

Summary
Autophagy was viewed until very recently primarily as a metabolic and intracellular biomass and organelle quality and quantity control pathway. It has now been recognized that autophagy represents a bona fide immunological process with a wide array of roles in immunity. The immunological functions of autophagy, as we understand them now, span both innate and adaptive immunity. They range from unique and sometimes highly specialized immunological effectors and regulatory functions (referred to here as type I immunophagy) to generic homeostatic influence on immune cells (type II immunophagy), akin to the effects on survival and homeostasis of other cell types in the body. As a concept-building tool for understanding why and how autophagy is intertwined with immunity, it is useful to consider that the presently complex picture has emerged in increments, starting in part from the realization that autophagy acts as an evolutionarily ancient microbial clearance mechanism defending eukaryotic cells against intracellular pathogens. In this review, we build a step-wise model of how the core axis of autophagy as a cell-autonomous immune defense against microbes evolved into a complex but orderly web of intersections with innate and adaptive immunity processes. The connections between autophagy and conventional immunity systems include Toll-like receptors (TLRs), Nod-like receptors (NLRs), RIG-I-like receptors (RLRs), damage-associated molecular patterns (DAMPs) such as HMGB1, other known innate and adaptive immunity receptors and cytokines, sequestasome (p62)-like receptors (SLR) that act as autophagy adapters, immunity related GTPase IRGM, innate and adaptive functions of macrophages and dendritic cells, and differential effects on development and homeostasis of T and B-lymphocyte subsets. The disease contexts covered here include tuberculosis, infections with human immunodeficiency virus and other viruses, Salmonella, Listeria, Shigella, Toxoplasma, and inflammatory disorders such as Crohn's disease and multiple sclerosis.
Keywords: autophagy, dendritic cells, T cells, AIDS, bacterial, inflammatory bowel disease
Introduction
The many presently recognized roles of autophagy in innate and adaptive immunity have been steadily increasing in complexity (1-4) (Fig. 1). The field has undergone rapid growth following the recognition of autophagy as (i) a specialized cytoplasmic system for direct elimination of intracellular bacteria (1, 5, 6), (ii) a contributor to major histocompatibility complex class II (MHC II) presentation of endogenously expressed antigens (7), and (ii) a host antimicrobial system targeted by specific bacterial adaptations to protect the invading microbe against autophagic elimination (8-11). The initial burst of mechanistic studies (5-8) was quickly followed by numerous studies further enriching the breath and depth of autophagy's role in immunity (12-18). The present explosive growth of the field occurred many years after the early observational report of autophagosome-like structures detected in polymorphonuclear leukocytes infected with rickettsiae (19) and the pioneering mechanistic work on the role of Beclin 1 [a key autophagy factor (20)] in antiviral defense (21) recently enriched by molecular links (22). The antiviral effects of autophagy have been extended to human immunodeficiency virus (HIV) (16) and roles for autophagy described in both cell-autonomous (11) and innate and adaptive immunity (23, 24) against HIV. This latest addition to the antimicrobial function of autophagy has been further strengthened by the findings that HIV proteins Nef and Env counter autophagy: Env inhibits autophagy induction in dendritic cells (DCs), thus inhibiting DC maturation and processing and presentation HIV antigen (Gag), whereas Nef blocks specifically the autophagic maturation step in infected macrophages, thus protecting HIV visions form degradation (11). In this review, we give a brief overview of autophagy as a fundamental biological process and provide an in-depth and step-wise update on connections between innate immunity and autophagy, including the specific aspects of microbial control in macrophages subjected to autophagic macrophage activation (APMA) as recently defined (3). Adaptive immunity aspects of autophagy have been recently comprehensively reviewed (2, 3, 14), and will be covered here when appropriate in the context of relationships affecting inflammatory and infectious diseases.
Fig. 1. Autophagy plays multiple roles in innate and adaptive immunity.
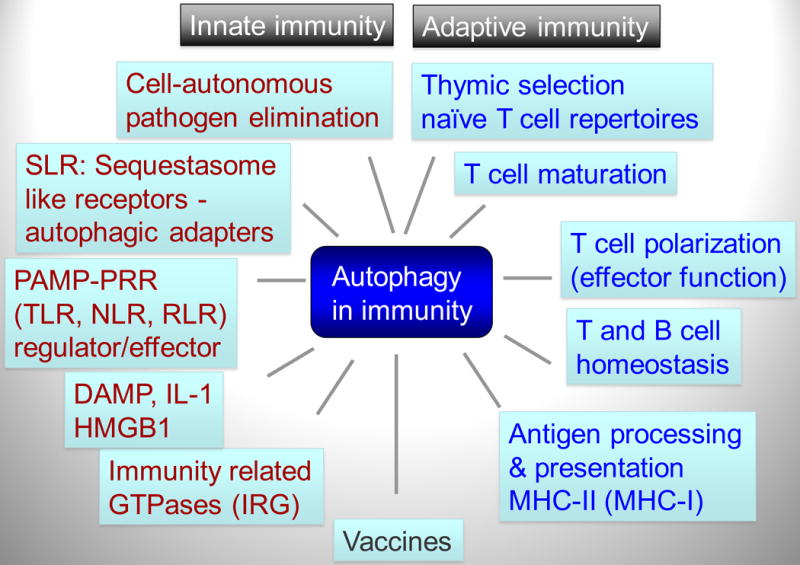
Autophagy functions within the innate immunity (red letters), adaptive immunity (blue letters), and their intersection (black font; e.g. vaccines). SLR, p62/Sequestasome like receptors that also function as autophagy adapters. PAMP, pathogen-associated molecular patterns. PRR, pattern recognition receptors. TLR, Toll-like receptors. NLR, Nod-like receptors. RLR, RIG-I-like receptors. DAMP, danger-associated molecular patterns. IRG, immunity-related GTPases.
Autophagy as a fundamental cell biology process
Autophagy, in the strict sense of the term, is a unique intracellular biomass quality and quantity pathway unrestricted by its target size or complexity. It is often referred to as macroautophagy when considered along with microautophagy and chaperone-mediated autophagy, which are among additional processes delivering cytosolic proteins and components to lysosomes for degradation. Autophagy is capable of removing by sequestration and degradation (25), or sometimes by secretion (26, 27) or extrusion (28, 29), a wide range of intracellular components ranging from protein aggregates to bulk cytoplasm and whole organelles such as mitochondria. Another distinguishing feature of the ‘strict sense’ autophagy is that its cytoplasmic targets are captured within organelles of endomembranous origin, termed autophagosomes, which subsequently mature into autolysosomes where the captured cargo is degraded or otherwise eliminated (Fig. 2). The physiological functions of autophagy (25) include providing a cell-autonomous source (by autodigestion of the cytosol) of energy and amino acids at times of cellular metabolic crisis or nutritional deprivation, prevention of cell death or senescence due to accumulation of faulty organelles and large macromolecular aggregates and the still debated potential cell death modality (30, 31). These classical roles of autophagy have recently been amended to include a wide range of innate and adaptive immunity functions (3). All cells rely on constitutive autophagy to carry out the basal housekeeping role of eliminating sporadically damaged organelles due to normal wear and tear, for example occasional depolarized mitochondria that cannot rejoin the mitochondrial network (32). The baseline housekeeping autophagy can be augmented by elicited autophagic responses to nutritional, differentiation, and danger signals (33). Autophagy in principle involves three morphological stages (Fig. 2): (i) initiation (formation of crescent membranes termed phagophores), (ii) elongation and closure (increase of the phagophore and its closure into a completed autophagosome containing the sequestered cargo), (iii) and maturation (conversion of autophagosomes into degradative organelles termed autolysosomes by fusion with late endosomal and lysosomal organelles or trafficking carriers). The keys to initiation of autophagy are the regulation of (i) Atg1 (in yeast) or its equivalent Ulk1 (in mammals) complexes (34) and (ii) the phosphatidylinositol 3-kinase (PI3K) hVPS34-Atg6 (Berlin 1) complex with Atg14 (complex I) and additional interacting components (35). To control autophagy in response to growth factors and nutritional signals (Fig. 2), the Atg1/Ulk1 complex is coupled to Tor complex 1 (TORC1). In mammals, mTORC1 during growth factor and nutrient-replete conditions associates with the Ulk1-Atg13-FIP200-Atg101 complex (with mammalian FIP200 being a functional equivalent of yeast Atg17), thus inhibiting autophagy. Upon receiving starvation signals, mTORC1 dissociates from the Ulk1-Atg13-FIP200-Atg101 complex, which appears to translocate (34) to (still elusive in mammalian cells) pre-autophagosomal membranes that may possibly involve rough endoplasmic reticulum (rear) (36) areas that can be visualized by a marker, DFCP-1 (37). There, the Ulk1 complex, in cooperation with the PI3K hVPS34 complex I and its lipid product phosphatidylinositol 3-phosphate (PI3P) along with the PI3P-binding effectors proteins WIPI-1 and WIPI-2 (equivalents of yeast Atg18) lead to the formation of nascent autophagosomes (34). More recent work has implicated additional intracellular compartments/organelles as contributors of membrane to nascent autophagosomes, in addition to the ER. These additional membrane sources are mitochondria (38), Golgi apparatus (39-41), and plasma membrane (42).
Fig. 2. Autophagy pathway in mammalian cells.
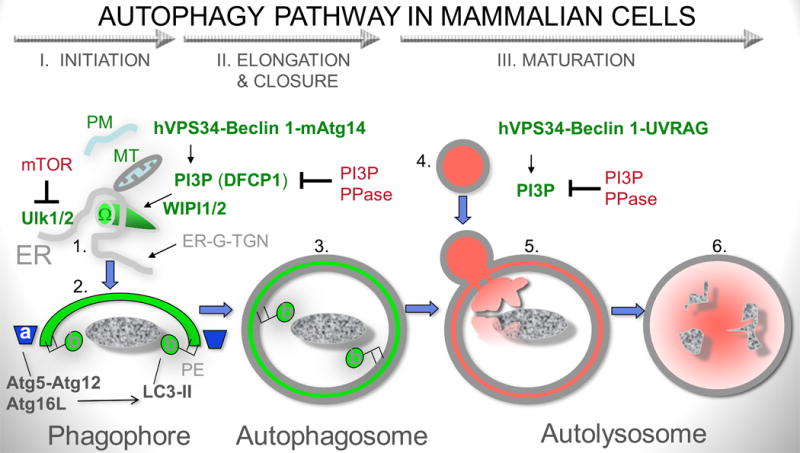
I. Phase I (initiation): mTOR (mammalian target of rapamycin)-dependent and mTOR-independent pathways can induce autophagy. mTOR works via Ulk1 and 2 (mammalian Atg1 orthologs) that in turn directs (most likely on the ER) appropriate protein and lipid transactions to allow formation of a nascent pre-phagophore (1) possibly corresponding to the well defined structure in yeast termed pre-autophagosomal structure (or phagophore assembly site; PAS). The pre-phagophore in mammalian cells may emerge from an ER structure termed omegasome (Ω). Contributions of the secretory pathway [ER-Golgi-trans Golgi network (ER-G-TGN)], plasma membrane (PM)-derived organelles and mitochondria (MT) have also been reported: PM, acts possibly as additional source of lipid bilayers or a source of regulatory effects within the endosomal system with which autophagosomes merge during maturation; MT may contribute as a major source of phosphatidylethanolamine (PE) where phosphatydilserine (PS) decarboxylase is located on the MT inner membrane to generate PE from PS for conjugation of LC3 (LC3-II) and as a source of regulatory reactive oxygen intermediates that may promote autophagosome formation; ER-G-TGN contributions are probably complex and affecting the process directly and indirectly at multiple stages. Another major system regulating autophagy is the phosphatidylinositol 3-kinase hVPS34, which generates phosphatidylinositol 3-phopshate (PI3P). DFCP is a protein with two FYVE (PI3P-binding) domains and serves as a marker for the merger of the hVPS34/PI3P systems and TOR/Ulk at the point of autophagy initiation at the ER. hVPS34 is in a complex (often referred to as complex I) with multiple proteins, with Beclin 1 being key for autophagy altogether, and mAtg14 being key specifically for autophagy initiation. WIPI1 and 2 (mammalian Atg18) bind to PI3P and likely facilitate anteretrograde (and retrograde for recycling) membrane and protein flow to the nascent phagosomes. PI3P phosphatases, such as Jumpy/MTMR14, modulate progression of autophagy by tempering PI3P-dependent processes. II. Phase II (elongation and closure): Autophagic membrane is elongated to from a structure known as sensu stricto phagophore (or isolation membrane) based on two conjugation systems: (a) Atg5-Atg12 in complex with Atg16L, which acts as an E3 enzyme (nomenclature borrowed form the ubiquitination system) topologically restricting and enzymatically facilitating (b) conversion of LC3 to LC3-II (lipidated at the C-terminus by PE). Atg16 is a Crohn's disease risk locus in human populations and has also been found in complexes with clathrin at the plasma membrane (not shown). Elongating phagophore, also known as isolation membrane (2) wraps around its targets and eventually closes to form a sealed double membrane organelle known as sensu stricto autophagosome (3). III. Phase III (maturation). Autophagosomes fuse with late endosomal and lysosomal organelles or intermediates in a process dependent on hVPS34 (in complex with Beclin 1 and UVRAG; referred to as complex II). This process is also subject to inhibition/modulation by PI3P phosphatases such as Jumpy/MTMR14. The fusion with lysosomal organelles heralds the dissolution of the inner membrane and formation of autolysosomes, where the degradation of the captured material occurs.
Autophagy and autophagy genes have been implicated in a broad spectrum of human health issues including Alzheimer's disease (43), Huntington's disease (44), Parkinson's disease diabetes (45-47), aging (48, 49), muscle atrophy and myopathies (50, 51), with additional roles in neural stem cells in adult brain (52, 53), liver (54, 55), antioxidant response (56), lipid metabolism (55), and cancer (57, 58). Autophagy has been associated through GWAS analyses with inflammatory illnesses such as Crohn's disease (59, 60) and through population genetic analyses with susceptibility to tuberculosis (61), confirming in an unbiased way our initially published work uncovering the role of autophagy in innate immunity defenses against Mycobacterium tuberculosis (5).
Autophagy in immunity: an overview
Autophagy participates in nearly all aspects of immunity, affecting both innate and adaptive immunity processes, with a term immunophagy referring to all such processes collectively (62). It is important to distinguish three principal types of contributions that autophagy imparts on the function of the immune system (Fig. 3): (i) Type I: Specialized autophagy immune processes that are performed by autophagic machinery at the cellular level. These include recognition, capture and elimination of intracellular pathogens (22, 63-66) via a process termed xenophagy (67) or delivery of antimicrobial products to intracellular niches where pathogens are ensconced, thus leading to their killing (68). These processes contribute to a recently defined specialized form of autophagic macrophage activation referred to as APMA (3), enhancement of the recognition of microbial products by innate immunity receptors such as intralumenal TLRs (69), and enhanced MHC II-restricted presentation of cytoplasmic self- or foreign antigens (2) or phagocytosed antigens (70). Autophagy is also a specialized effector mechanism downstream of innate immunity receptor stimulation (71) such as TLRs (18, 23, 72, 73) NLRs (74-76) and RLRs or intracellular stimulation by damage associated molecular patterns (DAMP; also known as alarmins) such as HMGB1 when it is displaced form the nucleus into the cytoplasm (77), or ATP (78, 79) and DNA complexes (80) released from damaged cells. Not only do autophagy or autophagy factors help induce, deliver and execute innate immunity responses, but they can also limit innate immunity responses and inflammation (15, 81, 82). Overall, autophagy in its specialized immune functions contributes to immune activation, acts as an effector and a regulator of innate and adaptive immunity responses, and eventually shuts down inflammation. This indicates that autophagy tracks along with a typically tightly regulated homeostasis of immune responses, where immune activation and elimination of offending insult is followed by downregulation and return to the basal level. (ii) Type II. Generic autophagy's role in cellular homeostasis. In this capacity, autophagy influences immune cells in the same way it affects any other cell type, e.g. a neuron or a muscle cell. Here autophagy is at the interface between cell survival and cell death (30), normally engaged in maintaining cellular viability and keeping intracellular organelles, such as mitochondria, in good repair and the cytosol free of toxic macromolecular aggregates. This determinant of the viability, fitness, and functionality affects immune cell types differentially. Some immune cells are more dependent on autophagy, such as T cells (83, 84). T cells may require autophagy for survival when growth factors are withdrawn, and as T cells mature, autophagy is the process executing the programmed reduction in the number of mitochondria that occurs upon naive T-cell exit from the thymus (85, 86). Other cell types are less sensitive, such as the conventional B cells (B2 cells) once they are past their developmental stages (87). (iii) Type III: Non-autophagic role of Atg factors. This is based on a separate group of activities that reflect non-autophagic participation of one or more of the Atg genes, when they exert their function other than through autophagy (13). These effects should not be confused with the role of canonical autophagy and the ‘day job’ of Atg factors and autophagy and immunity. Instead, they fall into the area of peripheral interactions and coordination between different functional systems.
Fig. 3. Three types of autophagy intersections with immune processes.
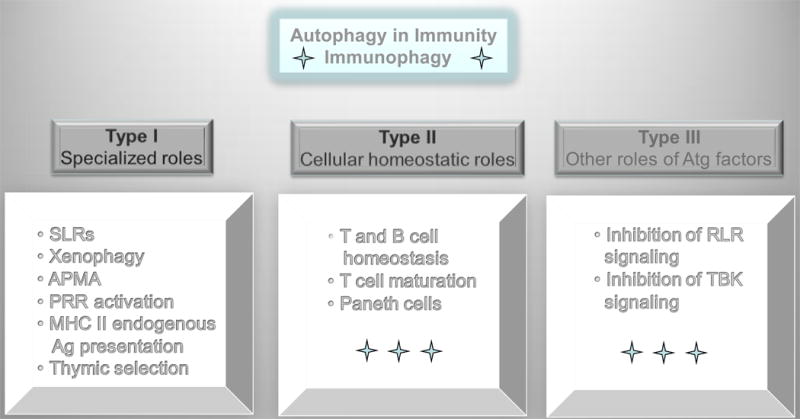
Type I immunophagy, encompasses specialized roles of autophagy in capturing, processing, or delivering microbes or microbial or endogenous immunologically active molecules. SLRs (p62/sequestasome-like receptors) that serve as bridges between microbial targets and autophagosomes. Xenophagy, process of direct capture and destruction of microbes in autolysosomes. APMA, autophagic activation of macrophages (a term describing the cumulative state of immunological activities and processes in macrophages induced for autophagy). PRR, pattern recognition receptors. Type II immunophagy refers to the role of autophagy in controlling cellular viability and general functionality of immune cells in ways that are not different than effects in all other cell types (e.g. neurons). Type III processes are those that are affected by isolated Atg factors but are not dependent on the execution of the entire autophagic pathway, as described in Fig. 2. For example, Atg5-Atg12 has been implicated in inhibition of RIG-I-like receptor (RLR) signaling, whereas Atg9 has been implicated in negatively regulating TBK1 as it contributes to type I interferon secretion.
New group of innate immunity receptors (SLRs) serve as autophagic adapters and recognize intracellular microbes for autophagic elimination
Direct autophagic capture of intracellular microbes occurs with a help of a new class of innate immunity receptors in the cytoplasm, termed here sequestasome (p62/SQSTM1)-like receptors (SLRs) (Fig. 4). SLRs are autophagic receptors/adapters that recognize targets earmarked for autophagy by ubiquitination tags and at the same time contain an LC3 interacting region (LIR). There are one or more LIRs in SLRs, and their interaction with one of the mammalian Atg8 paralogs (e.g. LC3B) brings the cargo into a nascent autophagosome (22, 63-66). Two out of three characterized SLRs, p62 (88), NBR1 (63), and NDP52 (63), play a role in autophagy of intracellular Salmonella (63, 66), Shigella (65), Streptococci (63), Listeria (64), and Sindbis virus (22). Since there are many proteins with putative LIR domains in the mouse and human genomes, there is much promise and work left for future developments in this area. Additional receptors, besides p62 and NDP52, are likely to play a role. Furthermore, the molecular entities on or associated with microbial targets that act as ubiquitinated tags for autophagic uptake remain to be characterized. At least two specific targets [VDAC1 and mitofusin (Mfn)] have been identified on stressed mitochondria (organelles originating from a Riskettsia-like symbiont) that are taken up into autophagosomes with p62's assistance. Intersections with regulatory systems such as BAG3, a member of the Bcl-2-associated athanogene (BAG) family that can redirect the poly-ubiquitinated cargo to autophagy (at present demonstrated only for aging-associated accumulation of large non-dissociable ubiquitinated cargo destined for degradation), may be of interest to explore given that bacteria and visions or their parts may have to be handled by redirecting proteolytic systems. Moreover, a possibility for tags other than ubiquitin on microbial targets for autophagy has been heralded by the identification of a lipid signal, diacylglycerol, associated with the recognition of a subset of Salmonella phagosomes earmarked for autophagy (89).
Fig. 4. SLRs (p62/sequestasome-like receptors) capture microbes or associated host structures for delivery to nascent autophagic organelles.
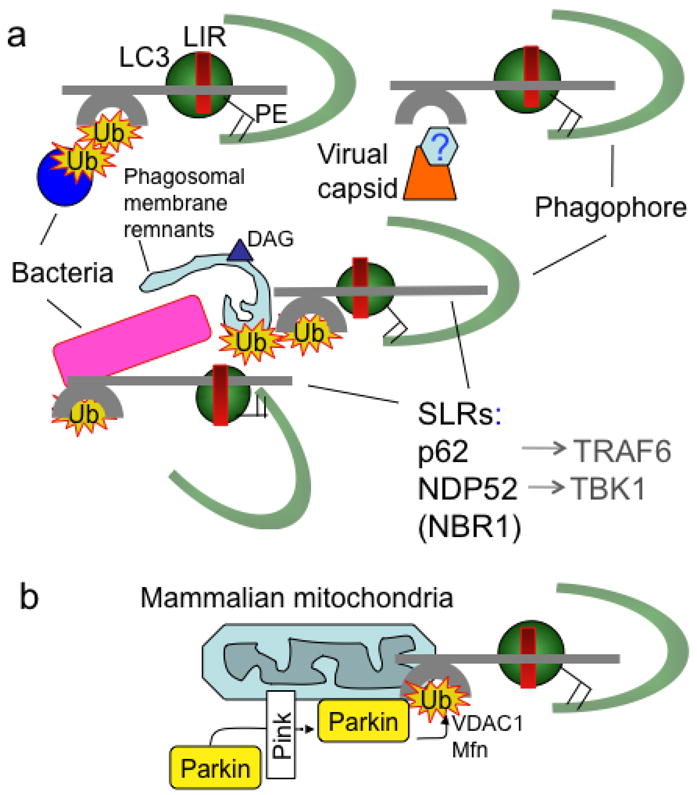
(A). Capture of microbes (bacteria, viral components/capsid) or parasitophorous vacuole membrane remnants by SLRs for autophagic degradation. (B). Capture of terminally depolarized/stressed/damaged mitochondria by SLRs for autophagic removal (“mitophagy”). SLRs include p62 sequestasome, NDP52, NBR1. All of these molecules serve as autophagic adapters by the virtue of interacting with LC3 (one of mammalian Atg8s) or other mammalian Atg8 paralogs, often through a motif termed LIR (LC3 interaction region, with a variation of the ‘WXXL’ motif: D/EW/FE/DXLI/V). SLRs also interact with proinflammatory signaling factors such as TRAF6 and TBK1. Ub, ubiquitin tags recognized by ubiquitin binding regions (e.g. UBA in p62 and NBR1 or zinc finger in NDP52). DAG (triangle), diacylglicerol, a lipid tag found on microbe-harboring phagosomal membranes as a signal for autophagic degradation that may act independently of ubiquitin tags (Ub). Hexagon labeled ‘?’, tags associated with viral capsids have not been identified. Green semicircle, phagophore. PE, phosphatidylethanolamine. Parking, ubiquitin E3 ligase recruited to stressed mitochondria by Pink, with ubiquitination of VDAC1 (a component of mitochondrial permeability transition pore and the most abundant mitochondrial outer membrane protein) and Mfn (mitofusin, a GTPase that controls mitochondrial dynamics and fusion). Note the striking similarity between removal of intracellular microbes (a) and mitochondria (b) by SLRs, possibly reflecting ancient evolutionary relationships since mitochondria evolved from α-proteobacterial (Rickettsia-like microorganisms) symbionts.
Generation and delivery of neoantimicrobial products by autophagy
Another function of SLRs (Fig. 5), at present demonstrated only for p62, is to gather cytoplasmic precursors for conversion in autolysosomes into anti-microbial products that upon delivery to cytoplasmic compartments harboring microbes transform them into autophagolysosomes, organelles with enhanced antimicrobial capacities relative to conventional phagolysosomes (68). As described for SLRs with M. tuberculosis, this pathogen resides inside a phagosome in infected macrophages. Instead of the microbe being captured by p62 and delivered to autophagosomes, p62 plays a role in antimicrobial action of autophagy in an indirect but rather intriguing process (68). The p62 harvests seemingly innocuous cytoplasmic proteins, such as ubiquitin or ribosomal precursor proteins, and delivers them to autophagosomes, where they are digested into peptides with new biological activities. Next, the autolysosomal peptide cargo is delivered to mycobacterial phagosomes where ribosomal and ubiquitin proteolytic fragments generated in autophagic organelles finally meet their targets and express antimicrobial properties that help kill intracellular M. tuberculosis (68).
Fig. 5. Autophagy generates neoantimicrobial peptides.
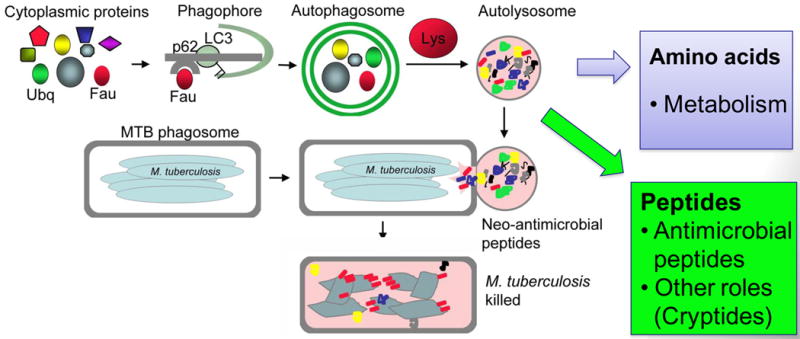
Autophagy has access to a variety of cytoplasmic proteins, such as FAU (a precursor for a ribosomal protein rpS30) and ubiquitin. Once sequestered into autophagosomes by an SLR, these proteins are subjected to proteolysis in autolysosomes. It is usually considered that proteins are degraded to amino acids and that amino acids are exported to the cytosol for nutritional purposes. However, autophagic organelles contain many proteolytic peptide intermediates, which when delivered by fusion with organelles (e.g. phagosomes) containing microbes (e.g. M. tuberculosis) act as antibacterial peptides. These have been termed neoantimicrobial peptides. However, the peptide mixture generated in autophagic organelles may harbor additional biological and signaling functions associated with potentially released or excreted peptides (termed cryptides).
Toll-like receptors and autophagy
Positive and negative interactions between autophagy machinery and innate immunity receptors including relationships upstream and downstream of pattern recognition receptors (PRRs), with Toll-like receptors (TLRs) being the first PRR subclass connected with autophagy in this capacity (90), albeit not without some initial controversy since not all investigators could detect the connection (82). Interactions between TLRs and autophagy include TLR2/TLR1 heterodimer (91), TLR3 (23), TLR4 (18, 23, 72, 73, 92), TLR7/8 (23), and indirectly TLR9 (80) in various cell types including macrophages, dendritic cells, and neutrophils (12, 18, 23, 69, 70, 72, 93-95) (Fig. 6). Signaling pathways connecting innate immunity receptors such as TLRs and autophagy have been recently uncovered (73, 92) (Fig. 6). A K63-linked ubiquitination of the Beclin 1 Lys117 residue, located within the BH3 domain of Beclin 1, dissociates the inhibitory Bcl-2 from the Beclin 1 complex and triggers autophagy (73). Autophagy activation by Beclin 1 ubiquitination downstream of TLR stimulation is executed by the E3 ligase TRAF6 and opposed by a deubiquitinating (Dub) enzyme A20 (73). A similar process may be linked to p62 recognition of microbial targets (65), since TRAF6 binds to p62. NF-κB activation downstream of TRAF6 induces expression of A20, a Dub, and thus may counter TLR-dependent induction of autophagy. Thus, the older reports of NF-κB being inhibitory to autophagy (96) now have a mechanistic basis (73). A recent study further implicates the signaling pathway leading to NF-κB activation but demonstrates that only IKK (a kinase upstream of NF-κB) is directly involved in induction of autophagy (97) and shows no strict NF-κB correlation with autophagy control. However, kinetic issues such as E3 followed by NF-κB followed by A20 action and the time needed for NF-κB to deploy its negative effect on autophagy can potentially explain variable observations and perhaps in part address the negative observations regarding TLR-dependent control of autophagy (82).
Fig. 6. Autophagy effectors and regulatory functions in the context of pattern recognition receptors (PRRs), pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs).
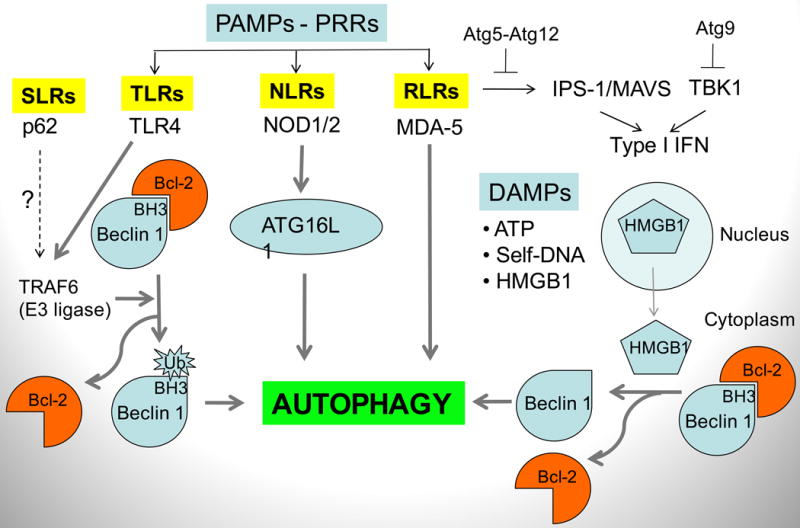
Autophagy intersects with innate immunity at the levels of PAMPs and PRRs in all innate immunity receptor categories: TLRs (Toll-like receptors), NLRs (Nod-like receptors), RLRs (RIG-I-like receptors). It also interacts with molecules signaling tissue damage (DAMP), such as ATP, HMGB1, chromatin/DNA complexes, released from damaged cells. Mechanisms: TLR4 recruits TRAF6 that acts as an ubiquitin E3 ligase, ubiquitinating a Lys residue within the BH3 domain of Beclin 1, thus dissociating Bcl-2, an inhibitor of Beclin 1 function in promoting autophagy. Nod2 is in complexes with Atg16L1 and affects autophagy as well as Atg16L1 recruitment to the point of microbial entry into the cell (not shown). MDA-5 induces autophagy, shown in the context of cell death in malignancies. HMGB1 (high-mobility group protein B1; a non-histone component of eukaryotic chromatin) is an archetypical DAMP/alarmin released form the nucleus into the cytosol and in dying cells (mostly during necrosis) is released extracellularly. In the extracellular milieu, it signals through different receptors in tissue repair (when not complexed with microbial products such as lipopolysaccharide (LPS) or alarmin cytokines such as IL-1β) or in anti-microbial inflammation (when complexed with LPS or IL-1β). In the cytosol, HMGB1activates autophagy by binding to Beclin 1 and dissociating its inhibitory partner Bcl-2. Atg gene products (Atg5, Atg9, and Atg12) have been implicated in negative regulation of proinflammatory responses leading to type I interferon (IFN) production. It may be a reflection of type III non-autophagic functions of Atg genes, but it can be also viewed as a negative feedback mechanism to inhibit pro-inflammatory reactions induced in part through help of autophagy and Atg factors.
NLRs and autophagy
Other innate immunity receptors work in concert with autophagy. Nod-like receptors (NLRs) signal to induce autophagy (Fig. 6), as first shown in the case of Drosophila's NLR-type pattern-recognition receptor PGRP-LE, where protective antibacterial autophagic response was induced in response to diaminopimelic acid-type peptidoglycan (74). In mammalian cells, Nod1 and Nod2 signal to induce autophagy and functionally interact with Atg16L1 (76). NOD2 recognition of bacterial muramyldipeptide induces autophagy in DCs, and this in turn supports adequate MHC II presentation (75). NOD2 and ATG16L1 have also been linked in the context of Crohn's disease (75, 76). When individuals carry a disease risk locus for either NOD2 of Atg16L1 this disrupts proper induction of autophagy, deviates bacterial handling by phagocytic cells, and disrupts antigen presentation by DCs and generation of MHC II-restricted CD4+ T responses (75, 76). It also appears that recruitment of Atg16L1 to the point of microbial entry at the plasma membrane is influenced by Nod1 and Nod2 (76). It is of interest that this latter finding tracks well with the recent studies showing that Atg16L1 at the plasma membrane (where it physically interacts with clathrin) is important for efficient autophagy induction, possibly by modulating membrane flux from the plasma membrane to phagophores (42). Of further interest is that a recent study explains the low phenotypic penetrance of the ATG16L1T300A allele as a risk locus for Crohn's disease using as a proxy an Atg16L1 hypomorph mouse model (98). This study has indicated a requirement for a triple hit – genetic predisposition in ATG16L1, viral infection, and presence of endogenous microbial flora in the gut, to generate ‘the perfect storm’ – resulting in inflammatory bowel disease (98).
RLRs and autophagy
RIG-I-like receptors (RLR) are negatively regulated by Atg5-Atg12 (81) and can activate autophagy (99). This suggests the existence of a negative regulatory feedback limiting the extent of type I IFN production and signaling (Fig. 6). Other autophagic components, such as Atg9, have been reported to negatively regulate trafficking, assembly and activation of TBK1 controlling type I interferon response elicited by intracellular double stranded DNA for which a specific PRR remains to be defined (15) (Fig. 6).
DAMP signaling and autophagy
Autophagy is a recognized effector downstream of systems sensing danger signals/alarmins, also known as damage-associated molecular patterns (DAMPs), such as ATP (78, 79) and self-DNA-containing complexes (80) (Fig. 6). DAMPs such as ATP can induce potassium fluxes and activation of inflammasome, with IL-1β as a key pro-inflammatory product that can induce autophagy (73, 100). The relationship of autophagy and IL-1β potentially extends to Crohn's disease, since in the absence of Atg16L1 in a mouse model of inflammatory colitis, abnormally increased IL-1β was one of the major findings (82). High mobility group box 1 (HMGB1) protein is another DAMP (101, 102) that, when found in the cytoplasm before extracellular release as a DAMP, has been implicated in the regulation of autophagy (77) (Fig. 6). During starvation, oxidation [possibly based on the known ROS released from mitochondria in starved cells (103)] of one of the three key cysteine residues, C106, promotes HMGB1 translocation to the cytoplasm from its normal nuclear localization where it plays other roles (101, 102). Once the C106 oxidized HMGB1 is in the cytosol, it needs its two other key cysteine residues (C23-C45) to be in a disulfide bridge to bind Beclin 1 and displace Bcl-2 thus promoting autophagy (Fig. 6). The mechanism and the net result of this HMGB1 action are akin to the effects of how BH3 mimetic peptides dissociate Bcl-2 from Beclin 1 or how JNK, DAPK-1, and TRAF6 modify either Bcl-2 or Beclin 1 to break up their interactions and induce autophagy (Fig. 6). Although unexpected at first blush, this action, assuming that HMGB1-induced autophagy plays a role primarily in cell survival, suggests a cell-protective role of HMGB1, which may help explain some of its contribution to aberrant cell survival in many cancer types (104). HMGB1, as an extracellular DAMP released from necrotic cells, can be proinflammatory via variety of receptors including TLR4 (101, 102), but when released from late-apoptotic cells (curiously, provided that it is properly oxidized at the same C106 that promotes its cytoplasmic translocation) is not proinflammatory but rather tolerogenic (105). Although pristine HMGB1 per se is not proinflammatory and is primarily a DAMP leading to regeneration of tissues in distress being a potent inducer of cell migration, it can complex with PAMPs (e.g. lipopolysaccharide) and IL1-β when it acts as a proinflammatory agent via binding partner's cognate receptors (101). The dichotomy of the extracellular HMGB1 function – when alone acting in tissue repair while when complexed with microbial products or IL-1β acting as a mediator of inflammation – fits with the roles of autophagy whereby cell survival is one of the principal outcomes, whereas immunophagy and inflammation is promoted when there are signs of microbial invasion.
Other immune receptors and autophagy
Autophagy is an effector downstream of cell surface receptors such as CD46-Cyt-1, which engages an adapter protein (GOPC) interacting with the key autophagy regulator, Beclin 1. This is reminiscent of the engagement of Beclin 1 complexes by MyD88 and TRIF downstream of TLRs (92). CD40 can also activate autophagy (106, 107). Since CD40 engages TRAF6, this pathway may include Beclin 1 modifications described for the TLR4 cascade (Fig. 6). Autophagy also factors in as a potentially key factor in activation and expansion of antigen-specific B cells. First, B-cell receptor (BCR) ligation induces autophagosome formation in B cells (80, 108), enhancing antigen processing and its presentation by MHC II to the helper T cell, which in turn provides CD40L for proper costimulatory signaling, B-cell expansion, and immunoglobulin class switching (108). Interestingly, BCR signaling leads to a strong recruitment of endosomal TLR9 (80) to organelles with internalized BCR, where it can interact with its ligands (normally pathogen DNA) taken up along with the BCR bound antigen; this system, evolved to protect against invading pathogens, when hyper-responsive may contribute to production of autoantibodies and pathology in systemic autoimmune disease.
Immunity related GTPases and autophagy
Autophagy is an anti-microbial effector of immunity related GTPases (IRG) (5, 109, 110) (Fig. 7). This includes the sole human sensu stricto IRG, IRGM (3, 109, 111, 112) and two out of the many murine IRGs, Irgm1 (LRG-47) (5, 109, 113), and Irga6 (IIGP1) (110). The mice and humans are at the opposite extremes of the spectrum in terms of the abundance of IRGs in their genomes. In the mouse, the copious nature of IRGs [24 genes including 20 complete sensu stricto IRGs and two pseudogenes (Irgm1-3; Irga1-8, Irgb1-10, and Irgd) in addition to two IRGs considered not to be immunity related (Irgc and Irgq)] has been uncovered only recently by genomic analyses (114). The same study uncovered a dearth of IRGs in the human genome, limited just to one IRGM (in addition to IRGC and IRGQ which are constant in all species but presumed not to be immunity-related) (114). Prior to these more recent studies, several murine IRGs (TGTP/Irgb6, IRG-47/Irgd, IGTP/Irgm3, GTPI/Irgm2, and IIGP1/Irga6) in addition to Irgm1 have been identified as contributors in defense against subsets of intracellular pathogens. Interestingly Irgm1, the ortholog-apparent of the sole human IRG, IRGM (115), affords the broadest protection against a variety of intracellular microbes tested in the mouse, but the mechanism of its action remained obscure for the most part (116, 117). The study that initially connected autophagy with IRGs did so in the mouse macrophage showing that Irgm1 was an IFN-γ-dependent effector and a regulator of autophagy in its function of eliminating M. tuberculosis in macrophages (5). This was followed by the demonstration that the human IRGM is required for IFN-γ-, rapamycin-, and starvation-induced autophagy in general terms, and in the context of controlling the intracellular M. tuberculosis in infected human macrophages (109). Shortly after that, unbiased human population genetic studies have established a role for autophagy in genetic predisposition to inflammatory bowel disease (specifically Crohn's disease) associated with single nucleotide polymorphisms in the ATG16L1 and IRGM genes (59). Finally, polymorphisms in the IRGM gene, with single nucleotide polymorphisms (SNPs) different from the Crohn's disease SNPs, have been reported as predisposing to tuberculosis in different human populations (61, 118).
Fig. 7. Summary of the known roles of human immunity related GTPase (IRG) IRGM and its murine ortholog Irgm1.
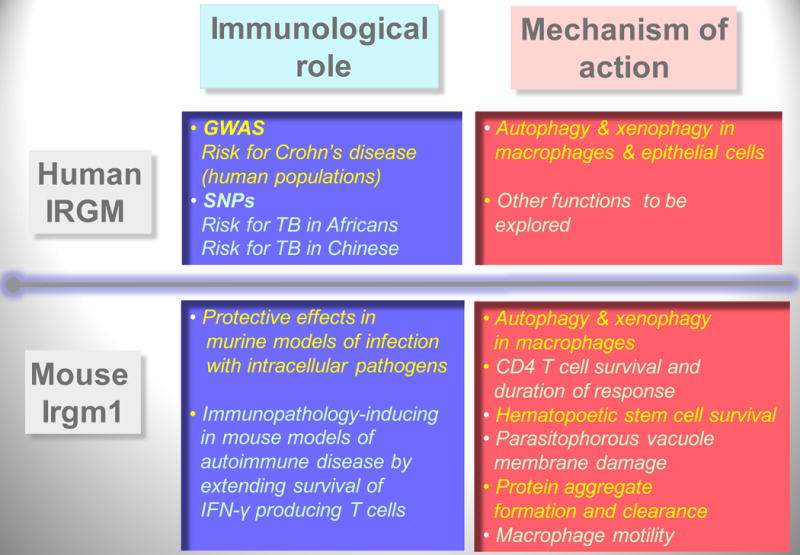
Roles, left boxes; mechanisms, right boxes. Humans have one sensu stricto IRG gene, IRGM. Mouse has 22 sensu stricto IRG genes, of which Irgm1 is believed to be orthologous to human IRGM, via the IRGM9 evolutionary intermediate gene in pre-simians. During primate evolution, IRGM has been pseudogenized (by Alu repeat insertion and termination codon mutations) but then reconstituted in anthropoids (humans, chimpanzees, and gorillas) under the expression control by an endogenous retroviral element ERV9. Murine Irgm1 is under control by IFN-γ, whereas the human gene IRGM is not controlled by IFN-γ but is needed for IFN-γ-induced autophagy. GWAS, Genome-wide association studies. SNPs, single nucleotide polymorphisms. TB, tuberculosis. Listed are findings from human population studies for IRGM as a risk locus for Crohn's disease (a high incidence form of inflammatory bowel disease) and a risk locus tuberculosis (two different SNPs) thus far identified in African and Chinese populations. Not shown is that GWAS studies have linked another autophagy gene, ATG16L1, with Crohn's disease predisposition. Note that many of the functions assigned to the murine Irgm1 fit directly or indirectly with autophagy or autophagy-associated processes.
Additional functions for the numerous murine IRGs have been proposed as alternatives or in addition to autophagy (119). These include the findings that IRGs associate with a wide variety of cellular membranes including lysosomal, Golgi, ER, or phagosomal locations (120-122), where they sometimes play a regulatory role in positioning of other IRGs on other membranes with yet to be identified biological outputs (119, 123). Murine IRGs can inflict parasitophorous vacuole membrane damage (124), and have been implicated in control cell death or survival (17, 113, 125). They have also been seen as affecting protein aggregate formation and clearance (119, 126). These functions are proposed to be other than autophagy (125-127) but can actually be seen as engaging the autophagic pathway, a view that is easily strengthened by the following: (i) the proposed IRG-dependent membrane damage is a principal signal for autophagic elimination of faulty organelles such as mitochondria, damaged parasitophorous vacuoles (125, 128), or pathogen-ruptured phagosomes (65); (ii) formation of toxic protein aggregates as cytoplasmic inclusions (119) are well recognized principal targets for clearance by autophagy in its cytoprotective role (129); (iii) effects of Irgm1 on cell survival and cellular homeostasis are one of the marquee biological roles of autophagy (25); and (iv) functional linkages have been found of IRGs other than Irgm1 or IRGM (e.g. Irga6) with autophagy factors such as Atg5 (110, 121). Nevertheless, the role of individual members of the large IRG family in the mouse is likely to be complex and exceedingly difficult to dissect in full, given the likely partial redundancies or interferences. For now, we are left with an Occam's razor explanation of the often mystifying variety of functions assigned to different murine IRGs as converging upon autophagy pathway. Paradoxically, the intersection between IRGs and autophagy is far easier to interpret in human cells, since the human genome encodes only three IRG genes, of which IRGM is the only human sensu stricto IRG defined as having an immunological function.
The in vivo immunological studies carried out in mice are nevertheless informative at organismal level and help illuminate contributions of IRGs through different cell types. The protective function of mouse Irgm1 against intracellular pathogens in vivo is likely a composite manifestation of Irgm1 roles in the key cell types involved, macrophages and T cells, well documented in vivo in the case of M. tuberculosis (130, 131). First, both human IRGM in human macrophages (109) and Irgm1 in murine macrophages (5) play direct roles in autophagic elimination of mycobacteria as an effector of activation by IFN-γ. This particular contribution should be considered as a specialized Type I immunophagy role as defined in the overview subsection. Second, murine Irgm1 plays a role in promoting autophagy as a cell survival mechanism and thus sustains the mature effector CD4+ T-cell viability (17, 131). Irgm1 also supports the viability and autophagy-dependent remodeling processes during differentiation of hematopoietic progenitor cells (113). These two phenomena fall under the Type II role of autophagy as a part of generic maintenance of cellular viability and homeostasis. As a postscript, Fang et al. (131) proposed in their study that Irgm1 protects T cells from what the authors considered to be autophagic cell death, although most mammalian systems and cell types rarely die from autophagy but rather are protected from cell death by autophagy (30). An apoptotic or necrotic cell death with features of autophagy often reflects a last ditch effort by autophagy to rescue the cell and can easily (but should not) be confused with cell death from autophagy (30). There is also a recent finding suggesting that Irgm1 may play a role in macrophage motility (132), which falls best under the Type III non-autophagic roles.
In Type II immunophagy, Irgm1 affects survival of mature effector CD4+ T cells by protecting them from IFN-γ inflicted apoptotic cell death (17, 133-135), thus delaying a homeostatic mechanism that otherwise limits the duration and strength of cell-mediated immunity response. This Irgm1-dependent persistence of IFN-γ-producing T cells can lead to pathologies when it comes to autoimmune disorders, as observed in the widely used model of multiple sclerosis [experimental autoimmune encephalomyelitis (EAE)], where Irgm1 ablation increases CD4+ T-cell death and ameliorates EAE pathology (17). Hence, at the level of T cells, Irgm1-supported autophagy is beneficial in infection by augmenting T-cell survival resulting in a durable response needed to control recalcitrant infection such as tuberculosis (even at the expense of causing type IV hypersensitivity). However, in the setting of autoimmunity and certain chronic inflammatory diseases, it can be detrimental. We predict that these phenomena will be associated with the effects of IRGM and possibly other autophagy genes in Crohn's disease and tuberculosis.
Concluding remarks: evidence for the role of autophagy in human immunity
One of the key issues is to understand whether and how the process of autophagy figures into innate and adaptive immunity in humans. The most telling evidence for the role of autophagy in human immunology entered the scene in the most unbiased way, by genome wide association screens (GWAS) for disease associations of polymorphisms in human populations (59). Among the key findings of this landmark GWAS study, polymorphisms in two autophagy-associated genes, ATG16L1 and IRGM, have been found to be in strong association with predisposition to Crohn's disease (59). These findings have since been replicated in different human populations as documented by 40 different publications (112). Further refinements in polymorphisms, haplotypes (136), and disease subgroup (Crohn's disease versus ulcerative colitis; adult versus childhood Chiron's disease) specificities have further affirmed the role of ATG16L1 and IRGM in inflammatory bowel disease. Moreover, IRGM is one of the three key loci linked with common diseases in humans identified in a recent comprehensive GWAS focused on the very common form of human genetic polymorphisms, i.e. copy number variants (CNVs) (137): IRGM CNVs were found to associate with Crohn's disease, HLA CNVs showed association with Crohn's disease, rheumatoid arthritis, and type 1 diabetes, whereas TSPAN8 CNVs showed association with type 2 diabetes. Importantly, IRGM polymorphisms predispose to tuberculosis in Chinese population (118) and in African population when, in the latter case, tuberculosis is caused by a specific M. tuberculosis complex member, M. africanum (61). These are key recent developments that bolster the significance of immunological role of autophagy in human disease and elevate immunophagy on par with the roles of autophagy in aging, cancer, metabolic disorders, myopathies, and neurodegenerative disorders.
Acknowledgments
This work was supported by NIH grants AI069345, AI42999, RC1AI086845, a grant from Crohn's & Colitis Foundation of America, and a grant from Bill and Melinda Gates Foundation. The author declares no conflict of interest.
References
- 1.Deretic V. Autophagy in innate and adaptive immunity. Trends Immunol. 2005;26:523–528. doi: 10.1016/j.it.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Munz C. Enhancing immunity through autophagy. Annu Rev Immunol. 2009;27:423–449. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- 3.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 6.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 7.Paludan C, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 9.Orvedahl A, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Gannage M, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyei GB, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA. 2009;106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deretic V. Autophagy in infection. Curr Opin Cell Biol. 2010;22:252–262. doi: 10.1016/j.ceb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saitoh T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinkins C, Arko-Mensah J, Deretic V. Autophagy and HIV. Semin Cell Dev Biol. 2010;21:712–718. doi: 10.1016/j.semcdb.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu H, et al. Genetic deficiency of Irgm1 (LRG-47) suppresses induction of experimental autoimmune encephalomyelitis by promoting apoptosis of activated CD4+ T cells. FASEB J. 2010;24:1583–1592. doi: 10.1096/fj.09-137323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rikihisa Y. Glycogen autophagosomes in polymorphonuclear leukocytes induced by rickettsiae. Anat Rec. 1984;208:319–327. doi: 10.1002/ar.1092080302. [DOI] [PubMed] [Google Scholar]
- 20.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 21.Liang XH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orvedahl A, Macpherson S, Sumpter R, Jr, Talloczy Z, Zou Z, Levine B. Autophagy Protects against Sindbis Virus Infection of the Central Nervous System. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanchet FP, et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32:654–669. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188:527–536. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schweers RL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor MP, Burgon TB, Kirkegaard K, Jackson WT. Role of microtubules in extracellular release of poliovirus. J Virol. 2009;83:6599–6609. doi: 10.1128/JVI.01819-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McPhee CK, Logan MA, Freeman MR, Baehrecke EH. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature. 2010;465:1093–1096. doi: 10.1038/nature09127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 37.Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hailey DW, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yen WL, et al. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J Cell Biol. 2010;188:101–114. doi: 10.1083/jcb.200904075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lynch-Day MA, et al. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci USA. 2010;107:7811–7816. doi: 10.1073/pnas.1000063107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Vaart A, Reggiori F. The Golgi complex as a source for yeast autophagosomal membranes. Autophagy. 2010;6:800–801. doi: 10.1091/mbc.E09-04-0345. [DOI] [PubMed] [Google Scholar]
- 42.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JH, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez-Vicente M, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meijer AJ, Codogno P. Autophagy: a sweet process in diabetes. Cell Metab. 2008;8:275–276. doi: 10.1016/j.cmet.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 46.Jung HS, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 47.Ebato C, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 48.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mammucari C, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Vergne I, et al. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 2009;28:2244–2258. doi: 10.1038/emboj.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 53.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 54.Komatsu M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 55.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 57.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 58.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parkes M, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Intemann CD, et al. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 2009;5:e1000577. doi: 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006;18:375–382. doi: 10.1016/j.coi.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 63.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 64.Yoshikawa Y, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 65.Dupont N, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 66.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 67.Levine B. Eating oneself and uninvited guests; autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 68.Ponpuak M, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010;32:329–341. doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 70.Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delgado M, et al. Autophagy and pattern recognition receptors in innate immunity. Immunol Rev. 2009;227:189–202. doi: 10.1111/j.1600-065X.2008.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 73.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yano T, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 76.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;6:409–411. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 77.Tang D, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Biswas D, Qureshi OS, Lee WY, Croudace JE, Mura M, Lammas DA. ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol. 2008;9:35. doi: 10.1186/1471-2172-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takenouchi T, et al. The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J Immunol. 2009;182:2051–2062. doi: 10.4049/jimmunol.0802577. [DOI] [PubMed] [Google Scholar]
- 80.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jounai N, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 83.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li C, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- 85.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 86.Mortensen M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA. 2010;107:832–837. doi: 10.1073/pnas.0913170107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller BC, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 88.Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 89.Shahnazari S, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8:137–146. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009;16:976–83. doi: 10.1038/cdd.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shin DM, et al. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol. 2010;12:1648–1665. doi: 10.1111/j.1462-5822.2010.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283:33175–33182. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mitroulis I, et al. Regulation of the autophagic machinery in human neutrophils. Eur J Immunol. 2010;40:1461–1472. doi: 10.1002/eji.200940025. [DOI] [PubMed] [Google Scholar]
- 94.Watts C, West MA, Zaru R. TLR signalling regulated antigen presentation in dendritic cells. Curr Opin Immunol. 2010;22:124–130. doi: 10.1016/j.coi.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 95.Yuk JM, et al. Bacillus calmette-guerin cell wall cytoskeleton enhances colon cancer radiosensitivity through autophagy. Autophagy. 2010;6:46–60. doi: 10.4161/auto.6.1.10325. [DOI] [PubMed] [Google Scholar]
- 96.Djavaheri-Mergny M, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–30382. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 97.Criollo A, et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010;29:619–631. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cadwell K, et al. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tormo D, et al. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell. 2009;16:103–114. doi: 10.1016/j.ccr.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.English L, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10:480–487. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86:573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 102.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 103.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tang D, Kang R, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Subauste CS, Andrade RM, Wessendarp M. CD40-TRAF6 and autophagy-dependent anti-microbial activity in macrophages. Autophagy. 2007;3:245–248. doi: 10.4161/auto.3717. [DOI] [PubMed] [Google Scholar]
- 107.Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Watanabe K, Tsubata T. Autophagy connects antigen receptor signaling to costimulatory signaling in B lymphocytes. Autophagy. 2009;5:108–110. doi: 10.4161/auto.5.1.7278. [DOI] [PubMed] [Google Scholar]
- 109.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 110.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bekpen C, Xavier RJ, Eichler EE. Human IRGM gene “to be or not to be”. Semin Immunopathol. 2010 doi: 10.1007/s00281-010-0224-x. in press. [DOI] [PubMed] [Google Scholar]
- 112.Brest P, et al. Autophagy and Crohn's disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr Mol Med. 2010;10:486–502. doi: 10.2174/156652410791608252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Feng CG, Weksberg DC, Taylor GA, Sher A, Goodell MA. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell. 2008;2:83–89. doi: 10.1016/j.stem.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bekpen C, et al. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005;6:R92. doi: 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bekpen C, et al. Death and resurrection of the human IRGM gene. PLoS Genet. 2009;5:e1000403. doi: 10.1371/journal.pgen.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taylor GA, Feng CG, Sher A. p47 GTPases: regulators of immunity to intracellular pathogens. Nat Rev Immunol. 2004;4:100–109. doi: 10.1038/nri1270. [DOI] [PubMed] [Google Scholar]
- 117.MacMicking JD. Immune control of phagosomal bacteria by p47 GTPases. Curr Opin Microbiol. 2005;8:74–82. doi: 10.1016/j.mib.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 118.Che N, et al. Identification of a novel IRGM promoter single nucleotide polymorphism associated with tuberculosis. Clin Chim Acta. 2010;411:1645–1649. doi: 10.1016/j.cca.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 119.Hunn JP, Howard JC. The mouse resistance protein Irgm1 (LRG-47): a regulator or an effector of pathogen defense? PLoS Pathog. 2010;6:e1001008. doi: 10.1371/journal.ppat.1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhao YO, Konen-Waisman S, Taylor GA, Martens S, Howard JC. Localisation and mislocalisation of the interferon-inducible immunity-related GTPase, Irgm1 (LRG-47) in mouse cells. PLoS One. 2010;5:e8648. doi: 10.1371/journal.pone.0008648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Khaminets A, et al. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol. 2010;12:939–961. doi: 10.1111/j.1462-5822.2010.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tiwari S, Choi HP, Matsuzawa T, Pypaert M, MacMicking JD. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P(2) and PtdIns(3,4,5)P(3) promotes immunity to mycobacteria. Nat Immunol. 2009;10:907–917. doi: 10.1038/ni.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hunn JP, et al. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J. 2008;27:2495–2509. doi: 10.1038/emboj.2008.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Martens S, et al. Disruption of Toxoplasma gondii Parasitophorous Vacuoles by the Mouse p47-Resistance GTPases. PLoS Pathog. 2005;1:e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 2009;5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Henry SC, et al. Balance of Irgm protein activities determines IFN-gamma-induced host defense. J Leukoc Biol. 2009;85:877–885. doi: 10.1189/jlb.1008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Howard J. The IRG proteins: a function in search of a mechanism. Immunobiology. 2008;213:367–35. doi: 10.1016/j.imbio.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 128.Yap GS, Ling Y, Zhao Y. Autophagic Elimination of Intracellular Parasites: Convergent Induction by IFN-gamma and CD40 Ligation? Autophagy. 2007;3:163–165. doi: 10.4161/auto.3655. [DOI] [PubMed] [Google Scholar]
- 129.Moreau K, Luo S, Rubinsztein DC. Cytoprotective roles for autophagy. Curr Opin Cell Biol. 2010;22:206–211. doi: 10.1016/j.ceb.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 131.Feng CG, et al. The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-gamma-induced cell death. Nat Immunol. 2008;9:1279–1287. doi: 10.1038/ni.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Henry SC, Traver M, Daniell X, Indaram M, Oliver T, Taylor GA. Regulation of macrophage motility by Irgm1. J Leukoc Biol. 2010;87:333–343. doi: 10.1189/jlb.0509299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dalton DK, Haynes L, Chu CQ, Swain SL, Wittmer S. Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J Exp Med. 2000;192:117–122. doi: 10.1084/jem.192.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon gamma is required for activation-induced death of T lymphocytes. J Exp Med. 2002;196:999–1005. doi: 10.1084/jem.20020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:123–128. doi: 10.1084/jem.192.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Craddock N, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]