

Abstract
BACKGROUND AND PURPOSE
We recently characterized LY2033298 as a novel allosteric modulator and agonist at M4 muscarinic acetylcholine receptors (mAChRs). Evidence also suggested a difference in the potency of LY2033298 at rodent relative to human M4 mAChRs. The current study investigated the basis for the species difference of this modulator and used this knowledge to rationalize its in vivo actions.
EXPERIMENTAL APPROACH
LY2033298 was investigated in vitro in CHO cells stably expressing human or mouse M4 mAChRs, using assays of agonist-induced ERK1/2 or GSK-3α phosphorylation, [35S]-GTPγS binding, or effects on equilibrium binding of [3H]-NMS and ACh. The in vivo actions of LY2033298 were investigated in a mouse model of amphetamine-induced locomotor activity. The function of LY2033298 was examined in combination with ACh, oxotremorine or xanomeline.
KEY RESULTS
LY2033298 had similar affinities for the human and mouse M4 mAChRs. However, LY2033298 had a lower positive co-operativity with ACh at the mouse relative to the human M4 mAChR. At the mouse M4 mAChR, LY2033298 showed higher co-operativity with oxotremorine than with ACh or xanomeline. The different degrees of co-operativity between LY2033298 and each agonist at the mouse relative to the human M4 mAChR necessitated the co-administration of LY2033298 with oxotremorine in order to show in vivo efficacy of LY2033298.
CONCLUSIONS AND IMPLICATIONS
These results provide evidence for species variability when comparing the allosteric interaction between LY2033298 and ACh at the M4 mAChR, and also highlight how the interaction between LY2033298 and different orthosteric ligands is subject to ‘probe dependence’. This has implications for the validation of allosteric modulator actions in vivo.
Keywords: allosteric interaction, antipsychotic, drug discovery, locomotor activity, muscarinic receptors, positive co-operativity, probe-dependence, radioligand binding, species variability, positive allosteric modulator
Introduction
Schizophrenia is a complex psychiatric disorder, affecting approximately 1% of the world's population, and symptoms are grouped into three major domains: (i) positive, (ii) negative and (iii) cognitive. Current drug therapy for schizophrenia is generally suboptimal, with negative and cognitive symptoms showing poorer outcomes than positive symptoms following treatment (Lieberman et al., 2005; Miyamoto et al., 2005; Murphy et al., 2006; Buckley and Stahl, 2007). This demonstrates an ongoing need for alternative and/or complementary approaches to the current clinical armamentarium in the treatment of schizophrenia, which consists predominantly of typical antipsychotics, such as haloperidol, atypical antipsychotics, such as clozapine, and third generation agents, such as aripiprazole (Miyamoto et al., 2005).
A growing number of studies have shown that cholinergic neurotransmission, mediated through the activation of M1 and M4 muscarinic acetylcholine receptor (mAChR) subtypes, may play a fundamental role in the underlying pathophysiology of schizophrenia (Bymaster et al., 2002; Langmead et al., 2008). There are compelling data suggesting that selective activation of the M4 mAChR, which is localized to the cortex, hippocampus and striatum – regions that are relevant to attention and learning (Hasselmo, 2006) – may be of particular benefit. For instance, M4 mAChR knockout mice show a phenotype that is delineated by hyperexcitability of the dopamine system (Gomeza et al., 1999; Felder et al., 2001; Zhang et al., 2002; Tzavara et al., 2004). Furthermore, genetic, biochemical and post-mortem imaging assays have found decreased M4 mAChR densities in prefrontal cortex and hippocampus of schizophrenic subjects (Scarr and Dean, 2008). Finally, a recent clinical trial found that xanomeline, a drug that preferentially activates both M4 and M1 mAChRs (Shannon et al., 2000; Stanhope et al., 2001; Bymaster et al., 2002), significantly improved scores in the positive and negative symptom scale (PANSS) for symptoms in schizophrenic patients (Shekhar et al., 2008); an effect probably mediated, in part, by activation of the M4 mAChR (Woolley et al., 2009).
Unfortunately, traditional approaches to selectively targeting mAChRs have not been successful, in large part due to the high degree of sequence conservation within the orthosteric (ACh-binding) site across the five mAChR subtypes. More substantial progress has been made through the identification of small molecule allosteric ligands of these receptors (Gregory et al., 2007; Conn et al., 2009). In particular, selective potentiators of ACh binding and function at the M4 mAChR have been disclosed, exemplified by 3-amino-5-chloro-6-methoxy-4-methyl-thieno(2,3-b)pyridine-2-carboxylic acid cyclopropylamide (LY2033298) and related compounds (Brady et al., 2008; Chan et al., 2008; Nawaratne et al., 2008; Leach et al., 2010).
We have recently performed a detailed characterization of the pharmacology of LY2033298, and found that it is both an allosteric modulator and an allosteric agonist at M4 mAChRs, that it possesses in vivo efficacy in models predictive of antipsychotic drug effects, and that its in vivo efficacy is substantially attenuated in M4 mAChR knockout mice (Chan et al., 2008; Nawaratne et al., 2008; Leach et al., 2010). Intriguingly, we also observed that the functional potency of the modulator is reduced at rodent M4 mAChRs relative to the human receptor, and that its in vivo behavioural effects in rodents can sometimes be difficult to observe, often requiring the co-administration of sub-effective doses of an orthosteric agonist to see potentiation (Chan et al., 2008; Leach et al., 2010). This latter finding may reflect pharmacokinetic limitations, but may also be indicative of a potential for species variation in allosteric sites. Moreover, this variation need not be manifested as a difference in binding affinity of the molecule, but can also arise due to differences in the degree of allosteric interaction (co-operativity) between the orthosteric and allosteric sites on a given species of receptor. Finally, the need to co-administer an orthosteric agonist with an allosteric modulator to provide sufficient ‘tone’ for assessing an allosteric effect will be impacted by the phenomenon of ‘probe dependence’. This is fundamental to the nature of allosterism and occurs when the allosteric ligand induces changes in the activity of one orthosteric ligand whilst imparting different or no effect on others (Kenakin, 2005; Leach et al., 2007).
The aim of the current study was to gain insights into the extent, nature and consequences of species differences and probe-dependence on the interaction between LY2033298 and orthosteric agonists at the M4 mAChR. We reveal that the mechanistic basis for species variation in the effects of this compound on ACh activity between human and mouse M4 mAChRs is due to differences in co-operativity, not affinity, and that the modulator can display marked probe-dependence with different agonists. However, we also showed that knowledge of these properties can be used to both predict and rationalize subsequent design of in vivo studies, and suggest that our findings are likely to be of relevance to translational studies of allosterism at other GPCR families.
Methods
Materials
LY2033298 and xanomeline were synthesized in house at Eli-Lilly (Indianapolis, USA). CHO Flp-In cells were purchased from Invitrogen (Carlsbad, USA). Hygromycin was purchased from Roche (Basel, Switzerland). AlphaScreen™ streptavidin donor beads and anti-IgG (Protein A) used for pERK1/2 and GSK-3α detection were obtained from Perkin Elmer (Massachusetts, USA), whereas the AlphaScreen SureFire phospho-ERK1/2 and GSK-3α reagents were generously donated by Drs Michael Crouch and Ron Osmond (TGR Biosciences, South Australia). Dulbecco's modified Eagle medium (DMEM) and foetal bovine serum (FBS) were purchased from Gibco (Gaithersburg, MD, USA) and JRH Biosciences, respectively. [3H]-N-methylscopolamine ([3H]-NMS; specific activity 72 Ci·mmol−1) and guanosine 5′-[γ-35S]triphosphate ([35S]-GTPγS) (<1000 Ci·mmol−1) were from PerkinElmer Life and Analytical Sciences. All other chemicals were from Sigma Chemical Co. (St Loius, MO, USA).
Cell lines
Generation, culture and maintenance of CHO Flp-In cells stably expressing the human M4 mAChR cell lines were as previously described (Nawaratne et al., 2008). For the mouse M4 mAChR cell line, CHO-K1 cells were purchased from the ATCC (American Type Culture Collection, MD) and transfected with cDNA encoding the mouse M4 mAChR, as described previously (Singer-Lahat et al., 1997), and were maintained in high glucose DMEM, containing 10% FBS, 16 mM HEPES and 200 µg·mL−1 geneticin (G418).
Radioligand binding assays
Equilibrium binding assays were performed using 30 µg (mouse M4 mAChR) or 15 µg (human M4 mAChR) of membrane per assay point. For interaction studies, competition between [3H]-NMS and ACh was determined in the presence of 100 µM Gpp(NH)p and the absence or presence of 10 µM of LY2033298. Membranes were incubated with [3H]-NMS (0.2 nM), ACh and LY2033298 for 180 min at 37°C in HEPES buffer (20 mM HEPES, 100 mM NaCl, 10 mM MgCl, pH 7.4). Non-specific binding was defined by 10 µM atropine. The reaction was terminated by rapid filtration onto GF/B grade filters using a Brandel cell harvester, followed by three washes with ice-cold NaCl (0.9%). Radioactivity was then measured by liquid scintillation counting.
Extracellular signal-regulated kinase 1/2 phosphorylation (pERK1/2) assay
Cells were seeded into 96-well transparent plates at 40 000 cells per well and grown overnight. Cells were then washed twice with PBS and incubated in serum free media at 37°C for at least 4 h (to allow FBS-stimulated pERK1/2 to subside). Initial ERK1/2 phosphorylation time course experiments were performed to determine the time at which ERK1/2 phosphorylation was maximal following stimulation by agonists (at 8 min). For determination of agonist stimulated concentration–response curves, cells were incubated with each agonist at 37°C for the time required to generate the maximal pERK1/2 response. For interaction studies, cells were incubated at 37°C with varying concentrations of agonist in the absence and presence of different concentrations of LY2033298. In all instances, the reaction was terminated by the removal of drugs and cell lysis with 100 µL of SureFire™ lysis buffer. Cell lysates were agitated for at least 3 min and then SureFire™ activation buffer was added (ratio of 4:1 v/v lysate : SureFire™ activation buffer). Under low light conditions, a 1:240 dilution of AlphaScreen™ beads : SureFire™ reaction buffer was prepared. This combination was then mixed with lysate mixture in opaque 384-well plates at a ratio of 6:5 v/v. The plates were then incubated for at least 1 h in the dark at 37°C, and the fluorescence signal was measured using a Fusion plate reader (Perkin Elmer). For all experiments, 10% FBS was used as a positive control.
Glycogen synthase kinase 3α (GSK-3α) phosphorylation assays
Stimulation of GSK-3α phosphorylation was performed as described for ERK1/2 phosphorylation assays with the following exceptions: after agonist-stimulation and lysis of cells with SureFire™ lysis buffer, a mixture of reaction buffer, activation buffer, dilution buffer (all as provided by the manufacturer) and AlphaScreen beads was prepared at a ratio of 90:10:40:1 and was added to cell lysates at a ratio of 7:5 in a 384-well opaque Optiplate™ under low light conditions, for a total volume of 12 µL per well. Plates were incubated in the dark at 37°C for 2 h before the florescence signal was measured on a Fusion-α™ plate reader (PerkinElmer) using standard AlphaScreen settings.
[35S]-GTPγS assay
Cell membranes, 50 µg (mouse M4 mAChR) or 15 µg (human M4 mAChR), were equilibrated for 75 min at 30°C with ligands in buffer (20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, pH 7.4) containing 1 µM GDP. [35S]-GTPγS (0.1 nM) was added to a final volume of 1 mL and membranes were incubated for 30 min. Termination of [35S]-GTPγS binding was by rapid filtration with a Brandel harvester onto GF/B filter paper followed by three washes with ice-cold 0.9% NaCl. Filter paper was dried and 4 mL UltimaGold added to each filter, before radioactivity was determined by liquid scintillation counting.
Locomotor activity testing in mice
Experiments were carried out on 118 male C57Bl6 mice (ARC Perth, Western Australia) weighing 25–30 g at the time of testing. The mice were housed under standard conditions in groups of four with free access to food and water. They were maintained on a 12 h : 12 h light/dark cycle (lights on at 0700 h) at a constant temperature of 21–22°C. All animal experimental procedures were approved by the Monash University Animal Ethics Committee and were performed in accordance with their guidelines.
Prior to testing, mice were habituated to the test room for at least 1 h. Mice received test drugs via either an i.p. injection (d-amphetamine sulphate, oxotremorine, xanomeline) or a s.c. injection (LY2033298). Locomotor activity was measured using an Animex® locomotor activity apparatus. This apparatus measures the changes in oscillator circuits as mice move across magnetic fields. The Animex® meter was pretuned and the sensitivity set at 40 µA before each test session. Subsequent to drug administration, mice were immediately placed in a perspex cage, which was mounted on top of the Animex® meter. Each animal was used only once. Locomotor activity was recorded in 10 min blocks for a period of 70 min and total activity counts were analysed at 40 min. Room temperature was recorded and ranged from 21 to 25°C. The doses of drugs employed in this study were as follows: d-amphetamine sulphate (5 mg·kg−1); xanomeline (0.1 mg·kg−1); oxotremorine (0.01 mg·kg−1) and LY2033298 (10 mg·kg−1). All doses refer to the salt forms. d-Amphetamine sulphate, oxotremorine and xanomeline were dissolved in saline, while LY2033298 was dissolved in 50% (v/v) pharmasolve, and 1.1% Tween 80 in D5W vehicle.
Data analysis
All data were analysed using Prism 5.03 (GraphPad Software, San Diego). Concentration–response data generated from ERK1/2 phosphorylation studies were normalized to the response generated by 10% FBS; GSK-3α phosphorylation data were normalized to the maximal response of each respective agonist in the absence of modulator; [35S]-GTPγS data were normalized to the maximal response generated by ACh. Data were then fitted to the following three-parameter Hill equation:
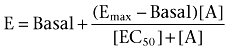 |
(1) |
where E is the pharmacological effect, [A] denotes the molar concentration of agonist, Basal denotes the basal response, Emax denotes the maximum agonist effect and EC50 denotes the molar agonist concentration that gives a response halfway between Basal and Emax. Concentration–response curves of ACh and LY2033298 at human and mouse receptors were also fitted to the following form of an operational model of agonism (Black and Leff, 1983) to obtain estimates of LY2033298 affinity and efficacy for the receptors:
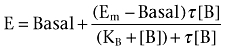 |
(2) |
where E is the pharmacological effect, [B] is the molar concentration of the allosteric (partial) agonist, Em is the maximal possible response, and KB and τ represent the equilibrium dissociation constant and the operational index of efficacy, respectively, of the allosteric agonist. For the analysis to converge, both ACh and LY2033298 concentration–response curves were globally fitted to Equations 1, for ACh, and 2 for LY2033298, with the Emax parameter from Equation 1 taken as an estimate of Em in Equation 2, and Basal shared between both data sets.
[3H]-NMS saturation and [3H]-NMS/ACh competition binding data were fitted to a one-site binding model, as described previously (Motulsky and Christopoulos, 2004) to obtain estimates of maximal density of binding sites (Bmax) and orthosteric agonist equilibrium dissociation constant (KI) respectively. For the determination of the binding co-operativity between ACh and LY2033298, the competition binding curves between ACh and [3H]-NMS in the absence and presence of LY2033298 were globally fitted to the following form of an allosteric ternary complex model (Lazareno and Birdsall, 1995; Christopoulos, 2000):
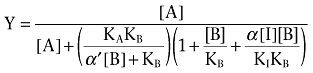 |
(3) |
where Y is fractional binding, [A], [B] and [I] are the concentrations of [3H]-NMS, LY2033298 and ACh respectively, KA, KB and KI are the equilibrium dissociation constants of [3H]-NMS, LY2033298 and ACh respectively, and α′ and α are the co-operativities between LY2033298 and [3H]-NMS or ACh respectively. Values of α (or α′) > 1 denote positive co-operativity; values < 1 (but >0) denote negative co-operativity, and values = 1 denote neutral co-operativity.
To determine the co-operativity between LY2033298 and various agonists in functional interaction studies, the EC50 values obtained for each agonist in the absence and presence of increasing concentrations of LY2033298 were used in the following equation (Christopoulos and Mitchelson, 1997; Christopoulos, 2000):
| (4) |
where pEC50 denotes the negative logarithm of the agonist EC50 values, pKB denotes the negative logarithm of the modulator KB value, logd is a fitting constant and αβ is a composite parameter, estimated as a single number, denoting the combined allosteric effect on affinity (α) and efficacy (β).
All parametric measures of potency, affinity, co-operativity and operational efficacy were estimated as logarithms (Christopoulos, 1998). Statistical analyses were by one-way analysis of variance (anova) followed by Dunnett's post-test, or by Student's t-test, as appropriate, with P < 0.05 taken as indicating significance. The data sets shown in Figure 4 were derived contemporaneously in a single experimental trial and therefore were analysed together, but for clarity, the data sets have been separated into two panels.
Figure 4.
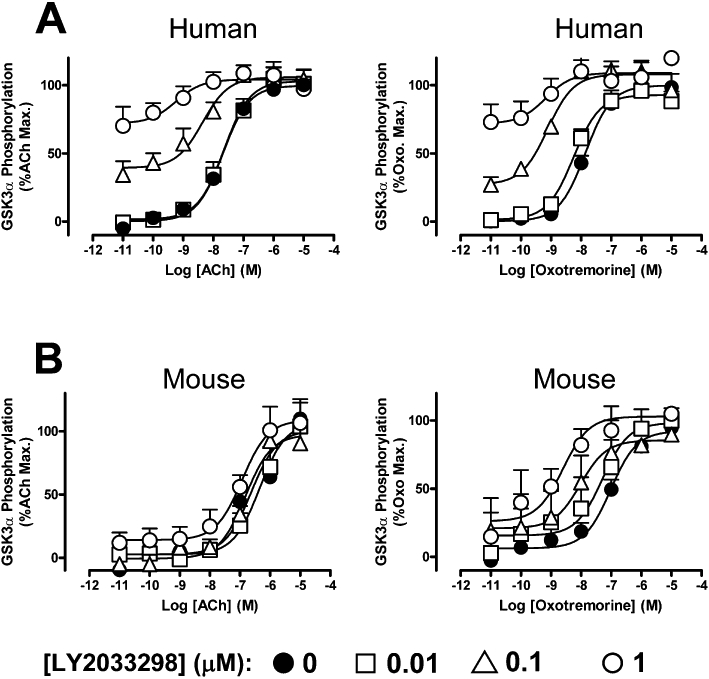
Effects of LY2033298 on the ability of ACh or oxotremorine to promote the phosphorylation of GSK-3α at the (A) human or (B) mouse M4 mAChR stably transfected in CHO cells. Data represent the mean of five experiments performed in duplicate.
All drug/molecular target nomenclature conform to the BJP's Guide to Receptors and Channels (Alexander et al., 2009).
Results
LY2033298 has similar affinities for, and displays robust agonism at, the human and mouse M4 mAChRs
Initial experiments determined the ability of LY2033298 and ACh to directly activate M4 mAChR by measuring agonist-induced ERK1/2 phosphorylation. This is an important, convergent, signalling pathway that has been implicated in regulating nervous system function, especially in association with brain regions implicated in learning and memory, that is, hippocampus, neocortex and cerebellum (Ortiz et al., 1995; Villarreal and Barea-Rodriguez, 2006). As shown in Figure 1, both the orthosteric agonist, ACh, and allosteric agonist, LY2033298, produced robust phosphorylation of ERK1/2 in CHO cells expressing the human or mouse M4 mAChRs. However, the potencies and maximal effects of each agonist were significantly reduced at the mouse relative to the human receptor (Figure 1; Table 1). This probably reflects the lower expression level of the mouse M4 mAChR (Bmax= 0.30 ± 0.04 pmol·mg−1, pKA= 9.74 ± 0.05; n= 3) relative to the human receptor (Bmax= 1.78 ± 0.52 pmol·mg−1, pKA= 9.51 ± 0.06; n= 3) in our cell lines, as determined by [3H]-NMS saturation binding assays. Importantly, the lower maximal effect of LY2033298 relative to ACh in both species allowed for the application of an operational model of agonism (Equation 2) to determine the affinity (pKB) and relative signalling efficacy (Logτ) of LY2033298 at both receptors. As shown in Table 1, an important finding from this analysis is that the allosteric ligand had essentially identical affinities for the allosteric site at the human and mouse receptors.
Figure 1.
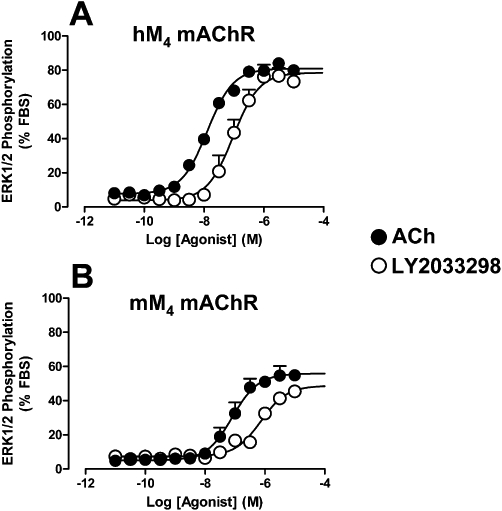
Effects of ACh or LY2033298 on ERK1/2 phosphorylation mediated by (A) human or (B) mouse M4 mAChRs stably transfected in CHO cells. Data points represent the mean ± SEM obtained from four to five experiments performed in duplicate.
Table 1.
Potency, maximal response and operational model parameters for agonist-mediated ERK1/2 phosphorylation
| Human M4 mAChR | Mouse M4 mAChR | |||
|---|---|---|---|---|
| Parameter | ACh | LY2033298 | ACh | LY2033298 |
| pEC50a | 7.89 ± 0.09 | 7.03 ± 0.09 | 7.09 ± 0.09* | 6.13 ± 0.12* |
| Emaxb | 80.9 ± 2.3 | 78.5 ± 2.9 | 55.9 ± 1.8* | 48.8 ± 2.9* |
| pKBc | n.a. | 5.39 ± 0.05 | n.a. | 5.49 ± 0.31 |
| Logτd | n.a. | 1.59 ± 0.05 | n.a. | 0.66 ± 0.22* |
Data represent mean ± SEM of four to five experiments performed in duplicate.
Significantly different (P < 0.05) from the corresponding value at the human receptor, as determined by Student's t-test.
Negative logarithm of the midpoint potency parameter.
Maximum agonist effect, expressed as a percentage of the ERK1/2 phosphorylation response elicited by 10% FBS.
Negative logarithm of the LY2033298 equilibrium dissociation constant, obtained from a global fit of an operational of agonism to the ACh and LY2033298 data sets.
Logarithm of the operational efficacy estimate.
n.a., not applicable.
LY2033298 has lower positive co-operativity with ACh at the mouse relative to the human M4 mAChR
Interaction studies were performed to determine whether the allosteric effect of LY2033298 on ACh differs between species. As shown in Figure 2A, LY2033298 (10 µM) resulted in an approximately 14-fold potentiation in the ability of ACh to inhibit the binding of [3H]-NMS at the human M4 mAChR (ACh pKi control = 5.84 ± 0.05; pKi in the presence of LY2033298 = 6.99 ± 0.06; n= 5), but only an approximately fivefold potentiation in the ability of ACh to inhibit the binding of [3H]-NMS at the mouse M4 mAChR (ACh pKi control = 5.74 ± 0.06; pKi in the presence of LY2033298 = 6.43 ± 0.07; n= 5). It was also noted that LY2033298 on its own caused a modest inhibition of the binding of [3H]-NMS at the human M4 mAChR, consistent with some negative co-operativity with the antagonist, but not at the mouse M4 mAChR. The data were also analysed according to an allosteric ternary complex model (Equation 3) to obtain an estimate of the binding co-operativity between ACh and LY2033298 at the M4 mAChR at each species. For this analysis, the pKB values of LY2033298 were fixed to those obtained from the operational model analysis (Table 1), and the log co-operativity between [3H]-NMS and LY2033298 (Logα′) at the mouse M4 mAChR was fixed to 0 (i.e. α′= 1). The resulting analyses yielded a Logα of 1.34 ± 0.02 (α= 22) for the interaction between ACh and LY2023398, and a Logα′ of −0.81 ± 0.20 (α′= 0.2) for the interaction between [3H]-NMS and LY2023398 at the human M4 mAChR. In contrast, the value of Logα for the interaction between ACh and LY2033298 at the mouse M4 mAChR was only 0.86 ± 0.09 (α= 7). The co-operativity factor between the modulator and agonist at the human receptor was in a similar range to that obtained previously for this interaction at the human M4 mAChR (Chan et al., 2008; Nawaratne et al., 2008; Leach et al., 2010), but significantly (P < 0.05) higher than that determined at the mouse receptor.
Figure 2.
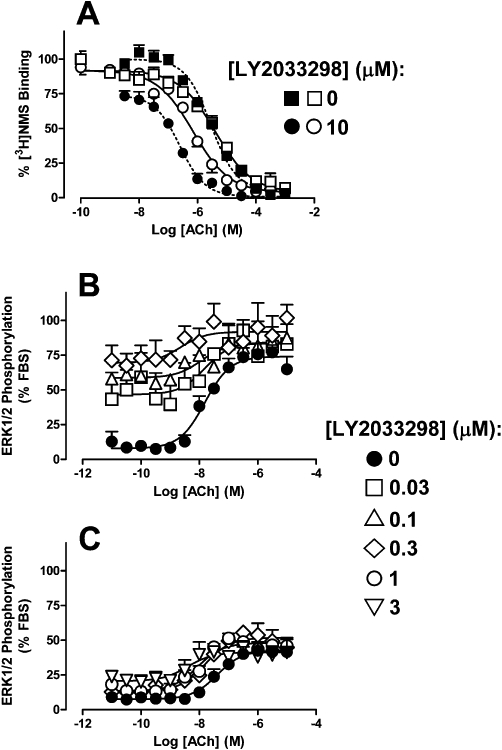
Effects of LY2033298 on the ability of ACh to (A) compete with the binding of the orthosteric antagonist, [3H]-NMS in membranes stably expressing the human (solid symbols) or mouse M4 (open symbols) mAChR, (B) promote the phosphorylation of ERK1/2 at the human M4 mAChR stably transfected in CHO cells, and (C) promote the phosphorylation of ERK1/2 at the mouse M4 mAChR stably transfected in CHO cells. Data points represent the mean ± SEM obtained from (A) five, (B) four or (C) five experiments performed in duplicate.
We next performed ERK1/2 interaction studies between LY2033298 and ACh to determine the extent of functional allosteric modulation between the two species of receptor. As shown in Figure 2B and C, the co-addition of ACh with LY2033928 resulted in both an increase in the basal responsiveness, due to allosteric agonism by LY2033298, and an increase in the potency of ACh, due to allosteric potentiation. However, it was also evident that the degree of functional potentiation was markedly lower at the mouse M4 mAChR than at the human M4 mAChR, with 0.3 µM LY2033298 producing a sevenfold potentiation of ACh at the human receptor, whereas ten times more modulator was required to potentiate the orthosteric agonist to a similar extent (10.5-fold) at the mouse receptor. Because the affinity of the modulator was unaltered between the two species of receptor, this finding is in agreement with the equilibrium binding assay suggesting a lower degree of positive co-operativity between LY2033298 and ACh at the mouse relative to the human receptor (see below for quantification).
The allosteric interaction between LY2033298 and other orthosteric agonists at the mouse M4 mAChR is characterized by ‘probe-dependence’
In contrast to orthosteric (competitive) interactions, allosteric interactions can vary markedly depending on the nature of the orthosteric ligand that is used as a ‘probe’ of receptor function (Kenakin, 2005; Leach et al., 2007). To assess whether such probe-dependence was manifest at the mouse M4 mAChR, we performed additional functional interaction studies with two other orthosteric mAChR agonists, oxotremorine and xanomeline. The former agent was chosen because it is centrally active and often used for in vivo studies of mAChR function (Witkin, 1989; Chan et al., 2008); the latter agent was chosen because it is M1/M4 mAChR-preferring and, as indicated in the Introduction, has shown clinical efficacy in reducing positive, negative and cognitive indices associated with schizophrenia. Figure 3 summarizes the results of these experiments, where it can be seen that substantially different degrees of potentiation were noted depending on whether the agonist was oxotremorine (Figure 3A) or xanomeline (Figure 3B).
Figure 3.
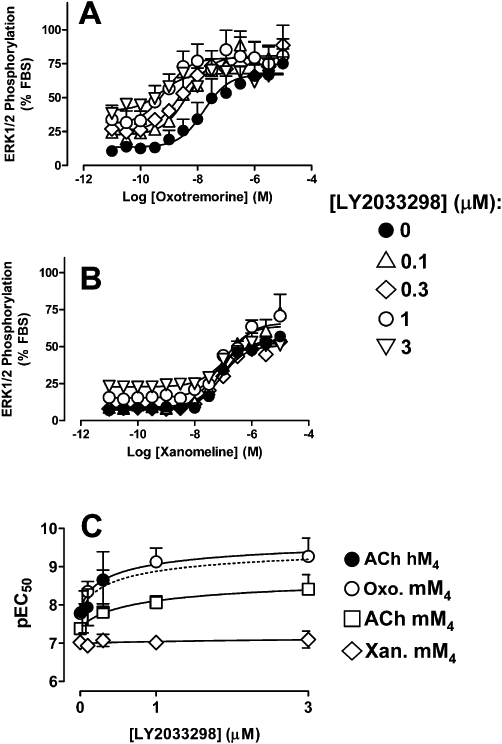
Effects of LY2033298 on the ability of (A) oxotremorine or (B) xanomeline to promote the phosphorylation of ERK1/2 at the mouse M4 mAChR stably transfected in CHO cells. Data represent the mean of (A) five or (B) four experiments performed in duplicate. (C) Nonlinear regression of the pEC50 values determined for ACh-, oxotremorine- or xanomeline-mediated ERK1/2 at the mouse M4 mAChR, or for ACh at the human M4 mAChR, in the absence or presence of LY2033298 according to an allosteric ternary complex model (Equation 4).
To obtain a quantitative estimate of the co-operativity between LY2033298 and each of the agonists investigated in our current study, we utilized the pEC50 values derived from the curves shown in Figures 2B,C and 3A,B in a nonlinear regression according to the allosteric model shown in Equation 4 (Figure 3C). By assuming a pKB of 5.49 for LY2033298 at the mouse M4 mAChR allosteric site, we obtained estimates that reflect the composite co-operativity on agonist binding (α) and intrinsic efficacy (β) (Aurelio et al., 2009; Leach et al., 2010), which are summarized in Table 2. A one-way anova, followed by Dunnett's post-test, indicated that, with the exception of the oxotremorine/LY2033298 co-operativity estimate, the values for ACh/LY2033298 and xanomeline/LY2033298 at the mouse M4 mAChR were significantly different (P < 0.05) from the value for ACh/LY2033298 at the human M4 mAChR. Thus, although we could not differentiate allosteric effects on affinity from effects on orthosteric agonist signalling efficacy, we were able to quantify the overall degree of probe-dependence between the modulator and each of the agonists.
Table 2.
Allosteric model co-operativity parameters (Logαβ) for the functional interaction between LY2033298 and orthosteric agonists at the M4 mAChR
| Agonist | ERK1/2 | GSK-3α | [35S]-GTPγS |
|---|---|---|---|
| ACh (human) | 1.75 ± 0.23 (56) | 2.29 ± 0.13 (194) | 1.97 ± 0.03 (93) |
| ACh (mouse) | 1.29 ± 0.06* (19) | 0.89 ± 0.36* (8) | 1.62 ± 0.08* (42) |
| Oxotremorine (human) | n.d. | 2.24 ± 0.51 (173) | n.d. |
| Oxotremorine (mouse) | 1.89 ± 0.09 (77) | 2.29 ± 0.17 (194) | 2.51 ± 0.06 *(326) |
| Xanomeline (mouse) | 0.18 ± 0.12* (1.5) | n.d. | 0.83 ± 0.01 *(7) |
Values represent the mean ± SEM from three to five experiments performed in triplicate and derived from application of Equation 2. Antilogarithms are shown in parentheses.
Significantly different (P < 0.05) from the corresponding value for ACh at the human receptor, as determined by one-way anova and Dunnett's post-test.
n.d., not determined.
Because we have recently shown that the magnitude of allosteric modulation of ACh by LY2033298 can vary with the signalling pathway (Leach et al., 2010), we also utilized a second whole cell functional assay, namely that monitoring the phosphorylation of GSK-3α, to determine the effects of the modulator on ACh in order to see whether the species variability is retained at another pathway; oxotremorine was included as a comparator. This signal transduction pathway was chosen because it has previously been implicated in playing a role in a number of CNS disorders, including schizophrenia (Lovestone et al., 2007). As shown in Figure 4, qualitatively similar results were obtained at this pathway to the findings made utilizing ERK1/2 phosphorylation as a functional readout. Application of Equation 4 to the data (n= 5) yielded the estimates of functional co-operativity summarized in Table 2. As expected, the magnitude of the positive co-operativity between each agonist and LY2033298 differed between pathways, but the fact that this co-operativity was markedly attenuated for the LY2033298/ACh pairing at the mouse receptor compared to the human receptor was retained; a one-way anova, followed by Dunnett's post-test, indicated that there was a significant difference (P < 0.05) between the Log(αβ) value for LY2033298/ACh at the mouse M4 mAChR compared to the other co-operativity factors determined in the GSK-3α experiments.
Finally, we investigated whether these differences in co-operativity and probe-dependence could be detected at the level of receptor-G protein coupling by determining the effects of LY2033298 on the ability of the M4 mAChR to promote [35S]-GTPγS binding to activated Gα proteins. Although this assay is performed in broken membranes under non-physiological conditions, it nonetheless represents an important proximal step in the process of GPCR-mediated signal transduction, can be performed at equilibrium, and can provide further support for the notion that probe-dependence is engendered at the level of the receptor conformation. Figure 5 shows the results of these experiments using LY2033298 against ACh, oxotremorine and xanomeline, and the derived co-operativity estimates are shown in Table 2. As with the whole cell functional studies, the co-operativity between ACh and LY2033298 was significantly lower at the mouse M4 mAChR than at the human M4 mAChR (P < 0.05).
Figure 5.
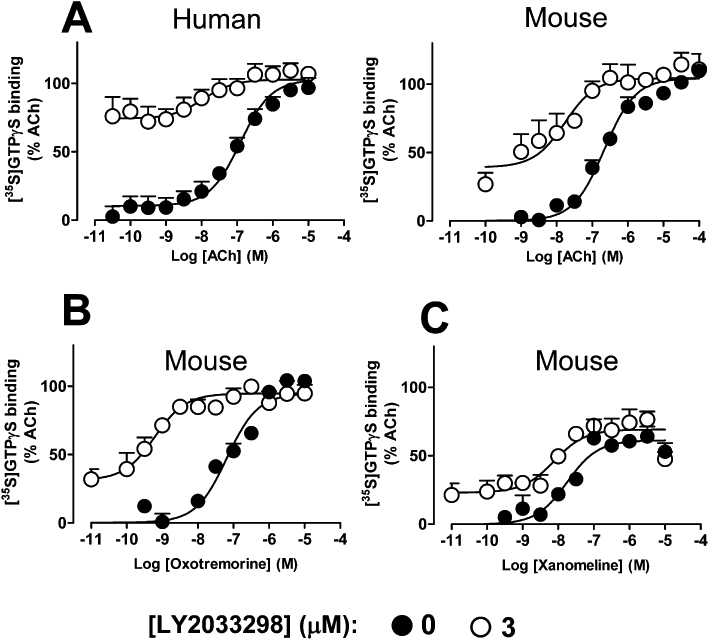
Effects of LY2033298 on the ability of (A) ACh (B) oxotremorine or (C) xanomeline, to promote the binding of [35S]-GTPγS to activated Gα proteins via the human or mouse M4 mAChR stably expressed in CHO cell membranes. Data represent the mean of three experiments performed in triplicate.
Determination of species variation in co-operativity and probe-dependence allows for prediction of the in vivo efficacy of LY2033298 in a mouse model of locomotor activity
The results from our in vitro studies raise a number of implications for the in vivo validation of allosteric modulator effects. For instance, the lower positive co-operativity between ACh and LY2033298 at the mouse relative to the human M4 mAChR suggests that the modulator may fail to show in vivo efficacy in a mouse model of mAChR activity if administered alone. Moreover, the success of exogenous co-administration of another orthosteric agonist to provide additional cholinergic tone will be predicated by the probe-dependence of the interaction between the agonist and the modulator.
The model chosen to assess in vivo activity in our study was the amphetamine-induced hyperlocomotor activity paradigm; this model has been commonly used as a screen to determine the efficacy of various agents in targeting and inhibiting central dopaminergic activity, and also for screening agents for activity as potential antipsychotic drugs (Geyer and Ellenbroek, 2003). Initial experiments established 5 mg·kg−1 as the appropriate dose of amphetamine required to induce a state of hyperlocomotor activity in mice, with this dose significantly increasing locomotor activity by approximately 1.5-fold above control (Figure 6). As prototypical comparators of efficacy in this model, we used the antipsychotic drugs, haloperidol (0.1 mg·kg−1) and clozapine (3 mg·kg−1), which both showed a significant reversal of the amphetamine-induced locomotor activity (Figure 6A).
Figure 6.
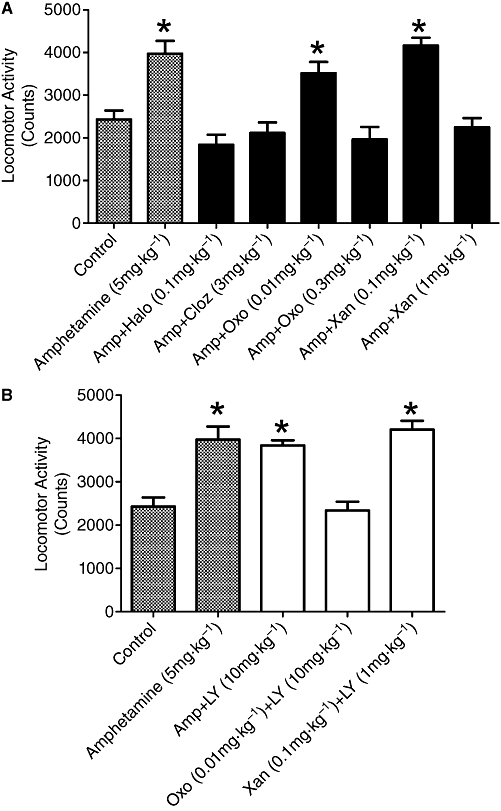
A. The effect of amphetamine alone and in combination with either haloperidol, clozapine, oxotremorine or xanomeline on locomotor activity measured as counts. Asterisks indicate significant differences from control untreated animals, P < 0.05, post hoc Dunnett's test. B. The effects of LY2033298 (10 mg·kg−1) in combination with amphetamine, amphetamine and oxtremorine, or amphetamine and xanomeline. Asterisks indicate significant differences from control untreated animals, P < 0.05, post hoc Dunnett's test. For clarity of comparisons locomotor activity in control untreated animals and amphetamine alone are included in the figure.
Sub-effective and effective doses of oxotremorine and xanomeline for subsequent interaction studies were determined and, as shown in Figure 6A, the sub-effective dose for oxotremorine was 0.01 mg·kg−1, while 0.3 mg·kg−1 oxotremorine significantly reversed amphetamine-induced locomotor activity. For xanomeline, the sub-effective dose was 0.1 mg·kg−1 while 1 mg·kg−1 readily reversed amphetamine-induced locomotor activity. For the interaction experiments the appropriate sub-effective doses of either oxotremorine or xanomeline were combined with LY2033298 (10 mg·kg−1) for their effects on amphetamine-induced hyperlocomotor activity. Figure 6B shows the results of these combination experiments. As predicted, LY2033298 (10 mg·kg−1) alone did not have a significant effect on amphetamine-induced locomotor activity. However, when combined with 0.01 mg·kg−1 oxotremorine, a significant (P < 0.05) decrease in locomotor activity was noted, virtually back to baseline levels. In contrast, but as expected, the combination of LY2033298 and xanomeline (0.1 mg·kg−1) had no effect on the amphetamine-induced locomotor activity.
Discussion and conclusions
There are two major findings from this study. The first is that the allosteric interaction between LY2033298 and ACh at the M4 mAChR is subject to species variability when comparing activity at human versus mouse receptors. The second finding is that the interaction between the modulator and different orthosteric ligands at the mouse M4 mAChR is subject to probe-dependence. Interestingly, the mechanistic basis of both phenomena is due to different co-operativities between the modulator and orthosteric ligands, as LY2033298 has similar affinity for the allosteric site on both the human and mouse M4 mAChRs. These findings have broad implications for the detection and validation of novel allosteric ligands for GPCRs, because they illustrate how allosteric mechanisms can lead to behaviours that can be misinterpreted as a lack of allosteric interaction and/or a lack of efficacy when attempting to translate in vitro findings to the in vivo situation.
The impact of species variability on the prediction of drug activity at human receptors based on pharmacology determined at animal receptors is well known. For instance, a single amino acid difference between human and rat 5HT1B receptors is sufficient to account for markedly different pharmacology between the two species (Hamblin et al., 1992). Similarly, WIN 51 708 is a potent antagonist at the rat NK1 receptor but not at the human receptor due to a few amino acid variations in the first extracellular loop and transmembrane domain 7 (Sachais and Krause, 1994). The advent of high throughput screening in recombinant systems has since led to a reversal in the order of screening preferences, with most initial screens performed on human receptors (Kenakin, 1996). Although this is a logical approach, promising candidates still need to be validated in appropriate in vivo systems, which almost invariably necessitates the use of animal models. For allosteric modulators, the challenge of validation is compounded by the fact that, unlike orthosteric sites, allosteric domains need not have evolved to accommodate endogenous ligands, and can thus show greater variation between receptor subtypes, and between species (Christopoulos and Kenakin, 2002). Although GPCR allosteric modulators have been identified in screens of activity at human receptors and subsequently shown to retain activity in animal models, what remains largely unknown is the number of allosteric modulator discovery programmes that have failed as a consequence of modest or lack of in vivo animal model efficacy. It is also possible that different behavioural assays may have different levels of sensitivity to allosteric modulation resulting in variable efficacy responses. Our study now highlights the fact that the basis of such potential ‘failures’ or unpredicted variable responses may be due to a lack of appreciation of the consequences of probe-dependence (i.e. differential co-operativity) in allosteric modulation.
In the initial discovery and characterization of LY2033298, we noted that the compound had reduced in vitro potency as a modulator at the rat when compared to the human M4 mAChR (Chan et al., 2008). This is in agreement with our current study at the mouse receptor. Importantly, the application of an operational model to the agonistic ERK1/2 response of LY2033298 allowed us to obtain functional estimates of its affinity at both the human and mouse M4 mAChRs in whole cells. A key finding was that these values were virtually identical, and similar to previously determined values at the human receptor (Nawaratne et al., 2008; Leach et al., 2010). This indicates, for the first time, that the species differences in the actions of LY2033298 do not arise from differences in the affinities for the allosteric site on the human relative to the rodent receptor. This was further validated using interaction studies at the level of radioligand binding as well as ERK1/2 phosphorylation (Figure 2), GSK-3α phosphorylation (Figure 4) and [35S]-GTPγS binding (Figure 5). The finding of different co-operativity with ACh, rather than different affinity, between species at the M4 mAChR also sheds new light on mutagenesis data from the initial study on LY2033298, where we identified two non-conserved residues in the third extracellular loop of the receptor that contribute to the potency of the modulator. In that study, substitution of the rat residues into the human receptor led to a loss of LY2033298 function, but substitution of the human residues into the rat receptor did not result in any gain of function (Chan et al., 2008), indicating that the phenomenon was likely to reflect a complex network of interactions (as expected for transmission of co-operativity) rather than a direct contribution of this region as an attachment point for LY2033298. This is also in agreement with a recent mutagenesis study that investigated the interaction of LY2033298 at the human M4 mAChR (Nawaratne et al., 2010).
Differential co-operativity between an allosteric modulator and orthosteric ligands at a given GPCR is also the mechanism underlying probe-dependence (Kenakin, 2005; Leach et al., 2007). The finding that LY2033298 displays such variations in co-operativity with orthosteric ligands has a number of important implications. The first relates to how much co-operativity is required to ensure an in vivo effect. This is clearly a major issue, but one whose answer is likely to vary with the receptor under investigation and the nature of the disease being targeted. For instance, the best-known allosteric drugs on the market remain the benzodiazepines, which act as allosteric potentiators of GABA binding at the GABAA ligand-gated ion channel. Classic studies on some of these compounds indicate that the degree of positive binding co-operativity with GABA is quite modest, for example less than fivefold (Tallman et al., 1978; Ehlert et al., 1982), but this is clearly sufficient to yield the desired therapeutic effect. In contrast, we have estimated a co-operativity factor for LY2033298 of sevenfold with ACh at the level of binding and values ranging from eight- (GSK-3α) to 42-fold ([35S]-GTPγS) at the level of function at the mouse M4 mAChR, but failed to see in vivo efficacy in a hyperlocomotor activity paradigm at the maximum LY2033298 dose of 10 mg·kg−1 administered alone. This suggests that the receptor system requires higher degrees of positive co-operativity between an allosteric modulator and ACh to appropriately engage the relevant neuronal circuitry (possibly due to insufficient basal ACh tone) and/or there is insufficient brain penetrance/receptor occupancy by the allosteric ligand. The finding that the affinity of LY2033298 for the allosteric site on the unoccupied receptor is in the micromolar range certainly contrasts to the benzodiazepines, which bind with nanomolar affinities to the GABAA receptor complex, and suggests that low occupancies by LY2033298 are likely to be achieved in the receptor compartment; this does not, however, rule out the possibility that greater co-operativity is also required to mediate a relevant effect.
The second implication of our finding of probe-dependence in the actions of LY2033298 is that the allosteric effect can indeed be unmasked in the in vivo setting by the judicious choice of an orthosteric agonist that displays sufficiently high co-operativity. As shown in Figure 3, LY2033298 potentiated oxotremorine-mediated responses by factors ranging from 77 to 326, depending on the functional assay, consistently higher than ACh. Accordingly, the combination of a sub-effective dose of oxotremorine with LY2033298 resulted in a significant reduction in amphetamine-induced hyperlocomotor activity (Figure 6B), indicating that LY2033298 was able to mediate a centrally active positive allosteric effect. This also explains why we noted in vivo efficacy of LY2033298 in our previous studies of conditioned avoidance responses in both the rat and the mouse (Chan et al., 2008; Leach et al., 2010), and prepulse inhibition in the rat (Leach et al., 2010) upon co-administration of sub-threshold doses of oxotremorine. Collectively, these results suggest that it is useful to determine the activity of lead allosteric drug candidates against a range of orthosteric probes at relevant receptor species prior to in vivo testing, as this may help in the discrimination of mechanisms underlying potential lack of efficacy and assist in providing proof-of-concept that the allosteric mechanism is still operative under appropriate conditions.
Perhaps a broader issue relating to the probe-dependence identified herein relates to the potential for combination therapies in the treatment of polygenic disorders, such as schizophrenia. We have previously found that the atypical antipsychotic, clozapine, is a negative allosteric modulator of the signalling efficacy of LY2033298 at the level of M4 mAChR-mediated ERK1/2 phosphorylation (Nawaratne et al., 2008). In the current study, we show that xanomeline, a direct M4 mAChR agonist that has clinical efficacy in treating cognitive episodes associated with schizophrenia, has almost neutral co-operativity with LY2033298. On the one hand, these findings may suggest that the combination of LY2033298 and other antipsychotic agents, such as clozapine and xanomeline, will not be a fruitful approach to targeting schizophrenia, but on the other hand it must be noted that these properties reflect only the actions at the level of the M4 mAChR. The clinical efficacy of clozapine is probably due to its interaction with a multitude of targets in the CNS (Roth et al., 2004), and it remains to be determined if its negative effects on LY2033298 signalling efficacy have substantial relevance to native tissue. Indeed, we have recently suggested that the ability of LY2033298 to act as a direct allosteric agonist of M4 mAChR signalling is likely to have only a minor influence on its actions in a native setting; the ability to allosterically potentiate ACh remains the predominant mechanism (Leach et al., 2010). Similarly, a finding of neutral co-operativity between LY2033298 and xanomeline only indicates a lack of synergy at the level of the receptor. It does not suggest any antagonistic interaction, nor does it rule out the possibility that positive interactions may still occur at the level of the intact organism due to other mechanisms. Nonetheless, the detection of probe-dependence is a vital tool in the ongoing process of novel drug discovery focusing on allosteric modulators of GPCRs, because it can assist in the setting of appropriate expectations with regards to the choice of combination regimens for validation and, perhaps, eventual therapeutic exploitation.
In conclusion, this study has identified differential co-operativity, not affinity, as the mechanistic basis for species variability in the actions of LY2033298 at the M4 mAChR that results in reduced efficacy in rodent behavioural assays. Moreover, we have found that different orthosteric agonists can display substantial probe-dependence with the same modulator at the same species of receptor. These findings are likely to be applicable to other GPCR families, and represent an important consideration when validating putative allosteric modulators in animal models and using the findings to infer possible impact on human clinical efficacy.
Acknowledgments
This work was funded by Program Grant 519461 of the National Health and Medical Research Council (NHMRC) of Australia. AC is a Senior, and PMS a Principal, Research Fellow of the NHMRC. We would like to thank Drs Michael Crouch and Ron Osmond (TGR Biosciences) for the generous gift of ERK1/2 and GSK-3α assay kits.
Glossary
Abbreviations
- [3H]-NMS
[3H]-N-methylscopolamine
- [35S]-GTPγS
guanosine 5′-[γ-35S]-triphosphate
- ERK1/2
extracellular signal-regulated kinases ½
- GSK-3α
glycogen synthase kinase 3α
- LY2033298
3-amino-5-chloro-6-methoxy-4-methyl-thieno(2,3-b)pyridine-2-carboxylic acid cyclopropylamide
- PANSS
positive and negative symptom scale
Conflicts of interest
CCF is an employee of Eli Lilly and Co. AC and PMS are members of the Scientific Advisory Board of Addex Pharmaceuticals, and have previously received funding from GlaxoSmithKline and Pfizer for studies of allosteric modulation.
Supplementary material
Supporting Information: Teaching Materials; Fig 1 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters J. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelio L, Valant C, Flynn BL, Sexton PM, Christopoulos A, Scammels PJ. Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-Substituted 2-Amino-3-benzoylthiophenes. J Med Chem. 2009;52:4543–4547. doi: 10.1021/jm9002582. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational Models of Pharmacological Agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, et al. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharmacol Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley P, Stahl SM. Pharmacological treatment of negative symptoms of schizophrenia: therapeutic opportunity or Cul-de-sac? Acta Psychiatr Scand. 2007;115:93–100. doi: 10.1111/j.1600-0447.2007.00992.x. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Felder C, Ahmed S, McKinzie D. muscarinic receptors as a target for drugs treating schizophrenia. Curr Drug Targets CNS Neurol Disord. 2002;1:163–181. doi: 10.2174/1568007024606249. [DOI] [PubMed] [Google Scholar]
- Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, et al. Allosteric modulation of muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci USA. 2008;105:10978–10983. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A. Assessing the distribution of parameters in models of ligand-receptor interaction: to log or not to log. Trends Pharmacol Sci. 1998;19:351–357. doi: 10.1016/s0165-6147(98)01240-1. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. Quantification of allosteric interactions at G protein-coupled receptors using radioligand binding assays. In: Enna SJ, editor. Current Protocols in Pharmacology. New York: Wiley and Sons; 2000. pp. 1.22.1–1.22.40. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Mitchelson F. Application of an allosteric ternary complex model to the technique of pharmacological resultant analysis. J Pharm Pharmacol. 1997;49:781–786. doi: 10.1111/j.2042-7158.1997.tb06112.x. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert F, Ragan P, Chen A, Roeske WR, Yamamura HI. Modulation of benzodiazepine receptor binding: insight into pharmacological efficacy. Eur J Pharmacol. 1982;78:249–253. doi: 10.1016/0014-2999(82)90246-1. [DOI] [PubMed] [Google Scholar]
- Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, et al. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001;27:2605–2613. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- Geyer M, Ellenbroek B. Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1071–1079. doi: 10.1016/j.pnpbp.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Zhang L, Kostenis E, Felder CC, Bymaster FP, Brodkin J, et al. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M4 muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci USA. 1999;96:10483–10488. doi: 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory K, Sexton PM, Christopoulos A. Allosteric modulation of muscarinic acetylcholine receptors. Curr Neuropharmacol. 2007;5:157–167. doi: 10.2174/157015907781695946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin M, Metcalf MA, McGuffina RW, Karpellsa S. Molecular cloning and functional characterization of a human 5-HT1B serotonin receptor: a homologue of the rat 5-HT1B receptor with 5-HT1D-like pharmacological specificity. Biochem Biophys Res Commun. 1992;184:752–759. doi: 10.1016/0006-291x(92)90654-4. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 2006;16:710–715. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. The classification of seven transmembrane receptors in recombinant expression systems. Pharmacol Rev. 1996;48:413–463. [PubMed] [Google Scholar]
- Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discov. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Watson J, Reaville C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol. 1995;48:362–378. [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopolous A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–289. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, et al. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35:855–869. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of Antipsychotic Drugs in Patients with Chronic Schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Killick R, Di Forti M, Murray R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007;30:142–149. doi: 10.1016/j.tins.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Duncan GE, Marx CE, Lieberman JA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry. 2005;10:79–104. doi: 10.1038/sj.mp.4001556. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Christopoulos A. Fitting Models to Biological Data Using Linear and Nonlinear Regression. New York: Oxford University Press; 2004. [Google Scholar]
- Murphy BP, Chung YC, Park TW, McGorry PD. Pharmacological treatment of primary negative symptoms in schizophrenia: a systematic review. Schizophr Res. 2006;88:5–25. doi: 10.1016/j.schres.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Nawaratne V, Leach K, Suratman N, Loiacono RE, Felder CC, Armbruster BN, et al. New Insights into the Function of M4 Muscarinic Acetylcholine Receptors Gained Using a Novel Allosteric Modulator and a DREADD (Designer Receptor Exclusively Activated by a Designer Drug) Mol Pharmacol. 2008;74:1119–1131. doi: 10.1124/mol.108.049353. [DOI] [PubMed] [Google Scholar]
- Nawaratne V, Leach K, Felder CC, Sexton PM, Christopoulos A. Structural determinants of allosteric agonism and modulation at the M4 muscarinic acetylcholine receptor: identification of ligand-specific and global activation mechanisms. J Biol Chem. 2010;285:19012–19021. doi: 10.1074/jbc.M110.125096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz J, Harris HW, Guitart X, Terwilliger RZ, Haycock JW, Nestler EJ. Extracellular signal-regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: regional distribution and regulation by chronic morphine. J Neurosci. 1995;15:1285–1297. doi: 10.1523/JNEUROSCI.15-02-01285.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth B, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Sachais B, Krause JE. Both extracellular and transmembrane residues contribute to the species selectivity of the neurokinin-1 receptor antagonist WIN 51708. Mol Pharmacol. 1994;46:122–128. [PubMed] [Google Scholar]
- Scarr E, Dean B. Muscarinic receptors: do they have a role in the pathology and treatment of schizophrenia? J Neurochem. 2008;107:1188–1195. doi: 10.1111/j.1471-4159.2008.05711.x. [DOI] [PubMed] [Google Scholar]
- Shannon HE, Rasmussen K, Bymaster FP, Hart JC, Peters SC, Swedberg MD, et al. Xanomeline, an M1/M4 preferring muscarinic cholinergic receptor agonist, produces antipsychotic-like activity in rats and mice. Schizophr Res. 2000;42:249–259. doi: 10.1016/s0920-9964(99)00138-3. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube SJ, Mallinkrodt C, et al. Selective Muscarinic Receptor Agonist Xanomeline as a Novel Treatment Approach for Schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Singer-Lahat D, Rojas E, Felder CC. A9 Fibroblasts Transfected with the M3 Muscarinic Receptor Clone Express a Ca2+ Channel Activated by Carbachol, GTP and GDP. J Membr Biol. 1997;159:21–28. doi: 10.1007/s002329900265. [DOI] [PubMed] [Google Scholar]
- Stanhope KJ, Mirza NR, Bickerdike MJ, Bright JL, Harrington NR, Hesselink MB, et al. The muscarinic receptor agonist xanomeline has an antipsychotic-like profile in the rat. J Pharmacol Exp Ther. 2001;299:782–792. [PubMed] [Google Scholar]
- Tallman JF, Thomas JW, Gallager DW. GABAergic modulation of benzodiazepine binding site sensitivity. Nature. 1978;274:383–385. doi: 10.1038/274383a0. [DOI] [PubMed] [Google Scholar]
- Tzavara E, Bymaster FP, Davis RJ, Wade MR, Perry KW, Wess J, et al. M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J. 2004;18:1410–1412. doi: 10.1096/fj.04-1575fje. [DOI] [PubMed] [Google Scholar]
- Villarreal J, Barea-Rodriguez EJ. ERK phosphorylation is required for retention of trace fear memory. Neurobiol Learn Mem. 2006;85:44–57. doi: 10.1016/j.nlm.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Witkin JM. Central and peripheral muscarinic actions of physostigmine and oxotremorine on avoidance responding of squirrel monkeys. Psychopharmacology. 1989;97:376–382. doi: 10.1007/BF00439454. [DOI] [PubMed] [Google Scholar]
- Woolley M, Carter HJ, Garlton JE, Watson JM, Dawson LA. Attenuation of amphetamine-induced activity by the non-selective muscarinic receptor agonist, xanomeline, is absent in muscarinic M4 receptor knockout mice and attenuated in muscarinic M1 receptor knockout mice. Eur J Pharmacol. 2009;603:147–149. doi: 10.1016/j.ejphar.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yamada M, Gomeza J, Basile AS, Wess J. Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice. J Neurosci. 2002;22:6347–6352. doi: 10.1523/JNEUROSCI.22-15-06347.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.