expressed in Xenopus oocytes and potassium currents were recorded using the two-microelectrode voltage clamp technique. Recovery from block by 10 propafenone derivatives with variable side chains, but a conserved putative pharmacophore, was analysed.
KEY RESULTS
We have identified structural determinants of propafenone derivatives that enable drug dissociation from the closed channel state. Propafenone and four derivatives with ‘short’ side chains were trapped in the closed channel. Five out of six bulky derivatives efficiently dissociated from the channel at rest. One propafenone derivative with a similar bulk but lacking an H-bond acceptor in this region was trapped. Correlations were observed between molecular weight and onset of channel block as well as between pKa and recovery at rest.
CONCLUSION AND IMPLICATIONS
The data show that extending the size of a trapped HERG blocker-like propafenone by adding a bulky side chain may impede channel closure and thereby facilitate drug dissociation at rest. The presence of an H-bond acceptor in the bulky side chain is, however, essential.
Keywords: arrhythmia, drug trapping, HERG, K-channels, propafenone, QT syndrome, voltage clamp, Xenopus oocyte
Introduction
Human ether-a-go-go related gene (HERG) channels conduct the rapid delayed rectifier K+ current (Sanguinetti et al., 1995; channel nomenclature follows Alexander et al., 2009). In the myocardium, this current accelerates the repolarization of the action potential (Tseng, 2001) and HERG channel inhibition delays repolarization and prolongs the cardiac action potential and QT interval (Haverkamp et al., 2000). The resulting prolongation of the QT interval can be associated with syncope and sudden death due to torsades de pointes ventricular tachycardia and ventricular fibrillation (Viskin, 1999; Keating and Sanguinetti, 2001). Several drugs have been withdrawn from the market due to HERG channel inhibition, including antiarrhythmics, antimicrobials, neuroleptics and antihistamines (Fermini and Fossa, 2003; Sanguinetti and Tristani-Firouzi, 2006).
Compared with other voltage-activated potassium (Kv) channels, HERG channels are remarkably sensitive to a large variety of structurally diverse drugs (Mitcheson and Perry, 2003). This ‘pharmacological promiscuity’ (Mitcheson, 2008) of HERG is apparently related to its structural and functional peculiarities. Most drugs inhibit HERG channels more efficiently in the open and/or inactivated state (for review, see Zou et al., 1997 and Sanguinetti and Tristani-Firouzi, 2006). There is evidence that drugs can be trapped in the channel pore (Mitcheson et al., 2000; Kamiya et al., 2001; Perry et al., 2004; Stork et al., 2007). The molecular mechanism of ‘drug trapping’ in HERG channels is currently unknown.
In a preceding study, we analysed the kinetics of HERG channel inhibition and drug dissociation of eight blockers. One group [1-[2-(6-methyl-2pyridyl)ethyl]-4-(4-methylsulfonyl aminobenzoyl)piperidine (E-4031), domperidone, terfenadine and bepridil] did not dissociate from the closed state at rest while channels almost completely recovered from block by amiodarone, cisapride, droperidol and haloperidol (Stork et al., 2007). No correlation between drug descriptors (size, volume, lipophilicity and accessible surface area) and the observed differences in drug trapping could, however, be established (Stork et al., 2007). We also gained preliminary insights into the molecular basis of trapping of the class 1C antiarrhythmic agent propafenone by docking a small set of compounds into protein homology models of the open and the closed state of the HERG channel (Thai et al., 2010).
In Thai et al. (2010), we classified a small set of propafenone derivatives as ‘trapped’ or ‘non-trapped’, based on theoretical considerations (i.e. ligand structures and docking studies with a homology model). These studies suggested a role of bulkiness in drug dissociation because the non-trapped derivatives analysed in this investigation carried a large substituent at the aniline nitrogen atom of the piperazine ring. This data set was, however, too small for linking drug trapping with molecular features of the compounds.
Here we have investigated the structural features of the ligand that could be linked to drug trapping, by making use of a systematically modified library of 10 propafenone derivatives. Propafenone itself is efficiently trapped in the HERG channel pore (Witchel et al., 2004). In particular, we studied propafenone derivatives with variable side chains while leaving the basic scaffold of the molecule (acylphenyloxypropanolamine) mostly unaffected (Figure 1).
Figure 1.
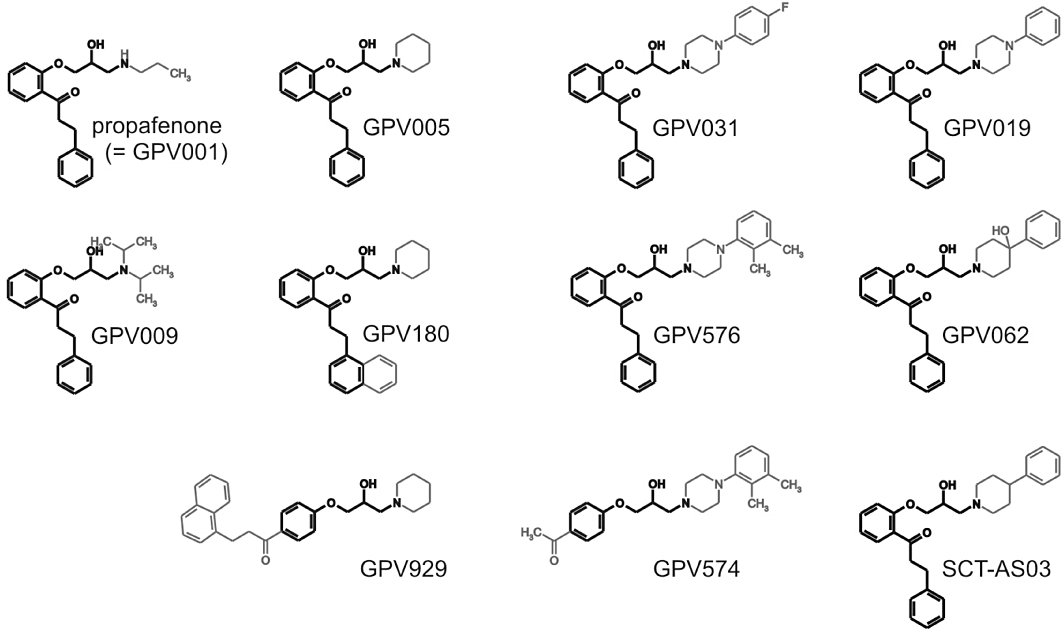
Chemical structures of propafenone and derivatives. All compounds were studied as hydrochlorides.
By analysing the kinetics of HERG inhibition and recovery from block we observed that more bulky derivatives tend to block the channels slowly, in a ‘use-dependent’ manner. At the same time, most bulky derivatives dissociated from the closed state at rest while propafenone and its less bulky derivatives were apparently trapped in the closed channel pore (Witchel et al., 2004). A bulky derivative (SCT-AS03) lacking the aniline-nitrogen in the side chain (i.e. carrying a phenylpiperidine instead of phenylpiperazine) displayed a slow onset of current inhibition and negligible recovery. Our data suggest that increased bulkiness and the H-bonding capabilities of the bulky moiety prevent appropriate channel closure, thereby enabling dissociation and recovery of the HERG channels from block.
Methods
Molecular biology
Preparation of stage V–VI oocytes from Xenopus laevis (NASCO, Fort Atkinson, WI, USA), synthesis of capped run-off complementary ribonucleic acid (cRNA) transcripts from linearized complementary deoxyribonucleic acid (cDNA) templates and injection of cRNA were performed as described previously (Sanguinetti and Xu, 1999). Complementary deoxyribonucleic acids of HERG (accession number NP_000229) and the mutants F656A and Y652A were kindly provided by Dr Sanguinetti (University of Utah, UT, USA).
Voltage clamp analysis
Currents through HERG channels were studied 1 to 4 days after microinjection of the cRNA using the two-microelectrode voltage clamp technique. The extracellular recording solution contained: 96 mM Na 2-(N-morpholino)ethanesulphonate, 2 mM K 2-(N-morpholino)ethanesulphonic acid, 2 mM CaCl2, 5 mM HEPES and 1 mM MgCl2, pH adjusted to 7.6 with methanesulphonic acid (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany).
Voltage-recording and current-injecting microelectrodes were filled with 3 M KCl and had resistances between 0.5 and 2 MΩ. Currents >3 µA were discarded to minimize voltage clamp errors. Ionic currents were recorded with a Turbo Tec 03X Amplifier (npi electronic, GmbH, Tamm, Germany) and digitized with a Digidata 1322A (Axon Instruments, Inc., Union City, CA, USA). The pClamp software package version 10.1 (Axon Instruments Inc.) was used for data acquisition. Microcal Origin 7.0 was employed for analysis and curve fitting.
A precondition for all measurements was the achievement of stable peak current amplitudes over periods of 10 min after an initial run-up period. All drugs were applied by means of the ScreeningTool fast perfusion system (npi electronic GmbH, Tamm, Germany) enabling solution exchange within 50–100 ms (Baburin et al., 2006). The oocytes were kept for 3 min at a holding potential of −80 mV to equilibrate drug diffusion. Use-dependent HERG channel block was estimated as peak tail current inhibition. The tail currents were measured at −50 mV, after a step to +20 mV. Cumulative concentration–inhibition curves were fitted using the Hill equation
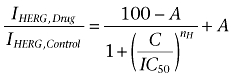 |
where IC50 is the concentration at which HERG inhibition is half-maximal, C is the applied drug concentration, A is the fraction of HERG current that is not blocked and nH is the Hill coefficient.
Recovery from channel block
Channel block was induced by applying 1 Hz trains of 14 pulses (conditioning train). Recovery from block at rest (holding potential −80 mV) was estimated after individual conditioning trains and subsequent rest periods of 10, 30, 60, 120, 210 or 330 s.
Data analysis
Data are presented as mean ± SEM from at least three oocytes from ≥2 batches; statistical significance of differences was defined as P < 0.05 in Student's unpaired t-test.
Materials
Propafenone and its derivatives GPV005, GPV009, GPV019, GPV031, GPV062, GPV180, GPV574, GPV576, GPV929 and SCT-AS03 (Figure 1; as hydrochlorides) were dissolved in dimethyl sulphoxide to prepare 10 mM stock solutions that were stored at −20°C. Drug stocks were diluted to the required concentration in extracellular solution on the day of each experiment. The maximal dimethyl sulphoxide concentration in the bath (0.1%) did not affect HERG currents.
Synthesis of novel propafenone derivatives
Propafenone and derivatives GPV005, GPV009, GPV019, GPV031, GPV062, GPV180, GPV574, GPV576 and GPV929 were synthesized as described previously (Chiba et al., 1996; Klein et al., 2002). Synthesis of SCT-AS03 was achieved as follows: 1-(2-(oxiranylmethoxy)phenyl)-3-phenyl-1-propanone (500 mg, 1.77 mmol) was dissolved in 2-propanol (5 mL). Then 4-phenylpiperidine (291 mg, 1.8 mmol) was added and the reaction mixture was stirred under reflux for 6 h. The solvent was evaporated and the residue was recrystallized from ethyl alcohol (5 mL). To prepare the hydrochloride, the product was dissolved in diethyl ether followed by addition of ethereal HCl (1 M). The precipitate was collected by filtration and provided 417 mg of the orange product with 53% yield. Physical characteristics as follows; 1H-NMR (200 MHz, CDCl3): δ= 7.72 (dd, 1 H, J= 7.7, 1.7 Hz), 7.50–7.39 (m, 1 H), 7.37–7.12 (m, 10 H), 7.07–6.93 (m, 2 H), 4.19–4.01 (m, 3 H), 3.67 (br s, 1 H), 3.45–3.30 (m, 2 H), 3.11–2.93 (m, 3 H), 2.85–2.70 (m, 1 H), 2.62–2.41 (m, 3 H), 2.40–2.21 (m, 1 H), 2.02–1.63 (m, 5 H). 13C-NMR(50 MHz, CDCl3): δ= 201.4 (Cq), 157.9 (Cq), 146.1 (Cq), 141.8 (Cq), 133.6 (CH), 130.6 (CH), 128.6 (CH), 128.6 (CH), 128.5 (CH), 128.4 (Cq), 126.9 (CH), 126.4 (CH), 126.0 (CH), 121.1 (CH), 112.8 (CH), 71.1 (CH2), 65.4 (CH), 61.3 (CH2), 56.1 (CH2), 52.9 (CH2), 45.8 (CH2), 42.4 (CH), 33.7 (CH2), 33.4 (CH2), 30.4 (CH2). MP: 102–103°C; MP (hydrochloride): 175–178°C. High Resolution Mass Spectrometry: Calculated: 444.2539; found: 444.2533.
Results
Propafenone library
The propafenone derivatives used in the present study are shown in Figure 1.
The basic side chain of propafenone (GPV001) was primarily altered by the replacement of the n-propylamino group by di-isopropylamino (GPV009), by 1-piperidinyl (GPV005) and by a series of 4-aryl-1-piperazinyl moieties with different substitution pattern on the aryl ring (unsubstituted GPV019, 4-fluoro GPV031, 3,4-dimethyl GPV576). Furthermore, we also used 4-phenyl-1-piperidinyl (SCT-AS03) and 4-hydroxy-4-phenyl-1-piperidinyl (GPV062) analogues.
The scaffold of GPV005 was further modified by the replacement of the 3-phenyl moiety by 1-naphthyl (GPV180) and a change in the substitution pattern of the central aromatic ring, where the 3(-(1-napthyl)-1-propionyl in the ortho position was shifted to the para position (GPV929). The scaffold of GPV576 was further modified by the replacement of the ortho-phenylpropionyl moiety by a para-acetyl group (GPV574).
Potency of HERG inhibition by propafenone derivatives
Human ether-a-go-go related gene channel inhibition by propafenone derivatives was studied in Xenopus oocytes by means of the two-microelectrode voltage clamp technique (see Methods). HERG channels were activated by a 300 ms depolarization to 20 mV (see Figure 2A). A subsequent repolarization to −50 mV induced a large tail current due to rapid recovery of HERG channels from inactivation. After pre-incubation of the oocytes with a given drug concentration, 0.3 Hz pulse trains were applied until steady-state was reached. Concentration–inhibition relationships for all tested compounds were estimated by plotting the steady-state values of current inhibition versus the cumulatively applied drug concentrations (Figure 2B and C). IC50 values for propafenone and its derivatives ranged from 0.77 to 5.04 µM (Table 1).
Figure 2.
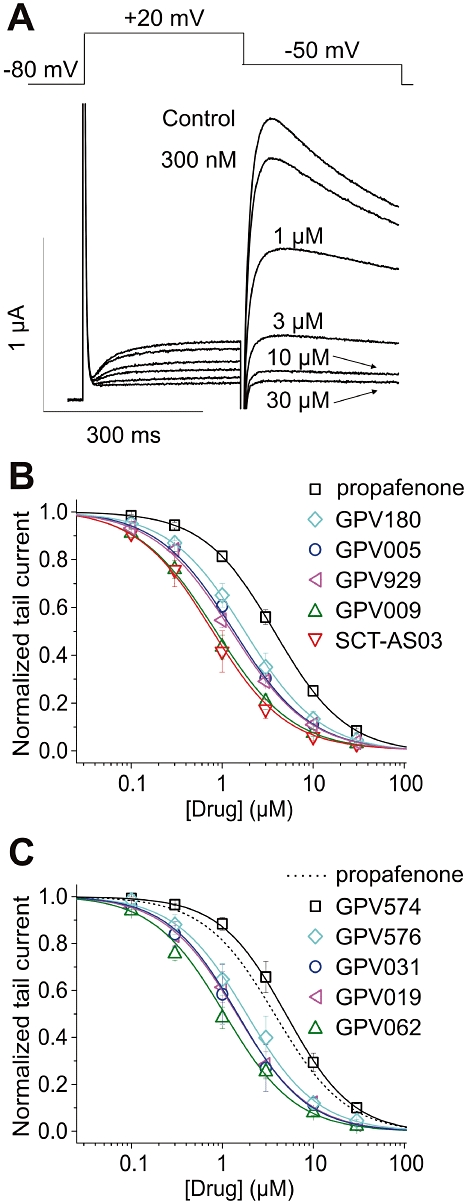
A, superimposed current traces recorded in the absence (control) and after attaining steady-state block with increasing concentrations of GPV009 (indicated in the figure). Voltage protocol is shown in the inset. B and C, concentration–response relationship for the block of human ether-a-go-go related gene tail current by propafenone and 10 derivatives.
Table 1.
Experimental data for propafenone and derivatives
| Compound | IC50 (µM) | Concentration (µM) | Time constant of block (s) | Recovery after 330 s (%) | Time constant of recovery (s) |
|---|---|---|---|---|---|
| Propafenone | 3.79 ± 0.240 | 10 | 0.44 ± 0.110 | 05.43 ± 12.240 | – |
| GPV005 | 1.34 ± 0.04* | 4 | 0.66 ± 0.11* | 05.68 ± 10.010 | – |
| GPV009 | 0.84 ± 0.08* | 3 | 1.39 ± 0.14* | 01.67 ± 07.220 | – |
| GPV019 | 1.42 ± 0.18* | 4 | 1.82 ± 0.14* | 59.00 ± 02.77* | 72.31 ± 16.53 |
| GPV031 | 1.46 ± 0.19* | 4 | 1.65 ± 0.18* | 69.25 ± 07.62* | 97.60 ± 27.25 |
| GPV062 | 1.05 ± 0.05* | 3 | 2.15 ± 0.27* | 46.41 ± 02.35* | 44.59 ± 16.51 |
| GPV180 | 1.78 ± 0.15* | 5 | 0.87 ± 0.15* | 00.58 ± 09.130 | – |
| GPV574 | 5.04 ± 0.540 | 15 | 0.48 ± 0.090 | 60.55 ± 11.98* | 61.09 ± 46.05 |
| GPV576 | 1.84 ± 0.24* | 6 | 1.58 ± 0.17* | 61.03 ± 06.91* | 84.74 ± 23.34 |
| GPV929 | 1.27 ± 0.05* | 4 | 1.00 ± 0.13* | 04.55 ± 05.200 | – |
| SCT-AS03 | 0.77 ± 0.11* | 2 | 2.39 ± 0.39* | 12.26 ± 04.290 | – |
Data are given as means ± SEM from at least three experiments. Drug concentrations (≈3·IC50) for studying onset of and recovery from HERG channel block were chosen to induce substantial and comparable current inhibition.
P < 0.05, significantly different from propafenone (unpaired Student's t-test).
Onset of HERG inhibition by propafenone derivatives
The onset of drug action was studied during trains of test pulses (1 Hz) applied after a 3 min equilibrium period in drug-containing solution, using the voltage protocol illustrated in the inset of Figure 2A. The development of HERG channel inhibition by propafenone and derivative GPV009 is illustrated in Figure 3A and B. The onset of channel inhibition was analysed by plotting the tail current amplitudes versus time (Figure 3C). While propafenone induced about 90% of steady-state inhibition during the 1st pulse, the onset of current inhibition by GPV009 was significantly slower (only 47.0% of steady-state inhibition during the 1st pulse). These differences prompted us to quantify the rate of onset of block (well-fitted with a mono-exponential function as exemplified in Figure 3C). To analyse the block onset for different derivatives, we applied different concentrations (≈ 3·IC50) to induce comparable current inhibition. This approach enabled us to compare the time courses of the onsets of block by compounds with different potencies. The time constants for all derivatives are given in Table 1.
Figures 3.
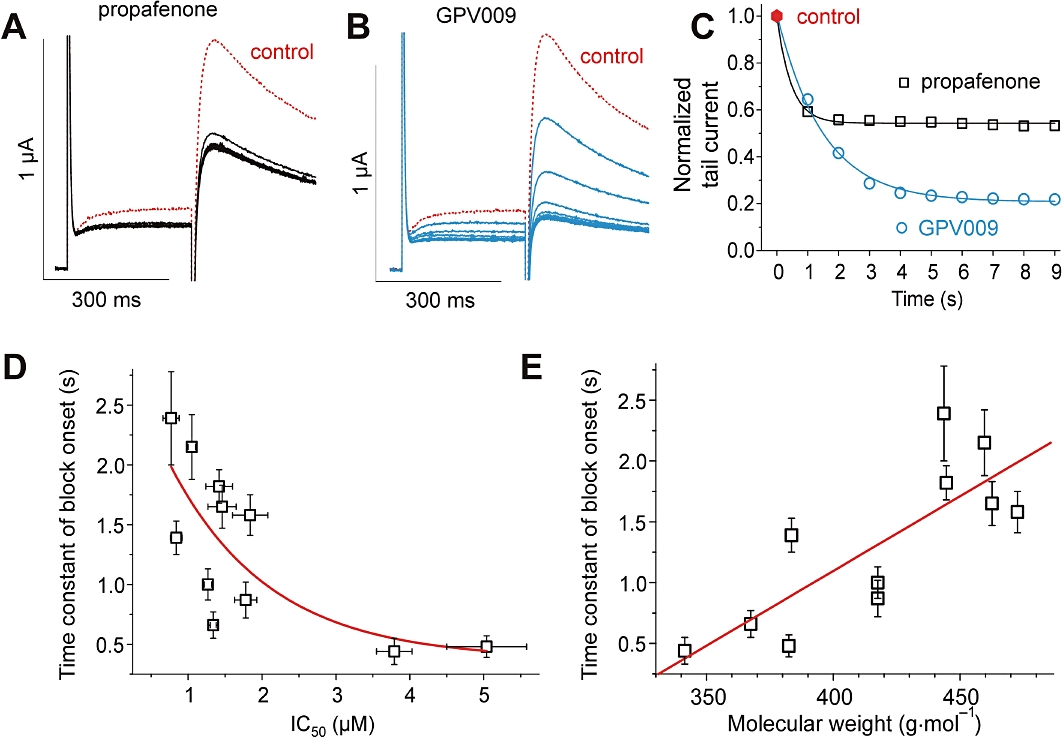
A–C, use-dependent block of human ether-a-go-go related gene currents by propafenone and GPV009. A and B, superimposed current traces during repetitive stimulation at a frequency of 1 Hz under application of 3 µM propafenone (A) or 3 µM GPV009 (B). Control currents are shown as dotted lines. C, peak tail current amplitudes are plotted versus time. D, exponential trend of the time constants of block development during a train of pulses (see Figure 3C, Table 1) versus the apparent affinities (IC50) of the studied propafenone derivatives. E, correlation (R= 0.8, P < 0.03) between the time constants of block development during a train of pulses and the size (molecular weight) of the studied propafenone derivatives.
Interestingly, a slower onset of channel inhibition was associated with higher apparent affinity (lower IC50 value). Thus, SCT-AS03 displayed the slowest onset with a τblock= 2.39 ± 0.39 s corresponding to the highest apparent affinity with an IC50= 0.77 ± 0.11 µM. The fastest block was induced by propafenone (τblock= 0.44 ± 0.11 s) followed by GPV574 (τblock= 0.48 ± 0.09 s) with corresponding IC50 values of 3.79 ± 0.24 µM and 5.04 ± 0.54 µM respectively (see Table 1 for details). This inverse relation between the time constants of block development and the apparent affinities is illustrated in Figure 3D. As shown for sodium channels (Courtney, 1980), we observed a correlation with the size (molecular weight) of the propafenone derivatives (Figure 3E).
Recovery from block at rest
Recovery from HERG channel block by propafenone derivatives (induced by 1 Hz pulse conditioning train, see Figure 4) was determined by applying test pulses after rest periods of 10, 30, 60, 120, 210 and 330 s. Each data point was measured after an individual conditioning train (see Methods). HERG channels blocked by propafenone and derivatives GPV005, GPV009, GPV180, GPV929 and SCT-AS03 did not recover at rest. This is exemplified for GPV009 in Figure 4C and D. Figure 4C illustrates three superimposed currents: (i) the current during the 1st pulse of the conditioning train in the presence of GPV009 (3 µM); (ii) a current during steady-state inhibition (14th pulse of the conditioning train); and (iii) the current after a 330 s rest period in the presence of drug. Figure 4D illustrates the gradual decline of the peak currents in GPV009 and the lack of recovery from block after 330 s.
Figure 4.
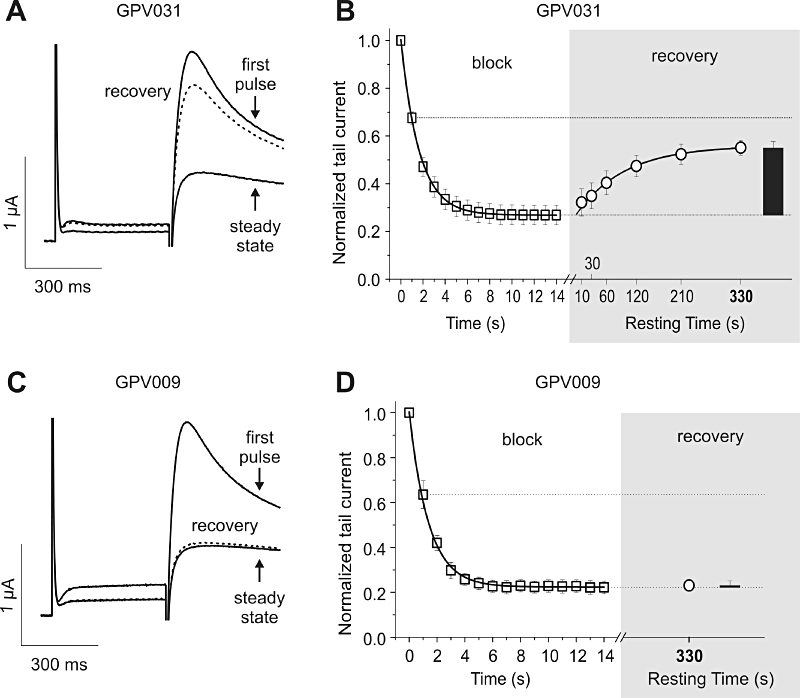
Recovery of human ether-a-go-go related gene channel from block at rest, in the continued presence of drug. Channel block was induced by 1 Hz pulse trains. Fourteen conditioning pulses were applied to reach steady state of inhibition by the particular compound. Single test pulses were applied after the indicated resting time at −80 mV. A and C, superimposed current traces of first and last (‘steady state’) pulse during a conditioning train after application of 4 µM GPV031 (A) and 3 µM GPV009 (C). Recovery current resulting from a single test pulse after 330 s resting time is depicted as dotted line. C and D, mean normalized tail current amplitudes in the presence of 4 µM GPV031 (B) and 3 µM GPV009 (D) are plotted against time. The section ‘block’ shows the development of inhibition during a 1 Hz pulse train. The grey highlighted section ‘recovery’ maps the amount of recovery after 330 s resting time as black bar, and additionally the recovery kinetics of GPV031 (B).
Mono-exponential recovery from block at rest was observed in the presence of GPV019, GPV031, GPV574 and GPV576 (shown in Figure 4A and B for compound GPV031, 4 µM).
The mean fractions of recovered HERG currents after a 330 s rest at −80 mV are given in Figure 5A and in Table 1. Negligible recovery (<13% after 330 s at rest) was observed in the presence of propafenone, GPV005, GPV009, GPV180, GPV929 and SCT-AS03 while substantial recovery was observed for GPV019, GPV031, GPV574 and GPV576 (range of recovery: 59.0–69.3%). An intermediate recovery value (46.4 ± 2.4%) was determined for GPV062.
Figure 5.
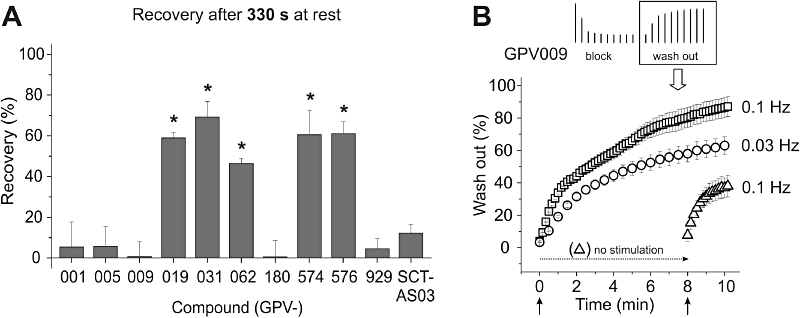
A, summary of the 330 s recovery fractions for the indicated compounds (GPV001 = propafenone). *Indicates statistically significant (unpaired t-test, P < 0.05) differences compared to propafenone. B, repetitive stimulation accelerates wash-out of GPV009 (and other trapped compounds, not shown in this figure). Human ether-a-go-go related gene channels were inhibited by a 1 Hz pulse train, as described in Figure 3. After reaching steady state of inhibition, the drug was washed out. During the wash-out process, pulses were applied with the indicated frequencies, starting either simultaneously with the beginning of wash-out or after 8 min of wash-out. Peak tail currents were normalized to control currents (amplitude before drug application) and plotted against time. More frequent stimulation (0.1 Hz) led to faster recovery during wash-out. Without pulsing (‘no stimulation’, −80 mV) channels barely recovered from block during wash-out.
Channel opening accelerates recovery from block during ‘wash-out’
As illustrated in Figure 5A, propafenone and its derivatives GPV005, GPV009, GPV180 GPV929 and SCT-AS03 did not recover from block during 330 s at rest. To study whether this lack of recovery results from drug trapping in the closed channel pore, we attempted to induce drug wash-out by repetitive channel opening (see Stork et al., 2007).
Human ether-a-go-go related gene currents were therefore inhibited by conditioning trains (14 pulses at 1 Hz). Drug wash-out was started immediately after the conditioning train by means of a fast perfusion system (Baburin et al., 2006). Repetitive pulsing enabled drug dissociation. This is illustrated in Figure 5B where the application of 0.1 Hz pulses after 8 min rest (without recovery) induced fast recovery from block by GPV009. Furthermore, pulse-induced recovery was frequency-dependent. Higher frequency pulsing (0.1 Hz) induced faster restoration of HERG currents than lower frequency pulsing (0.03 Hz, Figure 5B).
Evidence for interaction with putative binding determinants F656 and Y652
To study if the compounds interact with the putative binding pocket of propafenone, we analysed their action on selected HERG channel mutants. Alanine scanning mutagenesis revealed that the putative binding sites of most HERG inhibitors comprise three residues at the base of the selectivity filter (Thr623, Ser624 and Val625) and four on the S6 transmembrane helix (G648, Y652, F656 and V659) (Sanguinetti and Tristani-Firouzi, 2006; Mitcheson, 2008).
The binding pocket for propafenone comprises F656 and Y652. Alanine substitution of these residues markedly reduced HERG current inhibition by propafenone (Witchel et al., 2004). We compared the current inhibition of wild type (WT) and mutants F656A and Y652A by all compounds. Figure 6 shows representative currents recorded from oocytes expressing WT (Figure 6B), Y652A (Figure 6C) and F656A (Figure 6D) in the absence and presence of 15 µM GPV009. Figure 6E illustrates the effects of the propafenone derivatives on mutants Y652A and F656A. The drug concentrations were chosen to induce ≥90% inhibition of WT HERG channels (range: 15–70 µM). The mutant F656A channel almost completely lost sensitivity to all propafenone derivatives, while the Y652A channel showed a reduced sensitivity to propafenone and its derivatives.
Figure 6.
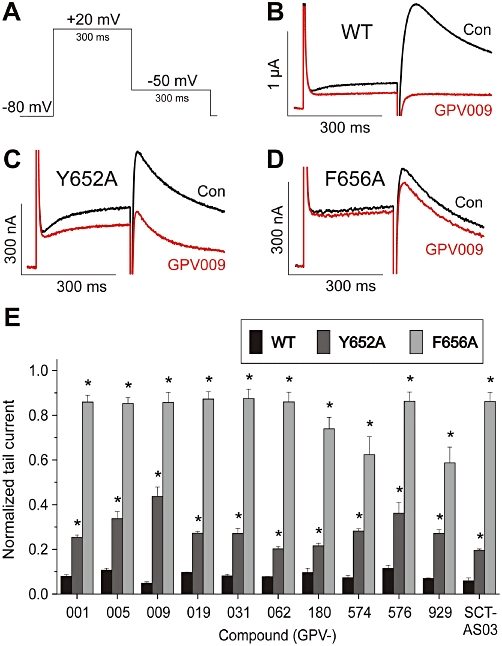
A, voltage pulse protocol for investigating wild type (WT)-, Y652A- and F656A-HERG channels. B–D, representative current traces of HERG and mutant HERG channels in the absence (Con) and presence (GPV009) of 15 µM GPV009. E, summary of mean current remaining with 50 µM propafenone (=GPV001), 15 µM GPV005, 15 µM GPV009, 15 µM GPV019, 15 µM GPV031, 15 µM GPV062, 15 µM GPV180, 70 µM GPV574, 25 µM GPV576, 15 µM GPV929 or 15 µM SCT-AS03. Inhibition of Y652A- and F656A-HERG by the indicated compounds was significantly weaker (unpaired t-test, P < 0.05) than inhibition of WT-HERG. HERG, Human ether-a-go-go related gene.
Discussion and conclusions
Recovery of voltage-gated ion channels from block after membrane repolarization is usually slower than the accumulation process and ranges from several seconds to several minutes. Physicochemical properties of the blocking molecule such as molecular weight (or size) (Courtney, 1980; Campbell, 1983), lipid solubility (Hille, 1977), stereo-specificity and the distribution of the molecule's charged groups have been shown to modulate recovery of sodium channels from block (Hille, 2001). Yeh and TenEick (1987) have shown that recovery from block in sodium channels can be accelerated by frequent pulsing to open the channels indicating that blockers can be trapped in the channel.
Evidence for drug trapping in HERG channels comes from ultra-slow recovery (or lack of recovery) at rest (e.g. Carmeliet, 1992; Mitcheson et al., 2000; Kikuchi et al., 2005; Kamiya et al., 2006). Stork et al. (2007) analysed the recovery kinetics of HERG channels from block by eight known HERG inhibitors that either dissociated at rest (amiodarone, cisapride, haloperidol and droperidol) or were completely trapped (bepridil, terfenadine, E-4031 and domperidone). Frequent depolarization during wash-out or reopening at hyperpolarization (mutant D540K) allowed the trapped drug molecules to escape from the channel (Stork et al., 2007).
The aim of the present study was to investigate whether structural changes in propafenone (a drug that is trapped in the closed channel, Witchel et al., 2004) would affect drug dissociation. To gain insights into determinants of drug trapping, propafenone was modified both in the vicinity of the basic nitrogen atom and at the central aromatic ring system (Figure 1).
All compounds inhibited HERG channels, with potencies (IC50) ranging from 0.77 ± 0.11 µM (SCT-AS03) to 5.04 ± 0.54 µM (GPV574) (Figure 2, Table 1). Structural modifications of propafenone affected the onset of HERG channel inhibition and recovery (Figures 2 and 5A, Table 1). Channel block by all derivatives was affected by mutations F656A and Y652A in the channel protein, suggesting that these compounds bind to the previously identified binding pocket of propafenone (Figure 6E, Witchel et al., 2004; Thai et al., 2010). Inhibition of mutant F656A by GPV929 and GPV574 was stronger than that by propafenone and the other derivatives. It is tempting to speculate that the ortho-phenylpropanone structure affects the orientation in the binding pocket, thereby making the drug-Phe656 interaction more favourable.
Open channel block by propafenone derivatives
Use-dependent HERG channel inhibition by propafenone derivatives is a hallmark of state-dependent drug binding. Arias et al. (2003) concluded that propafenone binds to the open and the inactivated states of the channel. Later studies revealed, however, that the non-inactivating HERG mutants S631A and S628C/S631C were also efficiently inhibited by propafenone indicating a less essential role of inactivation (Witchel et al., 2004).
With the exception of propafenone and GPV574, HERG inhibition gradually accumulated during the pulse trains. Pronounced first pulse inhibition observed for propafenone (Figure 3A and C) and GPV574 (data not shown) may reflect either resting state inhibition (initial block occurring during pre-incubation in drug) or fast inhibition of open channels. Our data support the second scenario where drug molecules rapidly enter and occlude the open channel. Hence, recovery from block by propafenone, GPV005, GPV009, GPV180 GPV929 and SCT-AS03 is induced by repetitive channel openings during wash-out (exemplified in Figure 5B for GPV009). These data suggest that access and dissociation of these compounds occurs via the open channel pore.
Relation between onset of channel block and IC50
Figure 3 illustrates substantial differences in block development induced by propafenone and GPV009. The initial rates of onset of HERG inhibition were comparable for both drugs (Figure 3C), while the steady-state level of block was deeper for GPV009. This pattern may reflect a decrease in the rate constant for GPV009 dissociation from the open channel state: a deeper level of channel inhibition (KD= k-/k+) at comparable initial rates of the reaction (k+[D]). An inverse relation between potencies (IC50) and the rate of block development was observed for most compounds (Figure 3D). At concentrations ≈3 × IC50 inducing comparable levels of channel block (see Table 1), current inhibition by SCT-AS03 (the most potent inhibitor, IC50= 0.77 ± 0.11 µM) occurred slowly (τblock= 2.39 ± 0.39 s) compared with the least potent inhibitor GPV574 (IC50= 5.04 ± 0.54 µM) which induced fast HERG inhibition (τblock= 0.48 ± 0.09 s). The inverse relation (Figure 3E) between the time constants of block development and the size of the molecules is in line with previous observations on sodium channels (Courtney, 1980).
Structural modifications of propafenone facilitate recovery of HERG channels from block
Ultraslow recovery from block at rest as indication for drug trapping was first observed in cardiac myocytes for rapid delayed rectifier K+ current block by methanesulfonanilides (dofetilide, MK-499) (Carmeliet, 1992). Direct evidence for drug trapping was provided by Mitcheson et al. (2000) using the HERG mutant D540K which reopens during pronounced hyperpolarization, thereby accelerating recovery of channels from block by MK-499. Similar observations were made for nifekalant, bepridil, E-4031, dofetilide (Kamiya et al., 2006) and propafenone (Witchel et al., 2004).
In a preceding study we reported that trapped compounds, such as bepridil, terfenadine, E-4031 and domperidone, cannot be washed out during 330 s (Stork et al., 2007) while repetitive stimulation during wash-out induced a rapid recovery of HERG channels from block (see also Figure 5B). These data support the hypothesis that trapped drugs may leave the channel through the open gate. From the study of Stork et al. (2007), it was not clear which physicochemical properties promoted drug dissociation from closed channels at rest. To get insights into the structure–dissociation relationship, we made use of systematically modified propafenone derivatives (a compound known to be trapped in HERG channels, Witchel et al., 2004). Propafenone and the five derivatives GPV005, GPV009, GPV180, GPV929 and SCT-AS03 (Figure 1) did not dissociate at rest (<13% during 330 s at −80 mV, Figures 4C,D and 5A). In contrast, channels recovered from block by GPV019, GPV031, GPV574 or GPV576 almost completely during a 330 s rest period (Figures 4A,B and 5A). In this context it would be conceivable that the bulky residue of these derivatives affects the closure of the channel gate according to a ‘foot in the door’ type mechanism of channel blockade. This mechanism was first hypothesized by Yeh and Armstrong (1978), and shown to be applicable to the block of HERG channels by chloroquine (Sanchez-Chapula et al., 2002) and Kv1.5 (Decher et al., 2006). We have therefore asked the question: What renders a compound a ‘foot’?
Role of physicochemical properties in drug trapping
The common structural modification of these compounds is the extension of the propafenone pharmacophore by a phenylpiperazine residue (Figure 1). An obvious feature of the phenylpiperazine residue is its bulkiness compared with the propylamino and piperidine side chains of propafenone and the other trapped derivatives. However, while the onset of block development correlated with the molecular weight (Figure 3E), no correlation between recovery from block and molecular weight was observed (Figure 7A; see Table 2 for physico-chemical values). Hence, the relatively small GPV574 was able to dissociate from the closed state while the bulky SCT-AS03 was efficiently trapped. This finding suggests that bulkiness per se does not explain recovery. Also no correlation between recovery and log P could be established (Figure 7B). Our data revealed, however, a significant correlation between dissociation of a given compound from the closed state (recovery at rest) and its pKa value (Figure 7C). This emphasizes that also the protonation state and/or the H-bond acceptor capabilities in this region of the molecules play a role.
Figure 7.
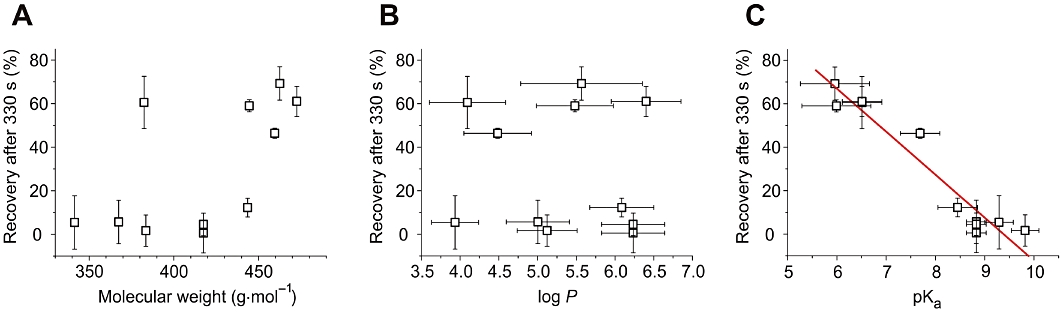
Correlations between the % of recovery after 330 s (Table 1) and different physicochemical properties (Table 2). A, no significant correlation with molecular weight. B, no correlation with log P. C, significant correlation (R=−0.96, P < 0.0001) with pKa.
Table 2.
Physicochemical properties of propafenone and derivatives
| Compound | Molecular weight (g·mol−1) | log P | pKa |
|---|---|---|---|
| Propafenone | 341.44 | 3.93 ± 0.30 | 9.29 ± 0.29 |
| GPV005 | 367.48 | 5.00 ± 0.41 | 8.83 ± 0.20 |
| GPV009 | 383.52 | 5.12 ± 0.39 | 9.82 ± 0.28 |
| GPV019 | 444.57 | 5.48 ± 0.50 | 5.99 ± 0.70 |
| GPV031 | 462.56 | 5.57 ± 0.79 | 5.96 ± 0.70 |
| GPV062 | 459.58 | 4.49 ± 0.44 | 7.69 ± 0.40 |
| GPV180 | 417.54 | 6.24 ± 0.41 | 8.83 ± 0.20 |
| GPV574 | 382.50 | 4.10 ± 0.49 | 6.51 ± 0.40 |
| GPV576 | 472.62 | 6.40 ± 0.50 | 6.51 ± 0.40 |
| GPV929 | 417.54 | 6.24 ± 0.49 | 8.83 ± 0.20 |
| SCT-AS03 | 443.58 | 6.09 ± 0.41 | 8.45 ± 0.40 |
Values were calculated using Advanced Chemistry Development (ACD/Laboratories) Software V8.14 (© 1994–2010 ACD/Laboratories, Toronto, Ontario, Canada).
Summary and outlook
An important finding of this study is that the extension of the propafenone pharmacophore by a phenylpiperazine moiety (Figure 1; GPV019, GPV031, GPV574 and GPV576) facilitates drug dissociation from the closed channel state (Figure 5A).
Our data suggest, however, that either the aniline-nitrogen atom or the basicity of N-1 of the piperazine plays a key role. Modification of the phenylpiperazine extension, for example, replacement of the aniline-nitrogen by a carbon atom (SCT-AS03) completely prevented drug dissociation (12% during 330 s, Table 1, recovery was induced by repetitive pulsing, data not shown). The addition of a hydroxyl group in this position (GPV062) partially restored drug dissociation (Table 1).
In scenario 1, the presence of an H-bond acceptor of the bulky side chain is essential. The aniline-nitrogen atom (GPV019, GPV031, GPV574 and GPV576) or the oxygen atom (GPV062) may form an H-bond with the channel locking mechanism (by affecting either the voltage sensor movement or the translation of this signal to the channel gate). In scenario 2, protonation may prevent drug dissociation (Figure 7C). To what extent an H-bond and/or charge prevent channel closure by a classical ‘foot in the door’ mechanism warrants further studies.
In summary, we have defined structural determinants of propafenone derivatives that mediate drug dissociation from the closed channel state. Besides increased bulkiness, changes in pKa and/or the capability of forming H-bonds, rigidity and conformation all may affect channel gating, thereby preventing appropriate channel closure and subsequently enabling dissociation and recovery of the HERG channels from block.
Acknowledgments
This work was supported by the PhD programme ‘Molecular Drug Targets’ of the University of Vienna and by a grant from The Austrian Science Fund (FWF; Project P19614-B11) to Steffen Hering. We are grateful to Annette Hohaus and Pakiza Rawnduzi for technical assistance. Andreas Windisch thanks the foundation ‘Burgenlandstiftung’ for patronage by means of a Theodor Kery prize.
Glossary
Abbreviations
- E-4031
1-[2-(6-methyl-2pyridyl)ethyl]-4-(4-methylsulfonyl aminobenzoyl)piperidine
- HERG
human ether-a-go-go related gene
- MK-499
(+)-N-[1′-(6-cyano-1,2,3,4-tetrahydro-2(R)-naphthalenyl)-3,4-dihydro-4(R)-hydroxyspiro(2H-1-benzopyran-2,4′-piperidin)-6-yl]methanesulfonamide] monohydrochloride
- QT
interval, QT interval of the electrocardiogram
- τ
time constant
- WT
wild type
Conflicts of interest
None.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C, Gonzalez T, Moreno I, Caballero R, Delpon E, Tamargo J, et al. Effects of propafenone and its main metabolite, 5-hydroxypropafenone, on HERG channels. Cardiovasc Res. 2003;57:660–669. doi: 10.1016/s0008-6363(02)00726-5. [DOI] [PubMed] [Google Scholar]
- Baburin I, Beyl S, Hering S. Automated fast perfusion of Xenopus oocytes for drug screening. Pflügers Arch. 2006;453:117–123. doi: 10.1007/s00424-006-0125-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell TJ. Importance of physico-chemical properties in determining the kinetics of the effects of Class I antiarrhythmic drugs on maximum rate of depolarization in guinea-pig ventricle. Br J Pharmacol. 1983;80:33–40. doi: 10.1111/j.1476-5381.1983.tb11046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet E. Voltage- and time-dependent block of the delayed K+ current in cardiac myocytes by dofetilide. J Pharmacol Exp Ther. 1992;262:809–817. [PubMed] [Google Scholar]
- Chiba P, Ecker G, Schmid D, Drach J, Tell B, Goldenberg S, et al. Structural requirements for activity of propafenone-type modulators in P-glycoprotein-mediated multidrug resistance. Mol Pharmacol. 1996;49:1122–1130. [PubMed] [Google Scholar]
- Courtney KR. Structure-activity relations for frequency-dependent sodium channel block in nerve by local anesthetics. J Pharmacol Exp Ther. 1980;213:114–119. [PubMed] [Google Scholar]
- Decher N, Kumar P, Gonzalez T, Pirard B, Sanguinetti MC. Binding site of a novel Kv1.5 blocker: a ‘foot in the door’ against atrial fibrillation. Mol Pharmacol. 2006;70:1204–1211. doi: 10.1124/mol.106.026203. [DOI] [PubMed] [Google Scholar]
- Fermini B, Fossa AA. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat Rev Drug Discov. 2003;2:439–447. doi: 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]
- Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, et al. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc Res. 2000;47:219–233. doi: 10.1016/s0008-6363(00)00119-x. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates Inc.; 2001. [Google Scholar]
- Kamiya K, Mitcheson JS, Yasui K, Kodama I, Sanguinetti MC. Open channel block of HERG K(+) channels by vesnarinone. Mol Pharmacol. 2001;60:244–253. doi: 10.1124/mol.60.2.244. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Nagatomo T, Abe H, Kawakami K, Duff HJ, Makielski JC, et al. Blockade of HERG cardiac K+ current by antifungal drug miconazole. Br J Pharmacol. 2005;144:840–848. doi: 10.1038/sj.bjp.0706095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Kaiser D, Kopp S, Chiba P, Ecker GF. Similarity based SAR (SIBAR) as tool for early ADME profiling. J Comput Aided Mol Des. 2002;16:785–793. doi: 10.1023/a:1023828527638. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS. hERG potassium channels and the structural basis of drug-induced arrhythmias. Chem Res Toxicol. 2008;21:1005–1010. doi: 10.1021/tx800035b. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Perry MD. Molecular determinants of high-affinity drug binding to HERG channels. Curr Opin Drug Discov Devel. 2003;6:667–674. [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Sanguinetti MC. Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J Gen Physiol. 2000;115:229–240. doi: 10.1085/jgp.115.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, de Groot MJ, Helliwell R, Leishman D, Tristani-Firouzi M, Sanguinetti MC, et al. Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol. 2004;66:240–249. doi: 10.1124/mol.104.000117. [DOI] [PubMed] [Google Scholar]
- Sanchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J Biol Chem. 2002;277:23587–23595. doi: 10.1074/jbc.M200448200. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Xu QP. Mutations of the S4-S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J Physiol. 1999;514(Pt 3):667–675. doi: 10.1111/j.1469-7793.1999.667ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Stork D, Timin EN, Berjukow S, Huber C, Hohaus A, Auer M, et al. State dependent dissociation of HERG channel inhibitors. Br J Pharmacol. 2007;151:1368–1376. doi: 10.1038/sj.bjp.0707356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai KM, Windisch A, Stork D, Weinzinger A, Schiesaro A, Guy RH, et al. The hERG potassium channel and drug trapping: insight from docking studies with propafenone derivatives. ChemMedChem. 2010;5:436–442. doi: 10.1002/cmdc.200900374. [DOI] [PubMed] [Google Scholar]
- Tseng GN. I(Kr): the hERG channel. J Mol Cell Cardiol. 2001;33:835–849. doi: 10.1006/jmcc.2000.1317. [DOI] [PubMed] [Google Scholar]
- Viskin S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–1633. doi: 10.1016/S0140-6736(99)02107-8. [DOI] [PubMed] [Google Scholar]
- Witchel HJ, Dempsey CE, Sessions RB, Perry M, Milnes JT, Hancox JC, et al. The low-potency, voltage-dependent HERG blocker propafenone – molecular determinants and drug trapping. Mol Pharmacol. 2004;66:1201–1212. doi: 10.1124/mol.104.001743. [DOI] [PubMed] [Google Scholar]
- Yeh JZ, Armstrong CM. Immobilisation of gating charge by a substance that simulates inactivation. Nature. 1978;273:387–389. doi: 10.1038/273387a0. [DOI] [PubMed] [Google Scholar]
- Yeh JZ, TenEick RE. Molecular and structural basis of resting and use-dependent block of sodium current defined using disopyramide analogues. Biophys J. 1987;51:123–135. doi: 10.1016/S0006-3495(87)83317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou A, Curran ME, Keating MT, Sanguinetti MC. Single HERG delayed rectifier K+ channels expressed in Xenopus oocytes. Am J Physiol. 1997;272(3 Pt 2):H1309–H1314. doi: 10.1152/ajpheart.1997.272.3.H1309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.