

Abstract
Pluripotency of embryonic stem (ES) cells is controlled by defined transcription factors1,2. During differentiation, mouse ES cells undergo global epigenetic reprogramming, as exemplified by X-chromosome inactivation (XCI) whereby one female X-chromosome is silenced to achieve gene dosage parity between the sexes3-5. Somatic XCI is regulated by homologous X-chromosome pairing6,7, counting8-10, and random choice of future active X (Xa) and inactive X’s. XCI and cell differentiation are tightly coupled11, as blocking one process compromises the other8,12 and dedifferentiation of somatic cells to induced pluripotent stem (iPS) cells is accompanied by X-reactivation2. Recent evidence suggests coupling of Xist expression to pluripotency factors13, but how the two are interconnected remains unknown. Here, we show that the Oct414 lies at the top of the XCI hierarchy and regulates XCI by triggering X-chromosome pairing and counting. Oct4 directly binds Tsix and Xite, two regulatory ncRNA genes of the X-inactivation center15,16, and also complexes with XCI trans-factors, Ctcf and Yy117, through protein-protein interactions. Depletion of Oct4 blocks homologous X-chromosome pairing and results in inactivation of both Xs in female cells. Thus, we have identified the first trans-factor that regulates counting and ascribed novel functions to Oct4 during X-chromosome reprogramming.
XCI has been reliably modeled in female mouse ES cells, which carry two Xa’s but will inactivate one when induced to differentiate into embryoid bodies (EB) ex vivo. Given tight linkage between XCI and differentiation2,8,11,12, XCI must in principle be regulated by factors that control pluripotency. Here, using bioinformatic analysis, we have surveyed the X-inactivation center (Xic) for pluripotency factor binding sites and found one Sox2 and two Oct4 motifs in a 15-kb region of Tsix and Xite 6,8,9 that contains two enhancers for Tsix, the antisense repressor of Xist18 (Fig. 1a,b). One Oct4 site occurs within the bipartite enhancer, adjacent to Ctcf-Yy1 binding motif, ‘Site E’, and ~1.0 kb downstream of Ctcf-Yy1 ‘Site D’ 17,19. Within Xite, a composite Oct4-Sox2 site resides within DNaseI hypersensitive sites of the 1.2-kb enhancer16,18.
Figure 1. Oct4 and Sox2 bind Tsix and Xite.
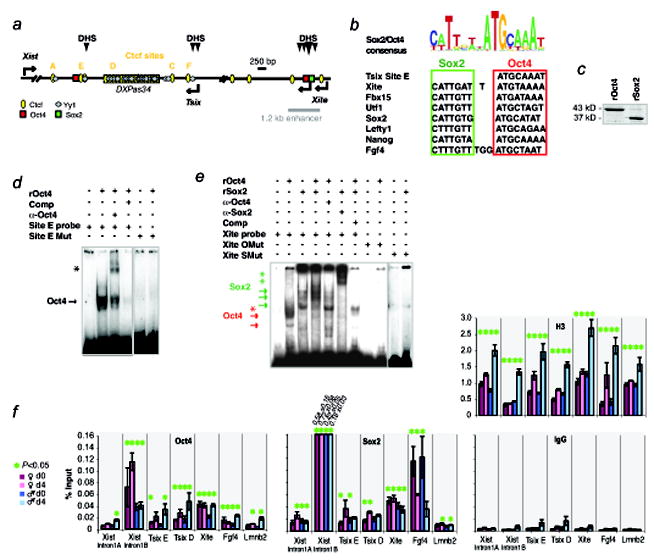
a, Map of Ctcf, Yy1, Oct4, and Sox2 sites within the 15-kb critical region of the mouse Xic responsible for counting, choice, and pairing.
b, The Sox2/Oct4 consensus28 and representative binding motifs within Tsix, Xite, and ES-specific genes, Fbx15, Utf1, Sox2, Lefty1, Nanog, and Fgf4.
c, Coomassie staining of purified recombinant Oct4 and Sox2 proteins (rOct4, rSox2).
d, Gel shift of Tsix site E motif using rOct4. Arrow, rOct4-DNA shift. Asterisk, supershifted rOct4-DNA. Comp, cold competitor added at 100x-fold molar excess.
e, Gel shift of the Xite motif using rOct4 and rSox2. Arrows, protein-DNA shifts. Asterisks, corresponding supershifts.
f, qChIP analysis of Oct4, Sox2, and positive control H3 at designated sites in d0 (day 0) and d4 (day 4) wildtype male and female ES cells. Averages of three independent biological replicates shown with standard error of the mean (S.E.M). Intron 1B results for Sox2 were off-scale, with actual averages +/- S.E.M shown. Statistical significance (P) of each result is calculated against the control IgG ChIP (background) using a paired, one-tailed t-test. *, P<0.05.
In electrophoretic mobility shift assays (EMSAs) using purified recombinant Oct4 protein (rOct4) and P32-labelled Tsix and Xite oligo-probes, we observed specific protein-DNA complexes that were supershifted by α-Oct4 antibodies (Fig. 1c,d,e). The interactions were competed away by excess cold oligos and were not detected when mutated probes were used. In contrast, recombinant Sox2 protein (rSox2) did not bind Tsix (data not shown) but specifically bound Xite (Fig. 1c,e). Mixing rSox2 and rOct4 together with Xite resulted in a further mobility shift, suggesting that the pluripotency factors co-occupied the Xite motif. The Sox2-Xite complex was supershifted by α-Sox2 antibodies, competed away by excess cold oligos, and abolished by Xite mutation. These results indicated that Oct4 and Sox2 specifically bind Tsix and Xite in vitro.
To investigate in vivo binding, we performed quantitative ChIP and observed binding at sites predicted by bioinformatic analysis and verified by EMSA (Fig. 1f). In both male and female ES cells, both Oct4 and Sox2 bound Tsix and Xite chromatin above background (IgG control ChIP). Because EMSA did not reveal direct binding of Sox2 to Tsix, the Sox2-Tsix complex may occur indirectly via known looping interactions between Tsix and Xite20. Although Xist intron 1B13 showed greatest pulldown by Oct4 and Sox2, ChIP levels at Tsix and Xite were comparable to that for positive control Fgf4. Binding to the unrelated Lmnb2 locus was low, as was binding to a control region ~600 bp upstream of intron 1B (intron 1A). These data demonstrated that Xite strongly binds Oct4 and Sox2 and that Tsix binds Oct4 in vivo.
Curiously, Oct4 and Sox2 sites occur near previously defined sites for Ctcf19,21,22 and Yy117,23,24 (Fig. 1a). Ctcf regulates X-chromosome pairing25 and, together with Yy1, controls XCI choice by transactivating Tsix17. Given Oct4 and Sox2’s proximity to Ctcf-Yy1 sites, we carried out GST pull-down experiments to determine if the factors interact. S35-labeled Oct4 bound GST-fused Ctcf but not Yy1 (Fig. 2a). To map binding domains, we tested Ctcf-GST fusions for binding to S35-labeled Oct4 and observed interaction through Ctcf’s Zn-finger region (aa284-583). To map Oct4 domains, we co-transfected Flag-Oct4 fusions with full-length Ctcf, co-immunoprecipitated (co-IP) with α-Flag antibodies, and observed interaction through Oct4’s N-terminus (Fig. 2b). Reciprocal native co-IPs in ES cells confirmed endogenous Ctcf-Oct4 interactions (Fig. 2c). We concluded that Oct4 and Ctcf interact in vitro and in vivo.
Figure 2. Oct4-Ctcf and Sox2-Yy1 interactions, and Tsix transcriptional activation.
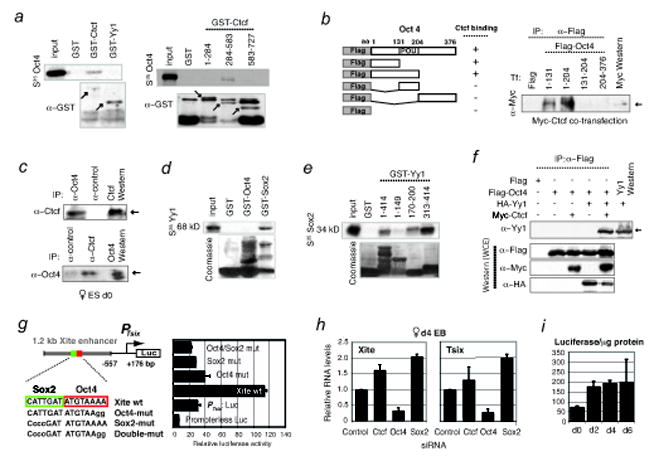
a, Testing mammalian GST fusions of full-length Yy1 and Ctcf or Ctcf domains (amino acids indicated) for interaction with S35-labelled Oct4. Lower panels, α–GST Western control. Arrows, specific protein fusions.
b, HEK cells were cotransfected with myc-Ctcf and indicated Flag-tagged Oct4 fragments. Whole cell extracts (WCE) were immunoprecipiated (IP) with α-Flag antibodies prior to Western blotting with α-myc antibodies. Arrow, Ctcf protein.
c, Reciprocal co-IP: Left, IP with α-Oct4 or control antibodies to test interaction with endogenous Ctcf; arrow, Ctcf detected by α-Ctcf Western analysis. Right, IP with α-Ctcf or control antibodies to test interaction with endogenous Oct4; arrow, Oct4 detected by α-Oct4 Western. Doublet bands and mobility differences may reflect isoforms due to post-translational modifications.
d, Yy1 (S35-labelled) binding to Sox2 but not Oct4 revealed by GST fusion analysis. Bottom, Coomassie staining to reveal GST fusion proteins.
e, Domain mapping of Yy1 binding to Sox2. Mobility differences may reflect post-translational modifications.
f, Overexpression and co-IP of Flag-Oct4, Myc-Ctcf, and HA-Yy1 in indicated combinations in HEK cells. Two 6-well plates of HEK cells were transfected with 8 μg of tagged constructs and harvested 35 hrs later for coIP analysis. Western blots of the co-IP (top panel) and whole cell extracts (bottom 3 panels) were performed using indicated antibodies.
g, Oct4 and Sox2 confer developmental specificity to Xite enhancer. Map of luciferase expression vector fused to the Tsix major promoter (PTsix) and the 1.2 kb enhancer. Mutations of the Oct4/Sox2 motifs in the enhancer compromised PTsix activity in transiently transfected male d0 ES cells. Error bars, 1 SD.
h, qRT-PCR for Xite and Tsix RNA after knocking down the indicated factors. Levels are normalized to β-actin levels. Error bars, 1SD.
i, Luciferase analysis of cells carrying the wildtype enhancer shows enhanced PTsix activity upon differentiation. Cells were differentiated in duplicate and luciferase levels were normalized to total protein levels.
Next, we tested if Sox2 directly interacts with Ctcf or Yy1. In GST-pulldown assays, S35-labeled Yy1 bound GST-Sox2 but not GST-Oct4 (Fig. 2d). Reciprocally, S35-labeled Sox2 bound full-length GST-Yy1 (Fig. 2e). Domain mapping showed that this interaction occurred through Yy1’s Zn-finger (aa 313-414) and HDAC domain (aa170-200). These data showed that Yy1 directly binds Sox2, whereas Oct4 directly binds Ctcf. Given that Yy1 interacts with Ctcf17 and Ctcf interacts with Oct4, we asked if Yy1 could indirectly interact with Oct4 by overexpressing tagged proteins in HEK cells. When Flag-tagged Oct4 was co-transfected with HA-tagged Yy1 alone, Flag-Oct4 did not efficiently co-IP Yy1; however, when Flag-Oct4 and HA-Yy1 were co-transfected with Myc-tagged Ctcf, Flag-Oct4 readily co-IP’d Yy1 (Fig. 2f; endogenous proteins were likely expressed at insufficient levels for detection). These results argued that Oct4 forms a multifactor complex in vivo and interacts with Yy1 through Ctcf. The complex may include Sox2 as well, as it interacts with Oct4 and Yy1.
Because of Ctcf and Yy1’s transcriptional roles at Tsix17 and Tsix’s unique temporal expression pattern15, the Oct4-Sox2 sites may also transactivate Tsix and regulate XCI through their intrinsic developmental specificity. Indeed, Oct4 and Sox2 were downregulated contemporaneously with Xite and Tsix, correlating with the upregulation of Xist (Fig. S1). Luciferase reporter assays showed that, whereas the 1.2-kb enhancer stimulated Tsix promoter activity almost 4-fold in ES cells, mutating the Oct4/Sox2 motifs abolished stimulation (Fig. 2g), supporting Oct4 and Sox2’s role in Tsix transactivation. Consistent with this, siRNA knockdown of Oct4 significantly reduced Tsix and Xite RNA levels in female ES cells (Fig. 2h). By contrast, knocking down Ctcf did not compromise expression, and knocking down Sox2 slightly increased RNA levels in this context (Sox2 may play a less crucial role than Oct4). Thus, Oct4 is required to transactivate Tsix in vivo. Previous genetic analysis suggested that the Xite enhancer is active in d0 cells, but acts principally during differentiation to maintain Tsix expression on one X and thereby ensure selection of only one Xa16. In support of this, stimulation of the Tsix-reporter fusion is enhanced ~3-fold from d2 to d6, the timeframe during which choice takes place (Fig. 2i). We propose that Oct4 transactivates Tsix/Xite and that its binding directly controls developmental timing of XCI.
What mechanistic aspects of XCI might Oct4 regulate? Given that X-chromosome pairing is one of the earliest events of XCI6,7 and that Oct4 sites occur within the Tsix/Xite pairing region6,25, we investigated whether Oct4 controlled pairing. We knocked down Oct4 by siRNA nucleofection of d2 female ES cells, harvested EB on d4, confirmed protein depletion (Fig. 3a), verified initiation of differentiation (Fig. S2), and measured normalized distances (ND) between the two Xic’s (Fig. 3b,c). In control d4 cells, ~22% of nuclei showed inter-Xic distances of < 0.1 ND (<1.0μ approximately). Oct4-knockdown cells displayed a significant reduction in such interactions, as < 7% of nuclei demonstrated ND <0.1 (Fig. 3c,d). The extent of reduction was similar to that observed in Ctcf knockdowns, a protein known to be required for pairing25. In contrast, knocking down Sox2 had no obvious effect on pairing – supporting the idea that, while Sox2 binds Xite and can transactivate Tsix, it may be functionally redundant with Oct4 and play a lesser role during XCI. We concluded that Oct4 is essential for X-X pairing.
Figure 3. Oct4 knockdown disrupts X-X pairing and results in aberrant Xist expression in female ES cells.
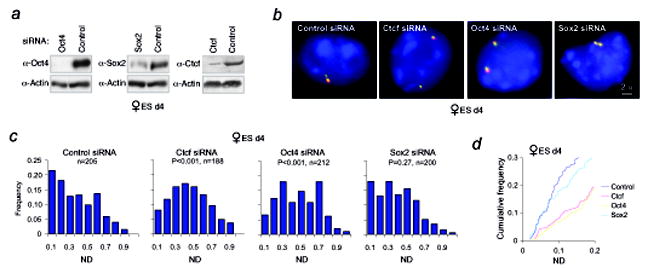
a, Western analysis confirms knockdown of indicated proteins in wildtype female ES cells nucleofected with siRNAs on d2 and harvested on d4.
b, Representative images of Xic –Xic localization in knockdown cells using a two-probe combination of pSxn (Tsix, green) and pSx9 (Xist, red). To ensure signal specificity, only those foci with overlapping red and green signals are scored.
c, Distribution of X-X distances in knockdown cells as indicated. n, nuclei counted. ND= Xic-Xic distance/d, where d = 2 × (nuclear area/π)0.5. The significance of the difference (P) between samples and control siRNA was calculated using the Kolmogorov-Smirnov (KS) test, a non-parametric test to determine whether two data-sets have a similar distribution (SPSS 13.0 software).
d, Cumulative frequency curves of the analysis carried out in panel c. ND 0.0-0.2 are shown.
Because pairing has been proposed to regulate counting and choice6,7, we next addressed whether Oct4 impacts these processes. In male cells, knocking down Oct4, Sox2, and Ctcf had no obvious effect on Xist expression during differentiation into EB (Fig. 4a,b). This result differs from a recent study reporting ectopic Xist expression when a tet-controlled, overexpressed Oct4 transgene is downregulated in male ES cells13. The difference might be attributed to experimental variation, such as use of different cell lines, methods of Oct4 repression, and methods of ES differentiation. Intriguingly, however, we observed an aberrant number of Xist RNA foci in female EB when Oct4 was knocked down (Fig. 4c). Not only did Oct4-deficient cells show a higher frequency of Xist upregulation but ~20% of Xist+ nuclei showed biallelic expression (Fig. 4c,d). Biallelic expression was not observed in control, Ctcf-knockdown, and Sox2-knockdown cells (Fig. 4c,d and data not shown). We excluded aneuploidy as a cause of two Xist foci, as DNA FISH -- carried out on the same nuclei -- demonstrated two Xic and two Chr.1 DNA signals and thereby confirmed 2X’s in a diploid background (Fig. 4c).
Figure 4. Ectopic Xist expression from both Xs in Oct4-deficient ES cells.
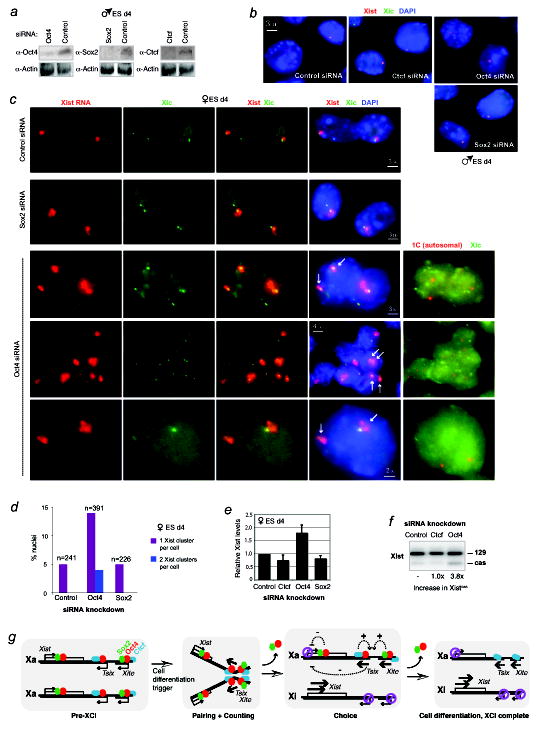
a, Western analysis confirms knockdown of indicated proteins in wildtype male ES cells nucleofected on d2 and harvested on d4.
b, RNA/DNA FISH analysis for Xist RNA (red) and Xic DNA (green) after indicated knockdowns in d4 male ES cells.
c, RNA/DNA FISH shows ectopic Xist upregulation in Oct4- but not Sox2- or control- female knockdown cells on d4. Three representative fields of Oct4-deficient cells are shown. Xist RNA, red. Xic DNA (pSxn probe), green. After RNA/DNA FISH for Xist/Xic, cells were hybridized with a Chr.1 (1C, red) and an Xic (green) probe, revealing that each nucleus with two Xist clusters is diploid. Note that original Xist RNA signals were destroyed in second-round FISH.
d, Percentage of cells displaying one versus two Xist RNA clusters on d4.
e, Quantitative RT-PCR of Xist RNA in d4 female knockdown cells.
f, Allele-specific RT-PCR of Xist in d4 female knockdown cells. Cas, M. castaneus allele.
g, Model: Dynamic and multifaceted regulation of XCI by Oct4. +, activating. -, repressive. See text for detailed discussion.
Realtime qRT-PCR corroborated elevated Xist expression in Oct4-knockdown cells (Fig. 4e). In the wildtype 16.7 line, XCI is normally skewed towards inactivating the 129 instead of the M. castaneus X-chromosome (evidenced by a 80:20 Xist RNA ratio) due to the ‘Xce’ modifier effect 26. We reasoned that, if there were increased biallelic Xist expression, we should observe elevated expression from the M. castaneus allele. Allele-specific RT-PCR showed that the castaneus allele displayed 4-fold elevated expression relative to the 129 allele (Fig. 4f). (Both alleles showed increased expression, but because of the Xce effect, the 129 allele maintained greater expression even as the castaneus allele proportionally increased.) Thus, RNA FISH and RT-PCR demonstrated that Xist becomes biallelically expressed in a fraction of female cells when Oct4 is deficient. This anomaly suggested a defect in counting that is consistent with the fact that the tightly linked process of pairing is also aberrant (Fig. 3).
To exclude the possibility that loss of pairing and ectopic Xist expression on d4 could be due to accelerated differentiation in Oct4-depleted cells, we performed nucleofection on d0 female cells and analyzed pairing on d1. Neither precocious pairing (Fig. S3a-c) nor premature Xist activation (Fig. S3d,e) was seen. On d1, female cells normally would not have initiated XCI, but ~4% of Oct4-knockdown cells and ~1% of Sox2-knockdown cells showed weak Xist upregulation (Fig. S3d,e). However, the Xist RNA clusters were generally sparse and diffuse (Fig. S3e, arrow) and distinctly different from Xist clusters of wildtype and Oct4-knockdown cells on d4. Therefore, loss of pairing and aberrant counting were not due to precocious differentiation caused by Oct4 depletion. We concluded that Oct4 not only transactivates Tsix/Xite but also regulates X-chromosome pairing and counting. Significantly, Oct4 is the first trans-factor identified for counting.
In summary, we have ascribed new functions to Oct4 and propose that Oct4 links cell differentiation to X-programming by controlling pairing and counting. Our data agree with a recent study proposing Oct4 as a regulator of XCI13. This study hypothesized that Oct4 binds Xist intron 1 and directly represses Xist; by contrast, our data argue that Oct4 binds and directly activates Xite/Tsix, which would in turn repress Xist. Direct effects on Xite/Tsix are supported by EMSA and transcription assays (Fig. 1,2), but concurrent Tsix downregulation and Xist upregulation in Oct4-knockdown cells (Fig. S4) are consistent with both models. Taken together, the two studies suggest that Oct4 may both activate Tsix/Xite and repress Xist in parallel. Our model (Fig. 4g) proposes that, in pre-XCI cells, Oct4 binds the 5’ ends of Xite and Tsix to transactivate the antisense transcript and thereby inhibit Xist. At the same time, Oct4 binds internal sites within Xist (and Tsix, as Xist and Tsix overlap), repressing Xist either directly or through Tsix. When cell differentiation is initially triggered, Oct4 – together with Ctcf – promotes X-X pairing through Xite/Tsix and ensures correct counting and mutually exclusive choice of Xa and Xi. On the future Xi, Oct4 binding would be lost as Oct4 levels decrease, Tsix is downregulated, and Xist is induced; on the future Xa, residual Oct4 would enable its transient persistence on Tsix, maintenance of Tsix expression, and inhibition of Xist in cis. Thus, Oct4’s intrinsic developmental specificity – active in pluripotent cells and downregulated in differentiating cells -- controls the timing of XCI by triggering pairing and counting. By molecularly linking XCI to differentiation through Oct4-Tsix/Xite interactions, our study reveals a complex network involved in epigenetic reprogramming of the X in stem cells.
METHODS SUMMARY
Cell Lines
The 16.7 female and J1 male ES lines, fibroblasts, and culture conditions have been described15.
EMSA
EMSAs were performed17 using rOct4 and rSox2 made from full-length mouse cDNAs cloned into pET-47 (Novagen), expressed in bacteria, purified by Nickel column, and desalted using a PD-10 column (GE). See full-length Methods for nucleotide sequences. Supershifts were carried out with α-Oct4 and α-Sox2 (Santa Cruz H134, 17320)
GST pulldowns and co-immunoprecipitations
Bacterial and mammalian GST fusions are described in the full Methods section. Two micrograms of DNA were transfected into HEK 293A cells (Lipofectamine 2000, Invitrogen), cells were harvested at 46-48 hours for GST-pulldown assays as described17. See full Methods and figure legends for co-IP details.
GST pulldowns and co-immunoprecipitations
Bacterial and mammalian GST fusions are described in the full Methods section. Two micrograms of DNA were transfected into HEK 293A cells (Lipofectamine 2000, Invitrogen), cells were harvested at 46-48 hours for GST-pulldown assays as described17. See full Methods and figure legends for co-IP details.
Chromatin Immunoprecipitations
ChIP analysis was carried out as described27 and the results averaged for three independent biological replicates. ChIP antibodies included α-Oct4 (Santa Cruz 8628), α-Sox2 (Santa Cruz 17320), normal goat IgG (Santa Cruz 2028), and α-H3 (Abcam 1791). qPCR was performed using an iCycler iQ real-time PCR detection system (Bio-Rad) by amplifying with primer pairs listed in the full-length Methods.
Luciferase Assays
NS11 (no promoter:luciferase), NS65 (-557/+176 Tsix promoter:luciferase), and NS135 (1.2-kb Xite enhancer plus -557/+176 Tsix promoter:luciferase) have been described18. Mutant vectors were constructed by PCR mutagenesis of NS135 to yield: Mutated Oct4 XiteL, 5’-CCAGGTCTGCATTGATATGTAAGGTAAGCACTTCTGTC-3’; and Mutated Sox2 XiteL, 5’-CCAGGTCTGCCCCGATATGTAAAATAAGCACTTCTGTC-3 ’. Doubly mutated Oct4 and Sox2, 5’-CCAGGTCTGCCCCGATATGTAAGGTAAGCACTTCTGTC-3’. Mutated nucleotides, underlined. 30μg of linearized construct were electroporated into male and female ES cells and G418-resistant colonies were pooled for the assay.
Knockdown analyses
Murine Oct4, Sox2, Ctcf, and control SMARTpool siRNAs (Dharmacon) were nucleofected at 0.8μM with the Amaxa Nucleofection System and harvested 24-48 hours later for expression or pairing analyses.
FISH and Pairing Assays
FISH and pairing analyses were performed as described previously6.
Supplementary Material
Acknowledgments
We thank N. Stavropoulos and J. Lopilato for their contributions to the early phase of this work. We also thank J. Zhao, J.A. Erwin, and J. Ahn for critical reading of the manuscript and all lab members for stimulating discussion. This work was funded by an NIH grant to J.T.L. (GM58839). J.T.L. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
STATEMENT OF CONTRIBUTIONS M.E.D. designed, performed, and analyzed the Tsix/Xite bioinformatics screen, EMSAs, tests of protein-protein interactions, luciferase assays, knockdowns, and qRT-PCR; S.S.S. designed, performed, and analyzed the pairing and counting experiments; and S.F.P. designed, performed, and analyzed the Xist bioinformatics screen and the qChIP assays. N.X. participated in initial pairing analyses. J.T.L. formulated, analyzed, and directed the study and wrote the paper.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Maherali N, et al. Directly Reprogrammed Fibroblasts Show Global Epigenetic Remodeling and Widespread Tissue Contribution. Cell Stem Cell. 2007;1(1):55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Wutz A, Gribnau J. X inactivation Xplained. Curr Opin Genet Dev. 2007;17(5):387–393. doi: 10.1016/j.gde.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Lucchesi JC, Kelly WG, Panning B. Chromatin remodeling in dosage compensation. Annu Rev Genet. 2005;39:615–651. doi: 10.1146/annurev.genet.39.073003.094210. [DOI] [PubMed] [Google Scholar]
- 5.Payer B, Lee JT. X Chromosome Dosage Compensation: How Mammals Keep the Balance. Annu Rev Genet. 2008 doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- 6.Xu N, Tsai CL, Lee JT. Transient homologous chromosome pairing marks the onset of X inactivation. Science. 2006;311(5764):1149–1152. doi: 10.1126/science.1122984. [DOI] [PubMed] [Google Scholar]
- 7.Bacher CP, et al. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat Cell Biol. 2006;8(3):293–299. doi: 10.1038/ncb1365. [DOI] [PubMed] [Google Scholar]
- 8.Lee JT. Regulation of X-chromosome counting by Tsix and Xite sequences. Science. 2005;309(5735):768–771. doi: 10.1126/science.1113673. [DOI] [PubMed] [Google Scholar]
- 9.Morey C, et al. The region 3’ to Xist mediates X chromosome counting and H3 Lys-4 dimethylation within the Xist gene. Embo J. 2004;23(3):594–604. doi: 10.1038/sj.emboj.7600071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monkhorst K, Jonkers I, Rentmeester E, Grosveld F, Gribnau J. X inactivation counting and choice is a stochastic process: evidence for involvement of an x-linked activator. Cell. 2008;132(3):410–421. doi: 10.1016/j.cell.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 11.Monk M, Harper MI. Sequential X chromosome inactivation coupled with cellular differentiation in early mouse embryos. Nature. 1979;281(5729):311–313. doi: 10.1038/281311a0. [DOI] [PubMed] [Google Scholar]
- 12.Silva SS, Rowntree RK, Mekhoubad S, Lee JT. X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0712136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarro P, et al. Molecular coupling of Xist regulation and pluripotency. Science. 2008;321(5896):1693–1695. doi: 10.1126/science.1160952. [DOI] [PubMed] [Google Scholar]
- 14.Nichols J, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95(3):379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 15.Lee JT, Lu N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell. 1999;99(1):47–57. doi: 10.1016/s0092-8674(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa Y, Lee JT, Xite X-inactivation intergenic transcription elements that regulate the probability of choice. Mol Cell. 2003;11(3):731–743. doi: 10.1016/s1097-2765(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 17.Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell. 2007;25(1):43–56. doi: 10.1016/j.molcel.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 18.Stavropoulos N, Rowntree RK, Lee JT. Identification of developmentally specific enhancers for Tsix in the regulation of X chromosome inactivation. Mol Cell Biol. 2005;25(7):2757–2769. doi: 10.1128/MCB.25.7.2757-2769.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao W, Huynh KD, Spencer RJ, Davidow LS, Lee JT. CTCF, a candidate trans-acting factor for X-inactivation choice. Science. 2002;295(5553):345–347. doi: 10.1126/science.1065982. [DOI] [PubMed] [Google Scholar]
- 20.Tsai CL, Rowntree RK, Cohen DE, Lee JT. Higher order chromatin structure at the X-inactivation center via looping DNA. Dev Biol. 2008 doi: 10.1016/j.ydbio.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohlsson R, Renkawitz R, Lobanenkov VV. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;7:520–527. doi: 10.1016/s0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- 22.West AG, Gaszner M, Felsenfeld G. Insulators: many functions, many mechanisms. Genes Dev. 2002;16(3):271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 23.Park K, Atchison ML. Isolation of a candidate repressor/activator, NF-E1, that binds to the immunoglobulin kappa 3’ enhancer and the immunoglobulin heavy-chain mu E1 site. Proc Natl Acad Sci USA. 1991;88:9804–9808. doi: 10.1073/pnas.88.21.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1. Biochim Biophys Acta. 1997;1332(2):F49–66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- 25.Xu N, Donohoe ME, Silva SS, Lee JT. Evidence that homologous X-chromosome pairing requires transcription and Ctcf protein. Nat Genet. 2007;39(11):1390–1396. doi: 10.1038/ng.2007.5. [DOI] [PubMed] [Google Scholar]
- 26.Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet. 2001;2(1):59–67. doi: 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- 27.Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. 2006;1(2):729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133(6):1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 29.Stavropoulos N, Lu N, Lee JT. A functional role for Tsix transcription in blocking Xist RNA accumulation but not in X-chromosome choice. Proc Natl Acad Sci U S A. 2001;98(18):10232–10237. doi: 10.1073/pnas.171243598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.