

Summary
Historically, the general secretory (Sec) pathway of Gram‐negative bacteria has served as the primary route by which heterologous proteins are delivered to the periplasm in numerous expression and engineering applications. Here we have systematically examined the twin‐arginine translocation (Tat) pathway as an alternative, and possibly advantageous, secretion pathway for heterologous proteins. Overall, we found that: (i) export efficiency and periplasmic yield of a model substrate were affected by the composition of the Tat signal peptide, (ii) Tat substrates were correctly processed at their N‐termini upon reaching the periplasm and (iii) proteins fused to maltose‐binding protein (MBP) were reliably exported by the Tat system, but only when correctly folded; aberrantly folded MBP fusions were excluded by the Tat pathway's folding quality control feature. We also observed that Tat export yield was comparable to Sec for relatively small, well‐folded proteins, higher relative to Sec for proteins that required cytoplasmic folding, and lower relative to Sec for larger, soluble fusion proteins. Interestingly, the specific activity of material purified from the periplasm was higher for certain Tat substrates relative to their Sec counterparts, suggesting that Tat expression can give rise to relatively pure and highly active proteins in one step.
Introduction
The export of proteins to the bacterial periplasm is a commonly used strategy in preparative protein expression (Georgiou and Segatori, 2005) and in many protein engineering applications (Smith, 1985; Francisco et al., 1993; Chen et al., 2001; Harvey et al., 2004). Since its discovery more than 25 years ago, the general secretory (Sec) pathway has been the primary vehicle for transporting proteins to the periplasm in biotechnological applications (Baneyx, 1999; Swartz, 2001; Baneyx and Mujacic, 2004). However, as only extended polypeptides can fit through the 5–8 Å diameter pore of the Sec apparatus (Van den Berg et al., 2004), it is imperative that protein substrates are maintained in an unfolded state via interactions with chaperones or by co‐translational protein synthesis (Schatz and Dobberstein, 1996; Driessen et al., 2001). For this reason, proteins exported via the Sec pathway attain their native, biologically active conformation only after they have been ‘threaded’ through the Sec pore. Consequently, numerous Sec‐targeted proteins are not readily exported due to premature folding in the cytoplasm (Benson et al., 1984; Schierle et al., 2003). This often results in blocking or jamming of the secretion apparatus in a manner that leads to the toxic accumulation of precursors of all secreted substrates in the cytoplasm of cells (Benson et al., 1984; Kiino and Silhavy, 1984; Hayhurst and Georgiou, 2001). Even when membrane translocation is successful, many proteins are susceptible to extensive degradation or cannot attain their native structure in the periplasmic space (Gentz et al., 1988; Feilmeier et al., 2000). Certain other complex proteins acquiring protein subunits or redox cofactors hinge on the formation of secondary or tertiary structure in the cytoplasm and thus are inherently incompatible with Sec export (Rodrigue et al., 1999; DeLisa et al., 2003).
Based on the above observations, it is clear that the Sec pathway is useful for the production of only a subset of proteins. As an alternative to the Sec system, we and others have explored the use of the twin‐arginine translocation (Tat) pathway for exporting heterologous proteins across the cytoplasmic membrane (DeLisa et al., 2003; Kim et al., 2005; Fisher et al., 2006; Bruser, 2007). Proteins routed via the Tat pathway are synthesized with N‐terminal signal peptides that bear a conserved, although non‐essential (DeLisa et al., 2002), S/T‐R‐R‐x‐F‐L‐K motif (Berks, 1996; Berks et al., 2000a). The Tat export in Escherichia coliminimally requires three integral membrane proteins, namely TatA (or TatE), TatB and TatC, which are predicted to form the translocase (Settles et al., 1997; Weiner et al., 1998; Berks et al., 2000b). While the precise mechanism of Tat‐dependent transport remains unresolved, recent evidence suggests a ‘channel‐less’ transport model whereby TatC pulls substrates through a patch of TatA on the lipid bilayer (Cline and McCaffery, 2007). Most importantly, whereas Sec‐dependent export is restricted to preproteins that have not yet reached a native conformation, the hallmark of the Tat pathway is its predilection for substrates that are correctly folded (Bruser et al., 2003; DeLisa et al., 2003; Fisher et al., 2006) with few exceptions (Richter et al., 2007). Based on this unique characteristic, it has been widely speculated that the Tat system will be a valuable tool for expressing heterologous proteins that attain a folded or partially folded conformation prior to reaching the translocation machinery. Moreover, as the Tat machinery does not typically accommodate unfolded proteins, poorly folded polypeptides will be effectively eliminated during protein production or in combinatorial library screening experiments. However, owing to its relative novelty and lower reported throughput compared with Sec (Settles et al., 1997; Berks et al., 2003), the potential of the bacterial Tat pathway for preparative expression of secreted proteins has not been fully explored. Here, we address a number of unresolved issues related to Tat‐dependent secretion of heterologous proteins.
Results
Identification of E. coli signal peptides that promote efficient Tat‐dependent transport
The E. coli genome is predicted to encode up to 34 Tat substrates based on the presence of an N‐terminal signal peptide containing a twin arginine motif (Robinson and Bolhuis, 2001; Dilks et al., 2003; Bendtsen et al., 2005). Of these, 27 have been validated experimentally based on their ability to export one or more reporter proteins through the Tat machinery (Tullman‐Ercek et al., 2007). Using maltose‐binding protein (MBP, encoded by malE) as a genetic reporter (see Fig. 1) along with a simple MacConkey maltose agar plate strategy (Blaudeck et al., 2003), we tested 29 Tat signal peptides for their ability to direct MBP to the periplasm. When E. coliHS3018 (a malE‐negative derivative of the strain MC4100) expressed a fusion between MBP and the Tat‐dependent signal peptide of trimethylamine N‐oxide reductase plus 10 amino acids of the mature TorA domain [ssTorA(+10)–MBP] cells appeared as dark red colonies on MacConkey maltose agar plates (Fig. S1A), indicating efficient periplasmic localization of functional MBP. On the contrary, plating HS3018 cells that lacked the tatC gene resulted in white colonies, indicating that ssTorA(+10)–MBP transport was dependent on a functional Tat pathway (Fig. S1A). We also observed complete loss of export when the consensus Arg–Arg residues in the ssTorA signal were mutated to Lys–Lys (Table 1). The Tat dependence for ssTorA(+10)–MBP was further confirmed by plating cells on minimal M9 agar supplemented with maltose as the sole carbon source (Fig. S1A) and by Western blot analysis of subcellular fractions (Fig. S1B).
Figure 1.
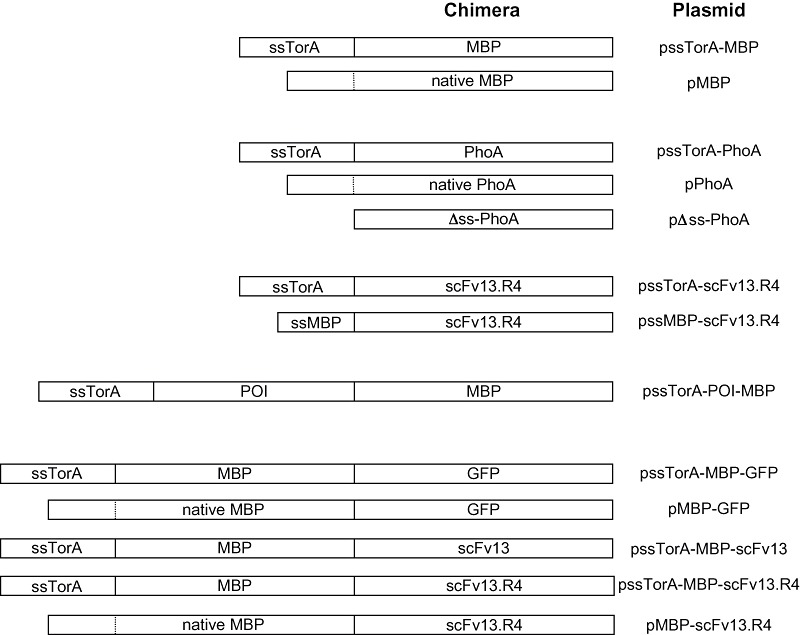
Schematic of expression constructs used in this study. Shown are the various chimeras generated for these studies along with the corresponding plasmid name (see Experimental procedures and Table 3 for more details). Dashed lines indicate cleavage site between native signal and mature domain for MBP and PhoA. POI includes one of the following: TrxA, GST, PhoA, GFP, Top7 and TraR. Drawing is not to scale.
Table 1.
Tat signal peptides that promote transport of MBP.
| Signal peptidea | HS3018 | HS3018 ΔtatC |
|---|---|---|
| AmiA | ++ | ++ |
| AmiC | ++ | ++ |
| CueO | ++ | − |
| DmsA | + | − |
| FdnG | + | − |
| FdoG | + | − |
| FhuD | ++ | ++ |
| HyaA | ++ | − |
| HybA | + | + |
| HybO | + | + |
| NapA | + | − |
| NapG | + | + |
| NrfC | + | + |
| SufI | ++ | − |
| TorA (RR) | ++ | − |
| TorA (KK) | − | − |
| TorZ | + | + |
| WcaM | + | − |
| YaeI | ++ | ++ |
| YahJ | + | + |
| YagT | + | − |
| YcbK | ++ | − |
| YcdB | ++ | − |
| YdcG | ++ | ++ |
| YdhX | ++ | − |
| YedY | + | + |
| YfhG | + | + |
| YnfE | ++ | − |
| YnfF | + | + |
Expression of each ssTat–MBP construct was from pBAD18‐Cm (Tullman‐Ercek et al., 2007).
++, bright red colonies, equivalent to malE+ cells streaked on MacConkey maltose agar plates; +, pale red colonies; −, white colonies; bold font, Tat specific.
Following the same strategy, we tested an additional 27 signal peptides for Tat export competence. As above, HS3018 cells were induced for expression of a putative Tat signal peptide plus six to ten amino acids of the mature protein fused in‐frame to MBP. Of these, a total of 14 (ssCueO, ssDmsA, ssFdnG, ssFdoG, ssHyaA, ssNapA, ssSufI, ssTorA, ssWcaM, ssYagT, ssYcbK, ssYedB, ssYdhX and ssYnfE) showed Tat‐dependent export as indicated by plating on MacConkey maltose agar plates (Table 1). The rest showed Tat‐independent transport in that they conferred a red appearance to both HS3018 and HS3018 ΔtatC cells, which was largely consistent with earlier analysis (Tullman‐Ercek et al., 2007). For the 14 Tat‐dependent signals, we observed varying levels of red colouration that we suspected was indicative of the relative quantity of MBP localized in the periplasm. Indeed, Western blot analysis of subcellular fractions isolated from cells expressing ssTorA–MBP (bright red) and ssFdnG–MBP (pale red) revealed much greater accumulation of ssTorA–MBP in the periplasmic fraction (data not shown). Based on this analysis, 8/14 Tat‐dependent signal peptides (CueO, HyaA, SufI, TorA, YcbK, YcdB, YdhX and YnfE) appeared to efficiently export MBP whereas the remaining six were less efficient targeting signals. Because ssTorA was found to efficiently export MBP and has been employed extensively in studies of the Tat system (Blaudeck et al., 2001; 2003; Perez‐Rodriguez et al., 2007; Strauch and Georgiou, 2007; Tullman‐Ercek et al., 2007), we chose this signal peptide for all further studies.
Translocation efficiency and processing of different MBP chimeras
Here and in many earlier studies employing ssTorA (Thomas et al., 2001; DeLisa et al., 2002; Tullman‐Ercek et al., 2007), a common practice for ensuring authentic and efficient substrate processing was to fuse the signal peptide plus several additional residues of the mature TorA protein to the recombinant protein‐of‐interest (POI). From a biotechnology standpoint, this will result in the undesirable outcome of a POI with an inauthentic N‐terminus because these extra residues remain following proteolysis of the signal peptide by signal peptidase I (SPase I). To determine whether these extra residues affected Tat transport efficiency or processing, we evaluated ssTorA fused to MBP with ten, six and zero residues of mature TorA [ssTorA(+10)–MBP, ssTorA(+6)–MBP and ssTorA(+0)–MBP]. Western blotting (Fig. 2A) and MacConkey maltose plating (Fig. 2B) revealed that each ssTorA signal was capable of efficiently routing MBP to the periplasm, with the +6 and +0 signals exhibiting slightly better export efficiency and significantly less insoluble accumulation compared with the +10 signal. For comparison, we also expressed MBP with its native Sec signal peptide and observed periplasmic accumulation that was comparable to that seen for the ssTorA constructs (Fig. 2A). One notable difference between the secretion pathways was that very little Sec‐targeted MBP accumulated in the cytoplasm or insoluble fraction, indicating that Sec export was more efficient than export via the Tat system, consistent with earlier findings (Settles et al., 1997; Berks et al., 2003). Despite this difference in transport efficiency, amylose affinity chromatography purification of the Tat‐ and Sec‐targeted constructs from the periplasm of 50 ml flask cultures resulted in ~5–10 mg l−1 of soluble MBP for each (Fig. 2C and Table 2).
Figure 2.
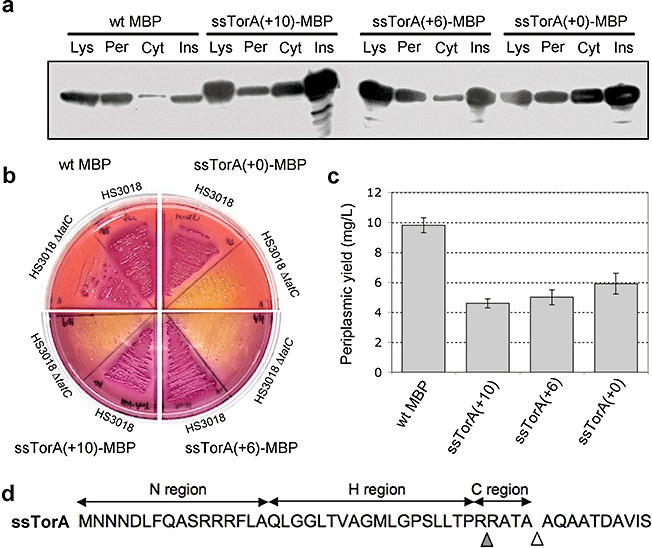
Intracellular localization, phenotype and yield of Tat‐targeted MBP. A. Western blot analysis of total soluble lysate (Lys), cytoplasmic (Cyt), periplasmic (Per) and insoluble (Ins) fractions from HS3018 (malE‐deficient derivative of MC4100) cells expressing wild‐type (wt) MBP and ssTorA–MBP with 0, 6 or 10 additional residues of mature TorA (+0, +6, +10). Western blots were probed with anti‐MBP primary antibody. B. HS3018 and HS3018 ΔtatC cells expressing the above constructs streaked on MacConkey agar media supplemented with 0.4% maltose. C. Yield of the above constructs following imac purification from the periplasm of HS3018 cells. Data represent the average of three replicate experiments. D. Amino acid sequence of the TorA (+10) signal peptide highlighting the primary signal peptidase cleavage site (white triangle) and the second cleavage site (grey triangle) as reported by N‐terminal sequencing.
Table 2.
Comparison of expression yields using the Tat or Sec export pathways.
| POI | Signal peptidea | Yield (mg l−1) | Tat/Sec ratio (%)b | Reference |
|---|---|---|---|---|
| E. coliexpressionc | ||||
| MBP | MBP | 9.8 | − | This study |
| TorA(+10) | 4.6 | 47 | This study | |
| TorA(+6) | 5.0 | 51 | This study | |
| TorA(+0) | 5.9 | 60 | This study | |
| GFP | PelB | nm | Nd | This study |
| TorA | 10–15 | Nd | Barrett et al. (2003) | |
| MBP–GFP | MBP | 0.4 | − | This study |
| TorA | 2.6 | 650 | This study | |
| scFv 26.10 | PelB | 1.0 | − | Ribnicky et al. (2007) |
| TorAd | 0.1 | 12 | Ribnicky et al. (2007) | |
| scFv13.R4 | MBP | 0.01 | − | This study |
| TorA | 0.06 | 600 | This study | |
| MBP–scFv13.R4 | MBP | 8.4 | − | This study |
| TorA | 0.5 | 6 | This study | |
| Human tPA (truncated) | StII | 140.0e | − | Kim et al. (2005) |
| TorA | 44.8e | 29 | Kim et al. (2005) | |
| S. lividansexpressionf | ||||
| S. lividansXlnB1 | XlnB1g | nr | − | Gauthier et al. (2005) |
| XlnCh | nr | 33 | Gauthier et al. (2005) | |
| S. lividansXlnB2 (truncated) | XlnB1 | nr | − | Gauthier et al. (2005) |
| XlnC | nr | 70 | Gauthier et al. (2005) | |
| S. equisimilisstreptokinase | Vsii | 4 | − | Pimienta et al. (2007) |
| XlnC | 0.1–0.2 | 4 | Pimienta et al. (2007) | |
| Human TNFa | Vsi | 23.0 | − | Schaerlaekens et al. (2004) |
| XlnC | 1.6 | 7 | Schaerlaekens et al. (2004) | |
| XyyZj | 1.4 | 6 | Schaerlaekens et al. (2004) | |
| Human IL‐2 | Vsi | 10.4 | − | Schaerlaekens et al. (2004) |
| XlnC | 4.8 | 46 | Schaerlaekens et al. (2004) | |
All signal peptides derived from E. coliunless otherwise noted. Sec‐dependent signal peptides are listed in italics.
Tat/Sec ratio calculated by dividing yield measured for Tat expression by the value measured for Sec expression. All values represent yield obtained from periplasmic or supernatant fraction.
All proteins expressed in E. coliwere purified from the periplasm.
Yields obtained using high‐cell E. colidensity fermentations.
Expression via the Tat system was achieved using strain FÅ113 that has a more oxidizing cytoplasm than wild‐type strains of E. coli.
All proteins expressed in S. lividanswere secreted and purified from the medium.
Sec signal peptide derived from Streptomyces venezuelae xylanase B1 (XlnB1).
Tat‐dependent signal peptide derived from S lividansxylanase C (XlnC).
Signal peptide derived from S. venezuelaeCBS762.70 subtilisin inhibitor (Vsi).
Signal peptide derived from S lividans tyrosinase (XyyZ).
nd, not determined; nm, not measurable (protein is completely undetectable); nr, not reported.
To assess whether the additional mature residues affected processing by SPase I, N‐terminal sequencing of the purified proteins was performed. As expected, the sequence KIEEGKLV corresponding to the authentic N‐terminal residues of mature MBP was obtained for Sec‐targeted MBP. Surprisingly, two sequences were obtained for each of the ssTorA constructs. The more abundant sequence corresponded to material that was processed after the predicted ATA cleavage site (Fig. 2D), yielding KIEEGKLV, AQAATDKI, and AQAATDAV for ssTorA(+0), ssTorA(+6) and ssTorA(+10) respectively. A second minor sequence of RATAAQAA for ssTorA(+6) and ssTorA(+10) or RATAKIEE for ssTorA(+10) was obtained. These minor sequences corresponded to material that was apparently degraded between Arg35 and Arg36, a location immediately prior to the ATA cleavage site and in the middle of the charged c‐region of the signal peptide.
Exceptional total expression for Tat‐targeted substrates
It was surprising to observe that the periplasmic accumulation of Sec‐ and Tat‐mediated MBP was comparable, especially considering that MBP is a native Sec substrate. To determine if this was specific to MBP, we next compared Sec and Tat export of another native Sec substrate, E. colialkaline phosphatase (PhoA). PhoA contains two disulfide bonds that are essential for folding into its native conformation (Sone et al., 1997). These disulfide bonds cannot form within the reducing cytoplasm of E. coli and, as a result, PhoA is incompatible with Tat transport under normal cellular conditions (DeLisa et al., 2003). However, using engineered strains that have a more oxidizing cytoplasm, such as E. coliFÅ113 (Bessette et al., 1999), protein disulfide bonds readily form in the cytoplasm of these cells, resulting in correctly folded PhoA that can be exported via the Tat system (DeLisa et al., 2003) (Fig. 3A and B). The efficiency of Tat‐mediated PhoA localization was comparable to that observed for native PhoA exported via the Sec pathway in either FÅ113 cells (Fig. 3A and B) or wild‐type DHB4 cells (data not shown).
Figure 3.
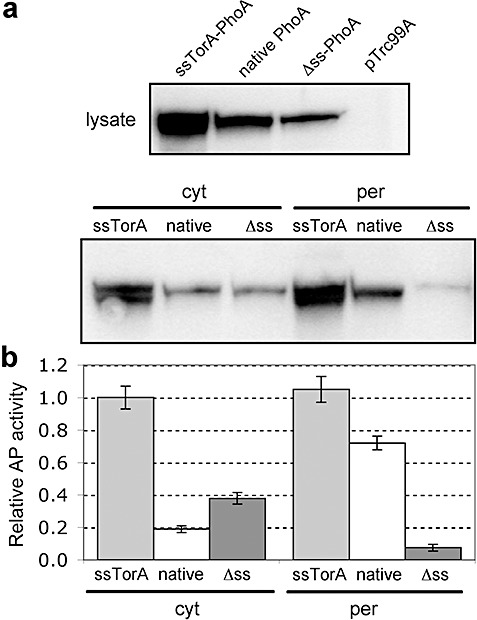
Intracellular localization and activity of ssTorA–PhoA. A. Western blot analysis of total soluble lysate (top panel) and cytoplasmic (cyt) and periplasmic (per) fractions (bottom panel) from FÅ113 cells expressing Tat‐targeted (ssTorA), Sec‐targeted (native) and cytoplasmic (Δss) PhoA and empty vector pTrc99A control cells. Western blots were probed with anti‐PhoA primary antibody. B. PhoA activity in the above cells and constructs assayed in both the cytoplasmic and periplasmic fractions. All data normalized to the activity measured for ssTorA–PhoA in the periplasmic fraction. Data represent the average of three replicate experiments.
Interestingly, inspection of whole‐cell lysates revealed a much greater total accumulation of ssTorA–PhoA relative to native PhoA or a version of PhoA expressed without an N‐terminal export signal (Δss‐PhoA, Fig. 3A). In fact, a similar phenomenon was observed above for MBP and previously for human tissue plasminogen activator (Kim et al., 2005). Using the mfold program (Zuker, 2003), we found that the mRNA covering the signal peptide‐coding region of torA folded into a stable stem‐loop structure (Fig. 4A) (ΔG = −37.10 kcal mol−1 at 37°C and physiological pH). Similar analysis of the 5′ regions encoding native phoA, Δss‐phoAor malEdid not yield a stem‐loop structural prediction (data not shown). Based on this analysis, we hypothesized that the difference in the amount of active protein could be explained by differences in the abundance of mRNA, akin to what was observed for the signal peptide‐coding region of native E. coli Tat substrate formate dehydrogenase N that folds into a stable mRNA secondary structure and mediates translational control (Punginelli et al., 2004). To examine this, we performed a quantitative real‐time PCR (RT‐PCR) and discovered that the amount of mRNA was nearly identical in cells expressing ssTorA–PhoA, native PhoA or Δss‐PhoA (Fig. 4B). Thus, we believe that the difference in PhoA protein levels is best explained by properties and interactions that occur at the protein level and not at the level of mRNA expression or stability as we had first hypothesized.
Figure 4.
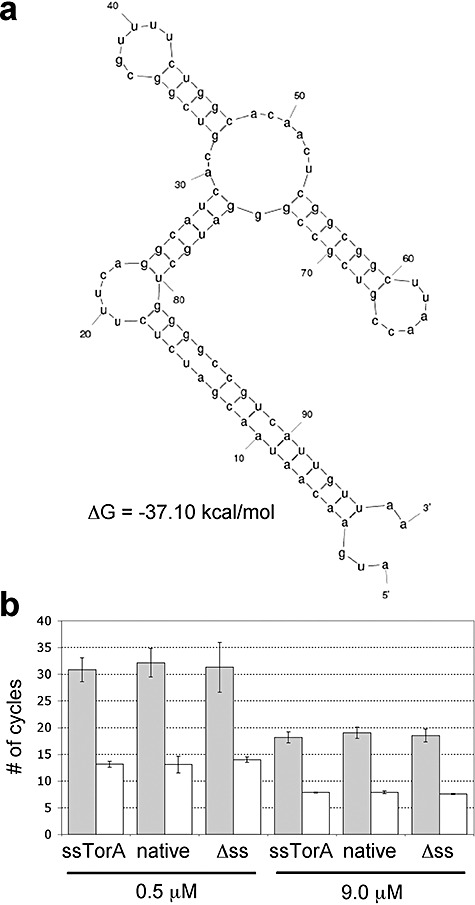
mRNA stability analysis of PhoA constructs. A. Predicted mRNA secondary structure and thermodynamic stability of sstorAtranscript using the mfold program (Zuker, 2003). B. Quantitative RT‐PCR analysis of mRNA isolated from FÅ113 cells expressing Tat‐targeted (ssTorA), Sec‐targeted (native) and cytoplasmic (Δss) PhoA. Grey bars indicate phoA levels while white bars correspond to 16s rRNA controls. Data for two different primer concentrations (0.5 and 9.0 µM) are shown and is the average of three replicate experiments.
MBP as a fusion partner for Tat‐transported proteins
Many proteins that accumulate as insoluble aggregates when they are overproduced in E. colican be rendered soluble simply by fusion to the C‐terminus of MBP (Kapust and Waugh, 1999). Moreover, fusion of a POI to either terminus of MBP enables facile purification of the fusion based on the affinity of MBP to cross‐linked amylose (di Guan et al., 1988). To determine if the broad utility of MBP as both a fusion partner and a reporter protein could be coupled with Tat transport, we examined a collection of POI–MBP fusions for their ability to be routed to the periplasm by the Tat pathway. Several proteins known to be capable of soluble expression in the E. colicytoplasm, namely E. coliglutathione S‐transferase (GST), E. colithioredoxin (TrxA), green fluorescent protein (GFP) and the de novo designed protein Top7, were fused in between ssTorA and MBP and expressed in E. coli. As expected, each of these constructs accumulated in the periplasm (Fig. 5A). It is noteworthy that most of the fusions accumulated to a lesser extent than ssTorA–MBP with the exception of the ssTorA–TrxA–MBP fusion that accumulated to a very high level in the periplasm. On the other hand, proteins that are known to misfold in the cytoplasm, such as E. coliPhoA or Agrobacterium tumefaciensTraR, were not localized in the periplasm (Fig. 5A). Transport to the periplasm for all of these constructs was blocked in a ΔtatCstrain (shown in Fig. 5A for ssTorA–MBP), indicating that each was exported in a Tat‐dependent manner. Thus, similar to our earlier observations (Fisher et al., 2006), export of these fusion proteins to the periplasm was dependent on correct folding and solubility of the POI in the cytoplasm and on an intact Tat system. To determine if transport of these MBP fusions conferred a mal+ phenotype to cells, we plated HS3018 cells expressing each of these constructs on M9 minimal maltose media. In general, the soluble POIs conferred growth in a Tat‐dependent manner while the insoluble POIs were incapable of restoring growth to cells (Fig. 5B), indicating that a simple plating strategy can be used to screen for correctly folded POI–MBP fusions. The one exception to this was the ssTorA–GFP–MBP fusion that was localized to the periplasm (Fig. 5A) but incapable of conferring growth to HS3018 cells (Fig. 5B).
Figure 5.
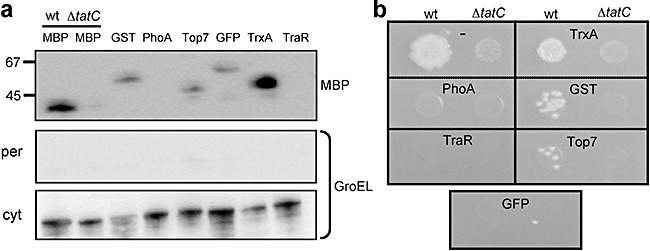
Intracellular localization and phenotype of Tat‐targeted MBP sandwich fusions. A. Western blot analysis of periplasmic (per; top panel) and cytoplasmic (cyt; bottom panel) fractions from HS3018 and HS3018 ΔtatC (lane 2 only) cells expressing ssTorA–MBP (lanes 1 and 2) and ssTorA–POI–MBP sandwich fusions of the following POIs: GST, PhoA, Top7, GFP, TrxA and TraR (lanes 3–8). Western blots were probed with anti‐MBP and anti‐GroEL primary antibody as indicated. GroEL was used to indicate the quality of each fractionation. B. Spot plating of cells and constructs described above on M9 minimal agar media supplemented with 0.4% maltose.
Export and purification of recombinant proteins using the Tat system
Next, we sought to take advantage of the MBP fusion partner for purifying Tat‐transported POIs from the periplasm of E. colicells. It should be noted that for these studies, we fused GFP to the C‐terminus of either ssTorA–MBP or native MBP as this orientation was likely to capitalize on the solubility‐enhancing properties of MBP (Kapust and Waugh, 1999). Earlier studies reported that Sec‐targeted MBP–GFP fusions were non‐fluorescent when exported via the Sec pathway (Feilmeier et al., 2000); thus, GFP export represents an example where the choice of the Tat system was expected to be advantageous over the Sec pathway. Indeed, purification of ssTorA–MBP–GFP from the periplasm using imac yielded 2.6 mg l−1 of pure material compared with only 0.4 mg l−1 of Sec‐targeted MBP–GFP (Table 2). More importantly, whereas the MBP–GFP was entirely non‐fluorescent in all fractions, the purified ssTorA–MBP–GFP was highly fluorescent in the cytoplasmic, periplasmic and purified fractions (data not shown), indicating that active GFP could be purified from the periplasm using a Tat, but not a Sec, targeting signal and an MBP fusion tag.
To determine whether the MBP fusion approach could be applied generally to Tat‐transported proteins, we expressed and purified single‐chain antibody fragments (scFvs) from the periplasm by fusion to the C‐terminus of ssTorA–MBP. This presented a challenging scenario because scFvs typically misfold in the reducing environment of the E. colicytoplasm where required disulfide bonds do not form. As a result, misfolded scFvs are not transported via the Tat system (DeLisa et al., 2003). However, scFvs fused to the C‐terminus of MBP can be expressed at high levels in the cytoplasm of E. coli as soluble and active proteins regardless of the redox state of the bacterial cytoplasm (Bach et al., 2001). The scFvs chosen for these studies were each specific for β‐galactosidase; the first was a wild‐type scFv that folds poorly in the cytoplasm (scFv13) and the second was a variant derived from scFv13 (scFv13.R4) that exhibits increased solubility in the cytoplasm (Martineau et al., 1998). Following expression of ssTorA–MBP–scFv13 and ssTorA–MBP–scFv13.R4 in wild‐type E. coli, we observed strong accumulation in the cytoplasmic and periplasmic fractions for both scFvs (Fig. 6A). Thus, consistent with previous studies (Bach et al., 2001), MBP–scFv fusions were soluble in the cytoplasm despite the fact that expression and folding occurred in a reducing environment. Moreover, as a result of this solubility, the scFv13 protein was competent for transport via the Tat pathway. Not surprisingly, the level of periplasmic accumulation for the better folding scFv13.R4 protein was markedly greater than scFv13, consistent with our earlier findings (Fisher et al., 2006). As ssTorA–MBP–scFv13.R4 exhibited greater accumulation in the periplasm, we chose this construct for subsequent purification studies. From the periplasmic fraction, we recovered 0.5 mg l−1 of soluble MBP–scFv13.R4, which was slightly greater than the yield reported for a different scFv that had been exported via the Tat pathway (Ribnicky et al., 2007). For comparison, we expressed and purified the scFv13.R4 as a C‐terminal fusion to full‐length MBP with its native Sec signal peptide (Fig. 6A) and recovered 8.4 mg l−1. Thus, for certain proteins, Tat export is less effective than Sec‐mediated export, which agrees with previous data comparing expression yields for scFvs and certain other enzymes exported via the Tat and Sec pathways (Table 2). Interestingly, despite the overall lower yield resulting from Tat export compared with Sec export, the specific activity of purified fusion protein recovered using the Tat pathway was ~30% greater on average relative to that recovered using the Sec pathway (Fig. 6B).
Figure 6.
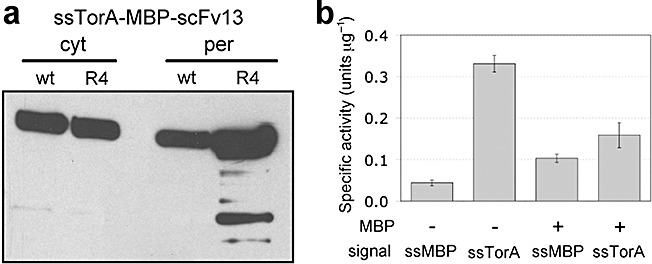
Intracellular localization and activity of Tat‐targeted MBP–scFv fusions. A. Western blot analysis of periplasmic (per) and cytoplasmic (cyt) fractions from HS3018 cells expressing ssTorA–MBP‐scFv13 and ssTorA–MBP–scFv13.R4. Western blots were probed with anti‐c‐myc primary antibody. B. In vitroβ‐galactosidase activity of whole‐cell lysates from E. coli959 cells incubated with purified periplasmic scFv13.R4 targeted through either Sec (ssMBP) or Tat (ssTorA) as an unfused protein (−) or as a fusion to MBP (+). Each sample was assayed in triplicate and data represent the average of three replicate purification and activity experiments.
Finally, it is noteworthy that when the same scFv13.R4 protein was expressed without the MBP fusion partner, the yield was only 0.01 mg l−1 for Sec export compared with 0.06 mg l−1 for Tat export (Table 2). The low yield of Sec‐exported scFv13.R4 was accompanied by a marked decrease in culture growth immediately following induction that was not seen when the same protein was exported via the Tat pathway (data not shown). Thus, similar to the results for GFP above, the inability to localize scFv13.R4 in the periplasm using the Sec pathway can be overcome by employing the Tat pathway. Also, the specific activity of the Tat‐exported scFv13.R4 was two to three times greater than that observed for the MBP fusion (Fig. 6B), suggesting that for some proteins exported via the Tat pathway, lower yields may be offset by greater activity.
Discussion
It has been previously reported that secretion via the Tat pathway results in lower levels of secreted protein as compared with Sec‐dependent secretion. This behaviour has been observed for a relatively small handful of secreted substrates expressed in E. colior Streptomyces lividans(see Table 2), leading one group to suggest that ‘the Tat apparatus is . . . orders‐of‐magnitude less efficient than the Sec system’ (Bruser, 2007). Surprisingly, we show comparable or improved periplasmic yield (Tat/Sec ratio > 47%; see Table 2) for a variety of substrates, including MBP, PhoA, GFP and an engineered scFv specific for β‐galactosidase. In fact, the only case where we observed significantly decreased export efficiency (~10‐fold) for the Tat system was following expression of larger proteins comprised of a fusion between MBP and POIs such as scFv13. Collectively, these results suggest that smaller, well‐folded substrates are more efficiently processed by the Tat pathway relative to larger, bulkier substrates. This observation is in line with the ‘channel‐less’ model of transport that suggests that protein fusions exceeding a characteristic or threshold length might be limited in Tat export (Cline and McCaffery, 2007). Another explanation for the less‐efficient export of larger proteins is that faster folding, as would be expected for smaller proteins, results in greater export efficiency (Ribnicky et al., 2007). It should be noted that our protein expression experiments were performed without any significant optimization, such as the coexpression of factors that are known to increase Tat throughput. Thus, we are optimistic that greater levels of export may be attained using high‐expression vectors and/or by coexpressing factors such as TatABC (Alami et al., 2002), PspA (DeLisa et al., 2004), TorD (Li, Chang and Lin et al., 2006) or DnaK (Perez‐Rodriguez et al., 2007), all of which have been shown to increase Tat translocation efficiency. This optimization will be especially important for commercial applications where titers in the 10–100 mg l−1 range or above are the benchmark. In that sense, it remains to be seen whether the Tat pathway will be competitive with the Sec pathway. Nonetheless, it is clear that the Tat pathway allows for the transport of folded proteins, thereby opening up a new window for secreted products.
Our studies revealed that Tat‐targeted MBP is an extremely versatile tool for expression and purification studies. For instance, Tat export of MBP was useful as a: (i) genetic reporter for screening or optimizing Tat export (e.g. via mutations to signal peptides or the TatABC machinery), (ii) non‐enzymatic reporter of fusion protein solubility, (iii) a solubility‐enhancing fusion partner and (iv) a facile affinity partner for protein purification. As a reporter for screening putative Tat signal peptides, we tested the mal phenotype on MacConkey medium containing maltose as a more rapid (overnight versus 2–3 days) and semi‐quantitative indicator of MBP transport via the Tat system that proved to be relatively consistent with earlier analysis (Tullman‐Ercek et al., 2007).
We further examined the N‐terminal processing of secreted MBP. As export is post‐translational, properties of the mature polypeptide are important in routing the protein to the appropriate Sec apparatus and in signal peptide cleavage. For example, a simple ≥ +1 charge to the N terminus of a mature protein abolished or drastically reduced routing through the Sec pathway without affecting the ability to transit the Tat pathway (Tullman‐Ercek et al., 2007). Other studies revealed that mutations in and around the signal peptide significantly altered substrate expression levels (Pratap and Dikshit, 1998; Robbens et al., 2006). Thus, to ensure fidelity with respect to routing and processing, it is common practice to include amino acids of the mature protein following the A‐X‐A SPase I cleavage site. However, our results indicated that similar N‐terminal processing by SPaseI occurred irrespective of whether 0, 6 or 10 residues of mature TorA were included in the fusion to MBP. Interestingly, two types of sequences were obtained for each of the ssTorA–MBP constructs. The more abundant sequence corresponded to protein that was processed after the predicted ATA cleavage site while a less abundant product corresponded to cleavage at a position five amino acids upstream of the ATA cleavage site (−5). Such proteolytic processing of ssTorA was observed previously (Genest et al., 2006) and while the cause of this is currently unknown, it is possible that redundant information in the signal peptide could lead to alternative cleavage sites processed by SPase I (Fikes et al., 1990). An alternative possibility is that this arginine pair is proteolytically cleaved by a protease such as OmpT, which exhibits a strong preference for cleavage between two basic residues (Lys and, especially Arg) in the P1 and P′1 positions of the substrate (Dekker et al., 2001).
We also demonstrated MBP to be an effective non‐enzymatic protein fusion reporter of Tat transport and as such, protein solubility. This was validated by our ability to recover HS3018 cells on maltose minimal media only when these cells expressed correctly folded POIs (e.g. GST, TrxA, Top7) fused to MBP; the same cells expressing MBP fusions with misfolded or unstable POIs (e.g. PhoA, TraR) exhibited a mal‐ phenotype. A notable exception was ssTorA–GFP–MBP, which was localized in the periplasm but did not confer growth to HS3018 cells. The ssTorA–GFP–MBP localized in the periplasm was fluorescent (data not shown), thus it appears that the GFP moiety folded correctly but somehow interfered with the function of MBP. As MBP must effectively bind maltose, change conformation and then engage the MalFGK2 complex in order to confer a mal+ phenotype, we suspect that GFP interferes with one or more of these steps and renders the screen ineffective.
Finally, we employed fusions in which MBP was positioned N‐terminally to ensure the solubility of the POI and determined that the yield of material recovered from the periplasm varied for different POIs. For instance, Tat‐secreted MBP–GFP yield was 6.5‐fold greater than that recovered using Sec export, whereas the yield of Tat‐secreted MBP–scFv13.R4 was 16.8‐fold less compared with Sec. Remarkably, for MBP–scFv13.R4, the specific activity of the purified material generated using the Tat pathway was 30% greater than for material obtained using the Sec pathway. We reported a similar phenomenon for export of a truncated version of human tPA when affixed with either a Tat or a Sec signal peptide (Kim et al., 2005). While the reasons for this difference in specific activity are not currently known, we speculate that periplasmic folding of certain Sec‐targeted substrates may be inefficient, resulting in a large quantity of partially folded, inactive protein in the periplasm. On the contrary, because the Tat system usually exports correctly folded proteins (Bruser et al., 2003; DeLisa et al., 2003; Fisher et al., 2006; Richter et al., 2007), the activity of material isolated from the periplasm represents proteins that have folded completely. Thus, despite the lower expression yields for certain substrates, a potential advantage of the Tat system is that purification from the periplasm can give rise to relatively pure and highly active proteins in one step.
Experimental procedures
Bacterial strains and plasmids
All strains and plasmids used in this study are listed in Table 3. Expression of ssTorA–MBP and all related variants was from plasmid pTrc99A unless otherwise noted. The signal peptide of TorA with an additional 0, 6 or 10 residues from the beginning of the mature portion of the TorA enzyme was PCR‐amplified using E. coliMC4100 genomic DNA and cloned into vector pTrc99A, yielding pssTorA(+0)–MBP, pssTorA(+6)–MBP and pssTorA(+10)–MBP respectively. Plasmids for the expression of ssTat–MBP fusions are described elsewhere (Tullman‐Ercek et al., 2007) as are constructs for expressing ssTorA–PhoA (DeLisa et al., 2003). Plasmids pPhoA and Δss‐PhoA were constructed by PCR amplification of native E. coliPhoA or PhoA lacking residues 1–22 and insertion of the resulting PCR products into pTrc99A. For expression of different ssTorA–POI–MBP fusion proteins, DNA encoding ssTorA and MBP was cloned separately in pTrc99A, yielding plasmid pTMM. Next, genes encoding various POIs were PCR‐amplified and cloned in between ssTorA and MBP in pTMM, yielding the final ssTorA–POI–MBP expression plasmids. PCR products were generated using the following as template: MC4100 genomic DNA for TrxA, GST and PhoA; plasmid pTGS encoding GFP (DeLisa et al., 2002); plasmid DNA encoding Top7 (Kuhlman et al., 2003) and A. tumefaciens TraR (Zhu and Winans, 2001). Plasmids pMBP–GFP and pMBP–scFv13 were created by first cloning native MBP into pTrc99A followed by in‐frame insertion of the gene encoding GFP or scFv13.R4 (Martineau et al., 1998). Plasmids pssTorA–MBP–GFP, pssTorA–MBP–scFv13 and pssTorA–MBP–scFv13.R4 were constructed by inserting the gene encoding GFP, scFv13 or scFv13.R4 after ssTorA–MBP (lacking a stop codon at the C‐terminus of MBP) in pTrc99A.
Table 3.
Bacterial strains and plasmids used in this study.
| Strain or plasmid | Relevant genotype or features | Reference or source |
|---|---|---|
| MC4100 | F‐ araD139Δ(argF‐lac)U169 flbB5301 deoC1 ptsF25 relA1 rbsR22 rpsL150 thiA | Laboratory stock |
| B1LK0 | MC4100 ΔtatC | Bogsch et al. (1998) |
| HS3018 | MC4100 malT(Con)‐1ΔmalE444 | Shuman (1982) |
| HS3018 ΔtatC | HS3018 tatC::Spec | Tullman‐Ercek et al. (2007) |
| DHB4 | F′lacIqpro/λ‐ ΔlacX74 galE galK thi rpsL phoRΔphoA(PvuII) ΔmalF3 | Laboratory stock |
| FÅ113 | DHB4 gor522. . . mini‐Tn10Tet trxB::Kan ahpC* | Bessette et al. (1999) |
| 959 | AMEF lacZgene | Messer and Melchers (1978) |
| pTrc99A | trcpromoter, ColE1 ori, Ampr | Amersham Biosciences |
| pMBP | Native MBP in pTrc99A | This study |
| pssTorA(+10)–MBP | ssTorA plus residues 1–10 of mature TorA fused to Δ(1–26)MBP in pTrc99A | This study |
| pssTorA(+6)–MBP | ssTorA plus residues 1–6 of mature TorA fused to Δ(1–26)MBP in pTrc99A | This study |
| pssTorA(+0)–MBP | ssTorA fused to Δ(1–26)MBP in pTrc99A | This study |
| pssTat–MBP | ssTat‐Δ(1–26)MBP in pBAD18‐Cm where ssTat is any putative E. coliTat signal peptide | Tullman‐Ercek et al. (2007) |
| pssTorA–PhoA | ssTorA‐Δ(1–22)PhoA in pTrc99A | DeLisa et al. (2003) |
| pPhoA | Native PhoA in pTrc99A | This study |
| pΔss‐PhoA | Δ(1–22)PhoA in pTrc99A | This study |
| pssTorA–scFv13.R4 | ssTorA fused to scFv13.R4 in pTrc99A | This study |
| pssMBP–scFv13.R4 | ssMBP fused to scFv13.R4 in pTrc99A | This study |
| pTMM | ssTorA fused to Δ(1–26)MBP with a mini MCS in between the signal and MBP, cloned in pTrc99A | This study |
| pssTorA–POI–MBP | ssTorA signal peptide–POI–Δ(1–26)MBP fusion in pTrc99A | This study |
| pssTorA–MBP–GFP | ssTorA‐Δ(1–26)MBP–GFP fusion in pTrc99A | This study |
| pMBP–GFP | Native MBP fused to GFP in pTrc99A | This study |
| pssTorA–MBP‐scFv13 | ssTorA‐Δ(1–26)MBP‐scFv13 in pTrc99A | This study |
| pssTorA–MBP–scFv13.R4 | ssTorA‐Δ(1–26)MBP–scFv13.R4 in pTrc99A | This study |
| pMBP–scFv13.R4 | Native MBP fused to scFv13.R4 in pTrc99A | This study |
Cell growth and protein expression
To test growth on maltose, E. coli HS3018 or its derivatives containing plasmids encoding MBP fusions were grown overnight, diluted, plated on either MacConkey agar plates containing 0.4% maltose or M9 minimal medium containing 0.4% maltose, and incubated at 37°C overnight or 48 h respectively. For liquid cultures, cells containing pTrc99A derivatives were grown overnight and then subcultured into fresh Luria–Bertani supplemented with 100 µg ml−1 ampicillin to a starting OD600 = 0.1 followed by incubation for 2–3 h at 30°C or 37°C with shaking. When cells reached an OD600 = 0.5, IPTG was added to a final concentration of 1 mM to induce protein synthesis and cells were grown an additional 4–6 h. Antibiotic selection was maintained for all markers on plasmids at the following concentrations: ampicillin, 100 µg ml−1 and chloramphenicol, 25 µg ml−1.
Cell fractionation
An equivalent number of cells were harvested following recombinant protein induction, pelleted by centrifugation and fractionated into cytoplasmic and periplasmic fractions by the ice‐cold osmotic shock procedure (Bogsch et al., 1998; DeLisa et al., 2003). The protocol was modified for the expression of disulfide bond‐containing proteins (e.g. PhoA) by treating intact cells with 100 mM iodoacetamide for 30 min prior to centrifugation to prevent spontaneous activation of free thiols (Derman and Beckwith, 1995). To analyse total cellular proteins, collected cells were centrifuged, re‐suspended in cold PBS containing 100 mM iodoacetamide and lysed by sonication. The insoluble fractions were removed by centrifugation (12 000 g, 10 min, 4°C), and soluble protein was quantified by the Bio‐Rad protein assay with BSA as standard. The quality of all fractionation experiments was confirmed by analysing the localization of a cytoplasmic marker protein, namely GroEL, using Western blot analysis.
Quantitative RT‐PCR
For RNA expression analysis, total RNA was extracted from cells using an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. The RNA extracted was digested with RNase‐free DNaseI for 15 min to remove contaminating DNA. The total mRNA of native PhoA, ssTorA–PhoA and Δss‐PhoA was detected by RT‐PCR using TaqMan One‐Step RT‐PCR Master Mix Reagents Kit (Applied Biosystems). Two microlitres of total RNA was used as a template for the amplification reaction. Probes and primer sets for PhoA were designed using Primer Express software (Applied Biosystems). The primer sequences specific for PhoA were: (i) forward: 5′‐AAAGGCGGAAAAGGATCGAT‐3′ and (ii) reverse: 5′‐GCCGCCAAGCGTAACATC‐3′; and the probe sequence specific for PhoA was: 6FAM‐CCGAACAGCTGCT TAACGCTCGTG‐TAMRA. For normalizing purposes, primer and probe sequences specific for 16s rRNA were also designed: (i) forward: 5′‐CCAGCAGCCGCGGTAAT‐3′ and (ii) reverse: 5′‐TGCGCTTTACGCCCAGTAAT‐3′; and the probe specific for 16srRNA sequence was: 6FAM‐CCGATTAACG CTTGCACCCTCCG‐TAMRA. The efficiency of the primers for amplification of both PhoA and 16srRNA sequences was tested by electrophoresis before performing the RT‐PCR. The programme of RT‐PCR was 50°C (2 min), 95°C (10 min), followed by 40 cycles of 95°C (15 s), 60°C (1 min).
Protein analysis
Western blotting was performed as previously described (DeLisa et al., 2003). All lanes of SDS‐12% PAGE were loaded with samples generated from an equivalent number of cells harvested from each experiment. The following primary antibodies were used: monoclonal mouse anti‐MBP (Sigma), monoclonal mouse anti‐PhoA (Sigma) and polyclonal rabbit anti‐GroEL (Sigma). The secondary antibody was goat anti‐mouse or goat anti‐rabbit horseradish peroxidase. All primary and secondary antibodies were diluted according to manufacturer's specifications. Membranes were first probed with primary and secondary antibodies and, following development, were stripped in Tris‐buffered saline/2% SDS/0.7 M β‐mercaptoethanol. Stripped membranes were re‐blocked and probed with anti‐GroEL antibody. PhoA activity was measured as described previously (Derman et al., 1993) while GFP activity was measured according to DeLisa and colleagues (2002). The scFv13 activity was quantified by measuring in vitro activation of a normally inactive β‐galactosidase variant known as AMEF β‐gal (Martineau et al., 1998). Briefly, lysate was prepared from 50 ml of cultures of E. colistrain AMEF 959 (Martineau et al., 1998) that express AMEF β‐gal. Next, 10 µl of lysate containing AMEF β‐gal was incubated with 10 µl of a purified scFv13.R4 chimera (e.g. ssTorA–MBP–scFv13.R4) for 3 h at 37°C in a 96‐well plate. To this, 100 µl of substrate (o‐nitrophenyl‐β‐D‐galactoside in Z buffer) was added to each well and incubated for 10 min at 37°C with shaking. The reaction was stopped by the addition of 50 µl of 1 M Na2CO3 and absorbance readings at 420 nm were recorded.
Purification and N‐terminal sequencing
For purification of his‐tagged proteins, standard imac was used according to manufacturer's specifications (Qiagen). For purification of all MBP constructs, periplasmic fractions were isolated from 50 ml bacterial cultures as described above and added to amylose affinity chromatography columns. Columns were prepared by adding 2 ml of amylase resin (New England Biolabs) to empty columns and washing three times with 5 ml of MBP buffer [20 mM Tris (pH 7.4), 200 mM NaCl, 1 mM EDTA, 10 mM 2‐mercaptoethanol]. Following addition of periplasmic fractions, columns were washed three times with 5 ml of MBP buffer. Bound proteins were eluted from the resin with elution buffer (MBP buffer + 10 mM Maltose). For N‐terminal sequencing, purified samples were separated using standard SDS‐PAGE protocols and electroblotted according to standard protocols. Briefly, proteins were transferred to polyvinyldifluoride (PVDF) membranes from SDS‐PAGE. Following Coomassie Blue staining of PVDF membranes, bands of interest corresponding in size to ssTorA–MBP or native MBP were excised with clean razor blades, transferred to sterile microcentrifuge tubes and stored at −70oC until sent to the Columbia University Medical Center Protein Core Facility for N‐terminal sequencing.
Acknowledgments
The authors would like to thank David Baker (Top7), Pierre Martineau (scFv13 and scFv13.R4) and Steve Winans (TraR) for plasmid DNA used in this study and Tracy Palmer and Howard Shuman for strains B1LK0 and HS3018 respectively. Funding was provided by an NSF CAREER Award (CBET #0449080) and a NYSTAR James D. Watson Award (both to M.P.D.).
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Phenotype and intracellular localization of Tattargeted MBP. (a) HS3018 (malE-negative derivative of E. coli MC4100) and HS3018 DtatC cells expressing ssTorAMBP plated as individual colonies on MacConkey (top) and M9 minimal (bottom) agar media supplemented with 0.4% maltose. (b) Western blotting of the cytoplasmic (cyt) and periplasmic (per) lysates of the above cells probed with anti-MBP primary antibody (top). Anti-GroEL primary antibody (bottom) was also used as a control to probe the fidelity of fractionation.
References
- Alami M., Trescher D., Wu L.F., Muller M. Separate analysis of twin‐arginine translocation (Tat)‐specific membrane binding and translocation in Escherichia coli. J Biol Chem. 2002;277:20499–20503. doi: 10.1074/jbc.M201711200. [DOI] [PubMed] [Google Scholar]
- Bach H., Mazor Y., Shaky S., Shoham‐Lev A., Berdichevsky Y., Gutnick D.L., Benhar I. Escherichia coli maltose‐binding protein as a molecular chaperone for recombinant intracellular cytoplasmic single‐chain antibodies. J Mol Biol. 2001;312:79–93. doi: 10.1006/jmbi.2001.4914. [DOI] [PubMed] [Google Scholar]
- Baneyx F. Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol. 1999;10:411–421. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- Baneyx F., Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22:1399–1408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- Barrett C.M., Ray N., Thomas J.D., Robinson C., Bolhuis A. Quantitative export of a reporter protein, GFP, by the twin‐arginine translocation pathway in Escherichia coli. Biochem Biophys Res Commun. 2003;304:279–284. doi: 10.1016/s0006-291x(03)00583-7. [DOI] [PubMed] [Google Scholar]
- Bendtsen J.D., Nielsen H., Widdick D., Palmer T., Brunak S. Prediction of twin‐arginine signal peptides. BMC Bioinformatics. 2005;6 doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson S.A., Bremer E., Silhavy T.J. Intragenic regions required for LamB export. Proc Natl Acad Sci USA. 1984;81:3830–3834. doi: 10.1073/pnas.81.12.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berks B.C. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- Berks B.C., Sargent F., De Leeuw E., Hinsley A.P., Stanley N.R., Jack R.L. A novel protein transport system involved in the biogenesis of bacterial electron transfer chains. Biochim Biophys Acta. 2000a;1459:325–330. doi: 10.1016/s0005-2728(00)00168-7. et al. [DOI] [PubMed] [Google Scholar]
- Berks B.C., Sargent F., Palmer T. The Tat protein export pathway. Mol Microbiol. 2000b;35:260–274. doi: 10.1046/j.1365-2958.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- Berks B.C., Palmer T., Sargent F. The Tat protein translocation pathway and its role in microbial physiology. Adv Microb Physiol. 2003;47:187–254. doi: 10.1016/s0065-2911(03)47004-5. [DOI] [PubMed] [Google Scholar]
- Bessette P.H., Aslund F., Beckwith J., Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA. 1999;96:13703–13708. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaudeck N., Sprenger G.A., Freudl R., Wiegert T. Specificity of signal peptide recognition in tat‐dependent bacterial protein translocation. J Bacteriol. 2001;183:604–610. doi: 10.1128/JB.183.2.604-610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaudeck N., Kreutzenbeck P., Freudl R., Sprenger G.A. Genetic analysis of pathway specificity during posttranslational protein translocation across the Escherichia coli plasma membrane. J Bacteriol. 2003;185:2811–2819. doi: 10.1128/JB.185.9.2811-2819.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogsch E.G., Sargent F., Stanley N.R., Berks B.C., Robinson C., Palmer T. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem. 1998;273:18003–18006. doi: 10.1074/jbc.273.29.18003. [DOI] [PubMed] [Google Scholar]
- Bruser T. The twin‐arginine translocation system and its capability for protein secretion in biotechnological protein production. Appl Microbiol Biotechnol. 2007;76:35–45. doi: 10.1007/s00253-007-0991-z. [DOI] [PubMed] [Google Scholar]
- Bruser T., Yano T., Brune D.C., Daldal F. Membrane targeting of a folded and cofactor‐containing protein. Eur J Biochem. 2003;270:1211–1221. doi: 10.1046/j.1432-1033.2003.03481.x. [DOI] [PubMed] [Google Scholar]
- Chen G., Hayhurst A., Thomas J.G., Harvey B.R., Iverson B.L., Georgiou G. Isolation of high‐affinity ligand‐binding proteins by periplasmic expression with cytometric screening (PECS) Nat Biotechnol. 2001;19:537–542. doi: 10.1038/89281. [DOI] [PubMed] [Google Scholar]
- Cline K., McCaffery M. Evidence for a dynamic and transient pathway through the TAT protein transport machinery. EMBO J. 2007;26:3039–3049. doi: 10.1038/sj.emboj.7601759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisa M.P., Samuelson P., Palmer T., Georgiou G. Genetic analysis of the twin arginine translocator secretion pathway in bacteria. J Biol Chem. 2002;277:29825–29831. doi: 10.1074/jbc.M201956200. [DOI] [PubMed] [Google Scholar]
- DeLisa M.P., Tullman D., Georgiou G. Folding quality control in the export of proteins by the bacterial twin‐arginine translocation pathway. Proc Natl Acad Sci USA. 2003;100:6115–6120. doi: 10.1073/pnas.0937838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisa M.P., Lee P., Palmer T., Georgiou G. Phage shock protein PspA of Escherichia coli relieves saturation of protein export via the Tat pathway. J Bacteriol. 2004;186:366–373. doi: 10.1128/JB.186.2.366-373.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker N., Cox R.C., Kramer R.A., Egmond M.R. Substrate specificity of the integral membrane protease OmpT determined by spatially addressed peptide libraries. Biochemistry. 2001;40:1694–1701. doi: 10.1021/bi0014195. [DOI] [PubMed] [Google Scholar]
- Derman A.I., Beckwith J. Escherichia coli alkaline phosphatase localized to the cytoplasm slowly acquires enzymatic activity in cells whose growth has been suspended: a caution for gene fusion studies. J Bacteriol. 1995;177:3764–3770. doi: 10.1128/jb.177.13.3764-3770.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman A.I., Puziss J.W., Bassford P.J., Jr., Beckwith J. A signal sequence is not required for protein export in prlA mutants of Escherichia coli. EMBO J. 1993;12:879–888. doi: 10.1002/j.1460-2075.1993.tb05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilks K., Rose R.W., Hartmann E., Pohlschroder M. Prokaryotic utilization of the twin‐arginine translocation pathway: a genomic survey. J Bacteriol. 2003;185:1478–1483. doi: 10.1128/JB.185.4.1478-1483.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen A.J., Manting E.H., Van Der Does C. The structural basis of protein targeting and translocation in bacteria. Nat Struct Biol. 2001;8:492–498. doi: 10.1038/88549. [DOI] [PubMed] [Google Scholar]
- Feilmeier B.J., Iseminger G., Schroeder D., Webber H., Phillips G.J. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol. 2000;182:4068–4076. doi: 10.1128/jb.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fikes J.D., Barkocy‐Gallagher G.A., Klapper D.G., Bassford P.J., Jr Maturation of Escherichia coli maltose‐binding protein by signal peptidase I in vivo. Sequence requirements for efficient processing and demonstration of an alternate cleavage site. J Biol Chem. 1990;265:3417–3423. [PubMed] [Google Scholar]
- Fisher A.C., Kim W., DeLisa M.P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin‐arginine translocation pathway. Protein Sci. 2006;15:449–458. doi: 10.1110/ps.051902606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco J.A., Campbell R., Iverson B.L., Georgiou G. Production and fluorescence‐activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc Natl Acad Sci USA. 1993;90:10444–10448. doi: 10.1073/pnas.90.22.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier C., Li H., Morosoli R. Increase in xylanase production by Streptomyces lividans through simultaneous use of the Sec‐ and Tat‐dependent protein export systems. Appl Environ Microbiol. 2005;71:3085–3092. doi: 10.1128/AEM.71.6.3085-3092.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest O., Seduk F., Ilbert M., Mejean V., Iobbi‐Nivol C. Signal peptide protection by specific chaperone. Biochem Biophys Res Commun. 2006;339:991–995. doi: 10.1016/j.bbrc.2005.11.107. [DOI] [PubMed] [Google Scholar]
- Gentz R., Kuys Y., Zwieb C., Taatjes D., Taatjes H., Bannwarth W. Association of degradation and secretion of three chimeric polypeptides in Escherichia coli. J Bacteriol. 1988;170:2212–2220. doi: 10.1128/jb.170.5.2212-2220.1988. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou G., Segatori L. Preparative expression of secreted proteins in bacteria: status report and future prospects. Curr Opin Biotechnol. 2005;16:538–545. doi: 10.1016/j.copbio.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Di Guan C., Li P., Riggs P.D., Inouye H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose‐binding protein. Gene. 1988;67:21–30. doi: 10.1016/0378-1119(88)90004-2. [DOI] [PubMed] [Google Scholar]
- Harvey B.R., Georgiou G., Hayhurst A., Jeong K.J., Iverson B.L., Rogers G.K. Anchored periplasmic expression, a versatile technology for the isolation of high‐affinity antibodies from Escherichia coli‐expressed libraries. Proc Natl Acad Sci USA. 2004;101:9193–9198. doi: 10.1073/pnas.0400187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayhurst A., Georgiou G. High‐throughput antibody isolation. Curr Opin Chem Biol. 2001;5:683–689. doi: 10.1016/s1367-5931(01)00266-6. [DOI] [PubMed] [Google Scholar]
- Kapust R.B., Waugh D.S. Escherichia coli maltose‐binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiino D.R., Silhavy T.J. Mutation prlF1 relieves the lethality associated with export of beta‐galactosidase hybrid proteins in Escherichia coli. J Bacteriol. 1984;158:878–883. doi: 10.1128/jb.158.3.878-883.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.‐Y., Fogarty E.A., Lu F.J., Zhu H., Henderson L.A., DeLisa M.P. Twin‐arginine translocation of active human tissue plasminogen activator in Escherichia coli. Appl Environ Microbiol. 2005;71:8451–8459. doi: 10.1128/AEM.71.12.8451-8459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L., Baker D. Design of a novel globular protein fold with atomic‐level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- Li S.Y., Chang B.Y., Lin S.C. Coexpression of TorD enhances the transport of GFP via the TAT pathway. J Biotechnol. 2006;122:412–421. doi: 10.1016/j.jbiotec.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Martineau P., Jones P., Winter G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 1998;280:117–127. doi: 10.1006/jmbi.1998.1840. [DOI] [PubMed] [Google Scholar]
- Messer W., Melchers F. The activation of mutant beta‐galactosidase by specific antibodies. In: Miller J.H., Reznikoff W.S., editors. Cold Spring Harbor Laboratory Press; 1978. pp. 305–315. [Google Scholar]
- Perez‐Rodriguez R., Fisher A.C., Perlmutter J.D., Hicks M.G., Chanal A., Santini C.L. An essential role for the DnaK molecular chaperone in stabilizing over‐expressed substrate proteins of the bacterial twin‐arginine translocation pathway. J Mol Biol. 2007;367:715–730. doi: 10.1016/j.jmb.2007.01.027. et al. [DOI] [PubMed] [Google Scholar]
- Pimienta E., Ayala J.C., Rodriguez C., Ramos A., Van Mellaert L., Vallin C., Anne J. Recombinant production of Streptococcus equisimilis streptokinase by Streptomyces lividans. Microb Cell Fact. 2007;6:20. doi: 10.1186/1475-2859-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J., Dikshit K.L. Effect of signal peptide changes on the extracellular processing of streptokinase from Escherichia coli: requirement for secondary structure at the cleavage junction. Mol Gen Genet. 1998;258:326–333. doi: 10.1007/s004380050738. [DOI] [PubMed] [Google Scholar]
- Punginelli C., Ize B., Stanley N.R., Stewart V., Sawers G., Berks B.C., Palmer T. mRNA secondary structure modulates translation of Tat‐dependent formate dehydrogenase N. J Bacteriol. 2004;186:6311–6315. doi: 10.1128/JB.186.18.6311-6315.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribnicky B., Van Blarcom T., Georgiou G. A scFv antibody mutant isolated in a genetic screen for improved export via the twin arginine transporter pathway exhibits faster folding. J Mol Biol. 2007;369:631–639. doi: 10.1016/j.jmb.2007.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter S., Lindenstrauss U., Lucke C., Bayliss R., Bruser T. Functional Tat transport of unstructured, small, hydrophilic proteins. J Biol Chem. 2007;282:33257–33264. doi: 10.1074/jbc.M703303200. [DOI] [PubMed] [Google Scholar]
- Robbens J., De Coen W., Fiers W., Remaut E. Improved periplasmic production of biologically active murine interleukin‐2 in Escherichia coli through a single aminoacid change at the cleavage site. Proc Biochem. 2006;41:1343–1346. [Google Scholar]
- Robinson C., Bolhuis A. Protein targeting by the twin‐arginine translocation pathway. Nat Rev Mol Cell Biol. 2001;2:350–356. doi: 10.1038/35073038. [DOI] [PubMed] [Google Scholar]
- Rodrigue A., Chanal A., Beck K., Muller M., Wu L.F. Co‐translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J Biol Chem. 1999;274:13223–13228. doi: 10.1074/jbc.274.19.13223. [DOI] [PubMed] [Google Scholar]
- Schaerlaekens K., Lammertyn E., Geukens N., De Keersmaeker S., Anne J., Van Mellaert L. Comparison of the Sec and Tat secretion pathways for heterologous protein production by Streptomyces lividans. J Biotechnol. 2004;112:279–288. doi: 10.1016/j.jbiotec.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Schatz G., Dobberstein B. Common principles of protein translocation across membranes. Science. 1996;271:1519–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- Schierle C.F., Berkmen M., Huber D., Kumamoto C., Boyd D., Beckwith J. The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J Bacteriol. 2003;185:5706–5713. doi: 10.1128/JB.185.19.5706-5713.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settles A.M., Yonetani A., Baron A., Bush D.R., Cline K., Martienssen R. Sec‐independent protein translocation by the maize Hcf106 protein. Science. 1997;278:1467–1470. doi: 10.1126/science.278.5342.1467. [DOI] [PubMed] [Google Scholar]
- Shuman H.A. Active transport of maltose in Escherichia coli K12. Role of the periplasmic maltose‐binding protein and evidence for a substrate recognition site in the cytoplasmic membrane. J Biol Chem. 1982;257:5455–5461. [PubMed] [Google Scholar]
- Smith G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Sone M., Kishigami S., Yoshihisa T., Ito K. Roles of disulfide bonds in bacterial alkaline phosphatase. J Biol Chem. 1997;272:6174–6178. doi: 10.1074/jbc.272.10.6174. [DOI] [PubMed] [Google Scholar]
- Strauch E.M., Georgiou G. A bacterial two‐hybrid system based on the twin‐arginine transporter pathway of E. coli. Protein Sci. 2007;16:1001–1008. doi: 10.1110/ps.062687207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz J.R. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotechnol. 2001;12:195–201. doi: 10.1016/s0958-1669(00)00199-3. [DOI] [PubMed] [Google Scholar]
- Thomas J.D., Daniel R.A., Errington J., Robinson C. Export of active green fluorescent protein to the periplasm by the twin‐arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol. 2001;39:47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- Tullman‐Ercek D., DeLisa M.P., Kawarasaki Y., Iranpour P., Ribnicky B., Palmer T., Georgiou G. Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J Biol Chem. 2007;282:8309–8316. doi: 10.1074/jbc.M610507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg B., Clemons W.M., Modis, Collinson I., Hartmann Y., Harrison E. X‐ray structure of a protein‐conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. et al. [DOI] [PubMed] [Google Scholar]
- Weiner J.H., Bilous P.T., Shaw G.M., Lubitz S.P., Frost L., Thomas G.H. A novel and ubiquitous system for membrane targeting and secretion of cofactor‐containing proteins. Cell. 1998;93:93–101. doi: 10.1016/s0092-8674(00)81149-6. et al. [DOI] [PubMed] [Google Scholar]
- Zhu J., Winans S.C. The quorum‐sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci USA. 2001;98:1507–1512. doi: 10.1073/pnas.98.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotype and intracellular localization of Tattargeted MBP. (a) HS3018 (malE-negative derivative of E. coli MC4100) and HS3018 DtatC cells expressing ssTorAMBP plated as individual colonies on MacConkey (top) and M9 minimal (bottom) agar media supplemented with 0.4% maltose. (b) Western blotting of the cytoplasmic (cyt) and periplasmic (per) lysates of the above cells probed with anti-MBP primary antibody (top). Anti-GroEL primary antibody (bottom) was also used as a control to probe the fidelity of fractionation.