

Abstract
In mammals, α-linked GlcNAc is primarily found in heparan sulfate/heparin and gastric gland mucous cell type mucin. α-N-Acetylglucosaminidases (αGNases) belonging to glycoside hydrolase family 89 are widely distributed from bacteria to higher eukaryotes. Human lysosomal αGNase is well known to degrade heparin and heparan sulfate. Here, we reveal the substrate specificity of αGNase (AgnC) from Clostridium perfringens strain 13, a bacterial homolog of human αGNase, by chemically synthesizing a series of disaccharide substrates containing α-linked GlcNAc. AgnC was found to release GlcNAc from GlcNAcα1,4Galβ1pMP and GlcNAcα1pNP substrates (where pMP and pNP represent p-methoxyphenyl and p-nitrophenyl, respectively). AgnC also released GlcNAc from porcine gastric mucin and cell surface mucin. Because AgnC showed no activity against any of the GlcNAcα1,2Galβ1pMP, GlcNAcα1,3Galβ1pMP, GlcNAcα1,6Galβ1pMP, and GlcNAcα1,4GlcAβ1pMP substrates, this enzyme may represent a specific glycosidase required for degrading α-GlcNAc-capped O-glycans of the class III mucin secreted from the stomach and duodenum. Deletion of the C-terminal region containing several carbohydrate-binding module 32 (CBM32) domains significantly reduced the activity for porcine gastric mucin; however, activity against GlcNAcα1,4Galβ1pMP was markedly enhanced. Dot blot and ELISA analyses revealed that the deletion construct containing the C-terminal CBM-C2 to CBM-C6 domains binds strongly to porcine gastric mucin. Consequently, tandem CBM32 domains located near the C terminus of AgnC should function by increasing the affinity for branched or clustered α-GlcNAc-containing glycans. The agnC gene-disrupted strain showed significantly reduced growth on the class III mucin-containing medium compared with the wild type strain, suggesting that AgnC might have an important role in dominant growth in intestines.
Keywords: Adhesion, Carbohydrate-binding Protein, Carbohydrate Function, Glycoprotein, Oligosaccharide, Clostridium perfringens, Glycosidase, Mucin
Introduction
In mammals, α-linked GlcNAc has been found as a repetitive disaccharide of glycosaminoglycans, such as heparin and heparan sulfate, and at the non-reducing terminus of O-glycans of mucin glycoproteins. α-GlcNAc also exists in lipopolysaccharides present on the surface of bacteria (1, 2) and phosphatidyl glycolipids in plants (3). Mucin containing peripheral α-GlcNAc, also called class III mucin, was characterized by paradoxical concanavalin A staining, which is a sequential histochemical process involving periodate reduction, sodium borohydrate reduction, and concanavalin A staining. This type of mucin was specifically detected within the gastric glands of the stomach, Brunner's glands of the duodenum, and the accessory glands of the pancreaticobiliary tract. The representative O-glycan structure of this type of mucin was determined to be GlcNAcα1,4Galβ1,3(GlcNAcα1,4Galβ1, 4GlcNAcβ1,6)GalNAc (4–6). A monoclonal antibody, HIK1083, that recognizes the non-reducing terminal α-GlcNAc was developed. Using this antibody, α-GlcNAc-containing glycans were found to be expressed on several tumor tissues, such as gastric adenocarcinoma, pancreatic ductal carcinoma, and adenocarcinoma of the uterine cervix (7–9). Human α1,4-N-acetylglucosaminyltransferase (α4GnT)3 that generates GlcNAcα1,4Galβ1R in class III mucin has been cloned and characterized (10). α4GnT mRNA was detected in the stomach and pancreas, confirming the restricted expression of this glycan. Recently, α-GlcNAc-containing O-glycans in the stomach were found to function as a natural antibiotic against Helicobacter pylori infection (11). The α-glucosylsterol synthase that forms α-glucosylsterol, an essential membrane component of H. pylori, was inhibited by α-GlcNAc-containing glycans, resulting in cell growth arrest (12–14).
We previously reported that Clostridium perfringens ATCC 10543 present in the gastrointestinal tract of humans possesses an enzyme that liberates the disaccharide GlcNAcα1,4Gal from O-glycans of class III mucins (15). This finding raised the question of how GlcNAcα1,4Gal is further processed. Searching the databases of the genomes of several strains of C. perfringens revealed that there are possible α-N-acetylglucosaminidases (αGNases) belonging to glycoside hydrolase family 89 (GH89) in the CAZy data base (see the CAZy Web site). Among GH89 members, only mammalian αGNases have been characterized; the enzymes are responsible for the degradation of heparin and heparan sulfate in lysosomes. Defective αGNase in humans causes Sanfilippo B syndrome (mucopolysaccharidosis IIIB), which is characterized by the accumulation of heparin and heparan sulfate (16–19). Recently, a GH89 αGNase from C. perfringens ATCC 13124 was structurally characterized as a model protein of human αGNase (20). Because αGNase from C. perfringens is a large multimodular protein containing a number of accessory domains in addition to the catalytic GH89 domain, a deletion protein containing an N-terminal GH89 domain was expressed and crystallized. The activity of this recombinant enzyme was only measured using synthetic monosaccharide substrates, such as GlcNAcα1pNP, and therefore the specificity toward natural substrates remains unresolved. Here, we report the expression and characterization of the full-length αGNase from another strain of C. perfringens (strain 13). The catalytic activity of the enzyme showed strict specificity toward class III mucin but not to the disaccharide unit of heparin and heparan sulfate. We also identify the function of the accessory domains that were found to increase affinity toward multivalent natural substrates.
EXPERIMENTAL PROCEDURES
Chemical Syntheses of Disaccharide Substrates
Galβ1pMP and GlcAβ1pMP were purchased from Kanto Chemical. GlcGlcNAcα1,4Galβ1pMP, GlcNAcα1,6Galβ1pMP, GlcNAcα1,3Galβ1pMP, GlcNAcα1,2Galβ1pMP, and GlcNAcα1,4GlcAβ1pMP were chemically synthesized by the method of Schmidt et al. (21). Each disaccharide was isolated by silica gel column chromatography and identified by NMR spectroscopy (supplemental Tables 1 and 2) using a JEOL JNM-ECA-600 (600-MHz) spectrometer and high resolution MS using a MarinerTM mass spectrometer (Applied Biosystems).
GlcNAcα1,4Galβ1pMP
The NMR analyses were carried out in CD3OD (1H NMR, 600 MHz; 13C NMR, 150 MHz). The correlation between the anomeric proton of the GlcNAc residue and the 4-position carbon of the Gal residue was observed by HMQC and HMBC NMR spectroscopic analyses. ESI-TOF-MS: calculated for C21H32NO12 m/z [M + H]+, 490.1919; found, 490.1891.
GlcNAcα1,6Galβ1pMP
The NMR analyses were carried in D2O (1H NMR, 600 MHz; 13C NMR, 150 MHz, 35 °C). The correlation between the anomeric proton of GlcNAc and the 6-position carbon of Gal was observed by HMQC and HMBC NMR spectroscopic analyses. ESI-TOF-MS: calculated for C21H32NO12 m/z [M + H]+, 490.1919; found, 490.1897.
GlcNAcα1,3Galβ1pMP
Hexa-O-Ac-GlcNAcα1,3Galβ1pMP was prepared as the precursor of GlcNAcα1,3Galβ1pMP. NMR spectra were measured in this step.
Hexa-O-Ac-GlcNAcα1,3Galβ1pMP
The NMR analyses were carried in CDCl3 (1H NMR, 600 MHz; 13C NMR, 150 MHz). The correlation between the anomeric proton of GlcNAc and the 3-position carbon of Gal was observed by HMQC and HMBC NMR spectroscopic analyses. The MS spectrum of GlcNAcα1,3Galβ1pMP was measured after the deprotection of hexa-O-Ac-GlcNAcα1,3Galβ1pMP. ESI-TOF-MS of GlcNAcα1,3Galβ1pMP: calculated for C21H32NO12 m/z [M+H]+, 490.1919; found, 490.1923.
GlcNAcα1,2Galβ1pMP
Hexa-O-Ac-GlcNAcα1,2Galβ1pMP was prepared as the precursor of GlcNAcα1,2Galβ1pMP. NMR spectra were measured in this step.
Hexa-O-Ac-GlcNAcα1,2Galβ1pMP
The NMR analyses were carried out in CDCl3 (1H NMR, 600 MHz; 13C NMR, 150 MHz). The correlation between anomeric protons of GlcNAc residue and 2-position carbons of Gal residue was observed by HMQC and HMBC of NMR spectroscopic analyses. The MS spectrum of GlcNAcα1,2Galβ1pMP was measured after the deprotection of hexa-O-Ac-GlcNAcα1, 2Galβ1pMP. ESI-TOF MS of GlcNAcα1,2Galβ1pMP: calculated for C21H32NO12 m/z [M+H]+, 490.1919; found, 490.1905.
GlcNAcα1,4GlcAβ1pMP
The NMR analyses were carried out in D2O (1H NMR, 600 MHz; 13C NMR, 150 MHz). The correlation between the anomeric proton of GlcNAc and the 4-position carbon of GlcA was observed using HMQC and HMBC NMR spectroscopic analyses. ESI-TOF-MS: calculated for C21H30NO13 m/z [M + H]+, 504.1712; found, 504.1707.
Cloning of αGNase from C. perfringens
Genomic DNA from C. perfringens strain 13 (22) was kindly provided by Dr. T. Shimizu. The DNA fragments of the probable αGNase gene (CPE0866) (nucleotides (nt) 76–6312 for full-length AgnC, nt 76–3399 for AgnCΔC, nt 2800–3618 for CBM(C2-C3), nt 2800–4044 for CBM(C2–C4), nt 2800–4485 for CBM(C2–C5), nt 2800–4875 for CBM(C2–C6), and nt 5404–5766 for FIVAR) were amplified by high fidelity PCR (Prime Star, Takara, Japan) using the genomic DNA of C. perfringens strain 13 and a pair of the following primers with restriction enzyme sites and the S-tag sequence: common forward primer for AgnC and AgnCΔC, CGGCGAGCTCGGTAGTGCAATTAAGGTAAGGGCATCA; reverse primer for AgnC with S-tag, CCGCTCGAGTTAGCTGTCCATGTGCTGGCGTTCGAATTTAGC; reverse primer for AgnCΔC with S-tag, CCGCTCGAGTTAGCTGTCCATGTGCTGGCGTTCGAATTTAGC; reverse primer for AgnCΔC without S-tag, CCGCTCGAGTTAAAGCTCTAAAAACTCAACATTTTC; forward primer for CBMs-GST, CGGCGAGCTCGGAAAGCCTGTAAGTAGTGAAACT; reverse primer for CBM(C2-C3), CCGCTCGAGCGAACCCGTCGATAACGTCTTCAT; reverse primer for CBM(C2–C4), CCGCTCGAGAATTTCTGCAATAGCTGCCCATGC; reverse primer for CBM(C2–C5), CCGCTCGAGAGCTGAAGCAAATCCTCCAACCCC; reverse primer for CBM(C2–C6), CCGCTCGAGGATTTCTCCAATTTTAACATTCTC; forward primer for FIVAR, CGGCGAGCTCGCTAAGGAAAAAGTAGAAAATGCA; reverse primer for FIVAR, CCGCTCGAGAGCCTTATTTATATTCTCTTCATT (underlined bases represent the restriction enzyme recognition sites). PCR products were treated with SacI and XhoI, ligated into the corresponding site of the pBluescript II KS+ vector (Stratagene), and sequenced. These inserts containing the S-tag sequence were excised by digestion with SacI and XhoI and ligated into the expression vector pCold-TF-DNA (Takara).
Expression and Purification of αGNase
The expression plasmid was designed to express the recombinant enzyme fused with a tandem His6 tag and trigger factor (TF) tag at the N terminus and a 15-amino acid S tag or GST tag at the C terminus. Escherichia coli BL21(λDE3) cells were transformed with each construct. The E. coli cells were grown in LB medium containing 50 μg/ml carbenicillin at 30 °C until the A600 reached 0.5. The cells were then cooled to 15 °C for 0.5 h, expression was induced by the addition of 0.1 mm isopropyl β-d-thiogalactopyranoside, and the cells were grown at the same temperature for 24 h. The E. coli cells were harvested and lysed in a buffer (50 mm sodium phosphate, pH 8.0, and 300 mm sodium chloride for His tag purification or 50 mm Tris-HCl, pH 7.5, 0.1% Triton X-100, and 0.1% β-mercaptoethanol for S tag and GST tag purification) containing 0.5 mm 4-(2-aminoethyl)benzenesulfonyl fluoride and 0.2% lysozyme from chicken egg (Nacalai Tesque). The cells were treated with intermittent sonication in an ice-water bath using a sonicator (Ultrasonic Generator US150, Nissei) at 40–50% output. The crude lysate was fractionated by centrifugation (20,000 × g, 4 °C for 15 min) and the supernatant was filtered through a 0.22-μm filter. The clear supernatant was applied to either of the two types of affinity columns for purification of the target protein. For S tag and His6 tag purification, S-protein-agarose beads (Novagen-Merck), and Ni2+-charged beads (Protino Ni 2000, Macherey-Nagel) were used, respectively. The His6-TF tag of the target protein was cleaved by Factor Xa (Novagen). For purification of GST-tagged proteins (CBMs-GST and FIVAR-GST), glutathione-Sepharose 4B (GE Healthcare) was used.
Protein concentrations were determined using the Bio-Rad protein assay kit. Protein purity was assessed by reducing SDS-PAGE followed by Coomassie Brilliant Blue staining. The purified recombinant protein yields were 5–15 mg/liter of culture.
Assay for αGNase
Activities for pNP-monosaccharide substrates (Sigma-Aldrich) were determined by measuring the release of p-nitrophenol and the absorbance at 420 nm. One unit of αGNase activity was defined as the amount of the enzyme releasing 1 μmol of p-nitrophenol/min. Activities for pMP-disaccharide substrates were assayed using HPLC. The reaction mixture consisting of 0.05–3 mm concentrations of the disaccharides and appropriate amounts of enzymes in 100 μl of PBS were incubated at 37 °C for the appropriate time. After termination of the reaction, the reaction mixtures were analyzed by HPLC. HPLC was carried out using a Hitachi D-7400 chromatography system equipped with a GL-7420 UV-PAD detector that is capable of simultaneously monitoring the absorbance between 210 and 400 nm. The reaction mixtures were separated using a reversed-phase column, Inertsil ODS-3 (0.46 × 25 cm, Shimadzu, Japan), under a constant flow (1.0 ml/min) of 5–20% acetonitrile (containing 0.1% TFA) over the course of 40 min at 40 °C. The pMP group was specifically detected at 280 nm.
Activity for natural O-glycans was determined using porcine gastric mucin (PGM) partially purified from crude PGM (Type III, Sigma-Aldrich) according to a previous method (6) (a gift from Dr. Kurihara). Released GlcNAc was detected/measured by TLC and HPLC. Silica gel TLC plates (Merck) were developed with 1-butanol/acetic acid/water (2:1:1, v/v/v), and the sugars were visualized using a diphenylamine-aniline-phosphoric acid reagent (23). HPLC was carried out using a normal phase column, NH2P50-E (0.30 × 20 cm, Asahi-Denka), under a constant flow at 0.5 ml/min of 70% acetonitrile over the course of 20 min at 40 °C. GlcNAc was monitored at 210 nm. A sandwich ELISA for α-linked GlcNAc on PGM was carried out using a commercially available ELISA kit using the HIK1083 monoclonal antibody (Kanto Chemical). The reaction mixtures containing 0.2% PGM and an appropriate amount of AgnC or AgnCΔC were incubated at 37 °C for 24 h and then loaded into the wells of a microtiter plate coated with the HIK1083 antibody and incubated at room temperature for 1 h. The wells were washed five times with 400 μl of PBS containing 0.05% Tween 20 (PBST), and the biotin-conjugated HIK1083 antibody was added. Colorimetric measurements were carried out according to the instruction manual.
The action of AgnC on the cell surface mucin was analyzed using adenocarcinoma AGS-α4GnT cells stably expressing GlcNAcα1,4Galβ1R as O-glycans on the cell surface (10). The cells were grown on Lab-Tek chamber slides (Nalge Nunc International) and fixed with 20% buffered formalin, pH 7.4, for 15 min. After washing with PBS, the fixed cells were incubated with 0, 10.2, or 40.2 milliunits of His-TF-tagged AgnCΔC in 500 μl of PBS at 37 °C for 24 h. For the living cells, 80% confluent cells were incubated in 500 μl of DMEM containing 0, 10.2, or 40.2 milliunits of His-TF-tagged AgnCΔC at 37 °C for 24 h. The cells were subjected to immunohistochemical analysis using the HIK1083 antibody as described previously (15). Fluorescein isothiocyanate-conjugated anti-mouse IgM was used as the secondary antibody, and Vectashield (Vector Laboratories) was used for mounting the slides. The immunolabeling was analyzed using a confocal laser-scanning microscope, LSM510 (Carl Zeiss).
Binding Assay of GST-tagged CBMs to Glycoproteins
Dot Blot Overlay Assay
10 μg of various glycoproteins (bovine submaxillary gland mucin, Type I-S (Sigma-Aldrich); human gastric mucin; crude PGM; and PGM) and glycosaminoglycans (chondroitin sulfate (Seikagaku Biobusiness Corp.) and heparan sulfate (Sigma-Aldrich)) were spotted onto a nitrocellulose membrane (28). Membranes were dried completely then blocked with 3% bovine serum albumin in PBST. The membranes were incubated with His-TF-CBMs-GST (1 μm protein in PBST) overnight at 25 °C. Blots were washed five times with PBST and then incubated with horseradish peroxidase (HRP)-conjugated anti-GST antibody (1:25,000; Nacalai Tesque) in PBS at 25 °C for 2 h. Membranes were washed five times with PBST, and the bound proteins were detected using the chemiluminescence reagent (ECL Plus, GE Healthcare).
Sandwich ELISA
PGM (0.5% in PBS) was incubated at 37 °C for 2 h in the wells of a microtiter plate coated with the HIK1083 antibody. The His-TF-CBMs-GST constructs were added to wells (1 μm protein in PBST) and incubated at 37 °C for 24 h. The wells were washed five times with PBST, and the HRP-conjugated anti-GST monoclonal antibody (1:35,000) was added and incubated at 25 °C for 1 h. The wells were washed five times with PBST, and the substrates for HRP were added. Colorimetric measurements were carried out according to the instruction manual.
Stereochemistry of the Hydrolysis Catalyzed by αGNase
1H NMR spectra were recorded on a JEOL JNM-ECA-600 spectrometer. The reaction mixture contained 5.0 mm GlcNAcα1pNP in a total volume of 500 μl of D2O-PBS (prepared by the addition of D2O into lyophilized PBS). After recording the reference spectrum (t = 0 min, 37 °C), 20 milliunits (50 μl in D2O-PBS) of the His6-TF-tagged AgnCΔC, prepared by substituting PBS to D2O-PBS using a Centricon 10 (Millipore), was added to initiate the reaction. The spectra were recorded at different time intervals (9 min to 18 h).
Construction of an agnC Null Mutant
C. perfringens strain 13 (a gift from Dr. T. Shimizu of Kanazawa University) was cultured at 37 °C in GAM medium (Nissui Pharmaceutical) or brain heart infusion medium (Sigma-Aldrich) under anaerobic conditions using Anaeropack (Mitsubishi Chemical).
The agnC gene was disrupted using the TargeTron gene knock-out system (Sigma-Aldrich). The sense-orientated intron insertion site was selected between nucleotides 303 and 304 of the agnC open reading frame (ORF) using the Sigma TargeTron algorithm (see the Sigma-Genosys Web site). Primers used to generate a 350-bp intron targeting sequence to this site of agnC ORF were IBS (AAAAAAGCTTATAATTATCCTTAGTGTACAAATATGTGCGCCCAGATAGGGTG), EBS1d (CAGATTGTACAAATGTGGTGATAACAGATAAGTCAAATATAATAACTTACCTTTCTTTGT), and EBS2 (TGAACGCAAGTTTCTAATTTCGATTTACACTCGATAGAGGAAAGTGTCT). The amplified 350-bp PCR fragment was then digested with HindIII and BsrGI and ligated into pJIR750ai (24). The resultant plasmid, named pJIR750agnC, was introduced into wild type C. perfringens strain 13 by electroporation (2.5 kV, 200 ohms, 25 microfarads). Transformants were plated onto BHI agar plates containing 15 μg/ml chloramphenicol. Colonies were PCR-screened for an intron-disrupted agnC gene using a pair of primers: TGGATAGAAGTTGACTTAGGTGGA and TCTTTCCTGCCCTTACATTATCA. A mutant shown to carry an agnC intron insertion was subcultured daily in GAM medium without chloramphenicol for 10 days to cure the intron-carrying donor plasmid pJIR750agnC. Curing was initially shown by lack of growth on chloramphenicol-containing GAM plates and then confirmed by Southern blotting, which demonstrated the presence of a single intron in the mutant. For Southern blotting, the genomic DNA from wild type or agnC null mutant was digested with EcoRI, separated by a 0.8% agarose gel, and transferred to a nylon membrane (GE Healthcare) for detection by an intron-specific digoxigenin-labeled probe. This probe was generated using primers IBS and EBS1d and a digoxigenin DNA labeling mix (Roche Applied Science). CSPD substrate (Roche Applied Science) was used for detection of digoxigenin-labeled hybridized probes according to the manufacturer's instructions.
αGNase Activity of Wild Type or agnC Null Mutant
Wild type or agnC null mutant was grown overnight at 37 °C in Duncan-Strong medium under anaerobic conditions. Each 40-ml culture was centrifuged at 4 °C for 10 min at 8,000 × g and then passed through a 0.22-μm syringe filter to exclude the remaining bacteria within the supernatant. The culture supernatant was collected and concentrated 43-fold using Amicon Ultra-15 centrifugal filter devices (50,000 molecular weight cut-off, Millipore, MA). The concentrated supernatant (2 μl) was incubated with 5 mm GlcNAcα1,4Galβ1pMP (4 μl) at 37 °C for 20 h, and the hydrolysis of substrate was analyzed by TLC as described above.
Growth of Wild Type or agnC Null Mutant in Minimal Medium Containing Mucin
Minimal medium was prepared as described previously (25). The medium containing 12 g of sodium acetate, 10 g of casamino acids (Difco-BD, NJ), 6.3 g of Na2HPO4, 4.1 g of NaH2PO4, 1.2 g of ammonium sulfate, 0.88 g of KH2PO4, 0.6 g of K2HPO4, 0.45 g of trisodium citrate, 0.06 g of adenine, 0.06 g of uracil, 0.04 g of guanine, 0.02 g of ferrous sulfate, 0.02 g of manganous sulfate, and 0.02 g of NaCl in a total volume of 1 liter of distilled waster was sterilized by autoclaving at 121 °C for 15 min. Prior to use, filter-sterilized solutions (0.2-μm pore size filters, Millipore) of MgSO4, cysteine-HCl, and Na2CO3 were added to final concentrations of 0.4, 0.5, and 4.4 g/liter, respectively, along with a filter-sterilized vitamin solution (2 ml/liter) (26). Carbohydrate sources in growth studies were prepared in 10 mm potassium phosphate buffer (pH 7.0) and comprised 20 mm glucose or 1% (w/v) PGM (Type III, Sigma-Aldrich); the former was sterilized by filtration and the latter by autoclaving. Each carbohydrate solution (10 ml) was mixed with equal volumes of minimal medium (10 ml) in sterile tubes. Overnight GAM cultures of wild type or agnC null mutant (1:20, v/v) were inoculated to various media and incubated anaerobically at 37 °C for 12 h. Growth of bacteria was monitored by removing 200-μl aliquots of each culture into a 96-well plate and measuring A620 using a microplate reader (Powerscan HT, DS Pharma Biomedical).
RESULTS
Identification of the Gene Encoding αGNase from C. perfringens
To identify αGNase acting on α-linked GlcNAc at the non-reducing terminus of O-glycans, we searched for genes homologous to the human αGNase (NAGLU) gene using the BLAST program. The search identified an open reading frame, CPE0866, in the genome of C. perfringens strain 13, which encodes a GH89 αGNase. Sequence analysis of CPE0866 using the SMART program (see the EMBL Web site) revealed the following features of the gene product (Fig. 1A). The protein is 2104 amino acids (aa) in length and contains a signal peptide at the N terminus (aa 1–25), a conserved catalytic GH89 domain (aa 245–906), six CBM32 domains (C1, aa 49–159; C2, aa 934–1063; C3, aa 1083–1206; C4, aa 1225–1348; C5 aa 1372–1495; C6, aa 1511–1625), two FIVAR domains (aa 1802–1849 and 1868–1922) and a fibronectin type 3 (FN3) domain (aa 2018–2102). The GH89 domain has 28% identity with the catalytic domain of human αGNase. Among the six CBM32 domains, C5 shows 42% amino acid identity with the CBM32 of β-N-acetylhexosaminidase (aa 12–126) from C. perfringens ATCC13124 and 39% identity with that of sialidase (aa 7–142) from the same strain (27–29). The other CBMs had slightly lower sequence identities (20–31%).
FIGURE 1.

Expression of the two forms of the recombinant AgnC. A, schematic representation of αGNase from C. perfringens strain 13. The numbering starts at a possible initiation codon. The two-headed arrows indicate the region of the prepared AgnC and AgnCΔC. Six CBM32 domains are depicted as C1–C6. B, SDS-PAGE of the recombinant αGNases overexpressed in E. coli. Lanes 1–3, AgnC; lanes 4–6, AgnCΔC. Lanes 1 and 4, crude extracts; lanes 2 and 5, His6-, TF-, and S-tagged forms; lanes 3 and 6, S-tagged forms, after removing His6 and TF tags by Factor Xa protease digestion.
Molecular Cloning and Expression of Full-length and Truncated Recombinant αGNases from C. perfringens Strain 13
To characterize the gene product of CPE0866 and elucidate the role of the CBM32s, expression of the full-length (AgnC; aa 26–2104) and C-terminal truncated (AgnCΔC; aa 26–1133) proteins using several conventional E. coli expression vectors was attempted. Expression of both proteins fused with His6 and TF tags at the N terminus and an S tag at the C terminus using the cold shock expression plasmid pCold-TF was successfully achieved. Expressed proteins were purified by either S tag or His6 tag affinity column chromatography and subsequently digested with Factor Xa to remove the His6 and TF tags. Purified AgnC and AgnCΔC gave a single protein band of 220 and 120 kDa, respectively, on SDS-PAGE (Fig. 1B). Because both of the purified proteins hydrolyzed the GlcNAcα1pNP substrate, we designated CPE0866 as agnC (accession number AB517031).
General Properties of the Recombinant αGNases
We determined the general catalytic properties of the recombinant enzymes using GlcNAcα1pNP as a substrate. Both AgnC and AgnCΔC were stable up to 37 °C and retained 80% activity after incubation at 50 °C for 3 h in PBS. The optimum temperature and pH for catalytic activity were determined as 50 °C and 7.0–7.5, respectively. These results suggest that this enzyme acts optimally under intestinal conditions (pH 7.0–7.5), which are noticeably different from the optimal conditions of human αGNase (i.e. pH 4.5–5.0), a lysosomal glycosidase (18, 19).
Substrate Specificities of the Recombinant αGNases
Initially, we examined the substrate specificity of AgnC using various pNP-monosaccharides as substrates. AgnC specifically released GlcNAc from GlcNAcα1pNP but did not act on the other substrates, such as GlcNAcβ1pNP, GalNAcα1pNP, Galα1pNP, Glcα1pNP, GlcAβ1pNP, Fucα1pNP, Fucβ1pNP, Galβ1pNP, Manα1pNP, and Xylα1pNP. AgnCΔC also showed the same specificity as AgnC. The kinetic parameters of AgnC and AgnCΔC for GlcNAcα1pNP were estimated (Table 1). The Km values of AgnC and AgnCΔC for GlcNAcα1pNP were 4.3 and 7.8 mm, respectively, which were significantly higher than the Km value measured for human αGNase (0.30 mm) (30). Because GlcNAcα1pNP is an artificial substrate, we chemically synthesized a new substrate GlcNAcα1,4Galβ1pMP using the method of Schmidt et al. (21), which has the same disaccharide structure as the non-reducing terminus of gastric mucin O-glycan. The purity and identity of the produced substrate were confirmed by 1H and 13C NMR and MS analyses (data presented under “Experimental Procedures”). The enzymatic hydrolysis was monitored by reversed-phase HPLC. AgnC and AgnCΔC hydrolyzed GlcNAcα1,4Galβ1pMP (Fig. 2B), and the product peak was confirmed to be Galβ1pMP by MS analysis (calculated mass, 286.1053; observed mass, m/z [M + Na]+ = 309.0945). The Km values of AgnC and AgnCΔC for the synthetic disaccharide were much lower than those for GlcNAcα1pNP: 200 and 280 μm, respectively. The kcat values were also higher than those for GlcNAcα1pNP: 2-fold for AgnC and 20-fold for AgnCΔC (Table 1). These results suggest that αGNase from C. perfringens recognizes aglycone structure.
TABLE 1.
Kinetic parameters of AgnC and AgnCΔC
| Km | kcat | kcat/Km | Specific activitya | |
|---|---|---|---|---|
| mm | s−1 | s−1mm−1 | μmol min−1mg−1 | |
| GlcNAcα1pNP | ||||
| AgnC | 4.3 | 0.69 | 0.16 | 0.10 |
| AgnCΔC | 7.8 | 0.74 | 0.095 | 0.10 |
| GlcNAcα1,4Galβ1pMP | ||||
| AgnC | 0.20 | 1.24 | 6.2 | 0.20 |
| AgnCΔC | 0.28 | 6.19 | 22.1 | 2.82 |
a Specific activities for GlcNAcα1pNP and GlcNAcα1,4Galβ1pMP were measured at 10 and 3 mm, respectively.
FIGURE 2.

Substrate specificity of AgnC. Chemically synthesized pMP-disaccharides containing α-GlcNAc were incubated with AgnC and analyzed by HPLC. A and F, standard Galβ1pMP and GlcA1βpMP; B–E and G, GlcNAcα1,4Galβ1pMP, GlcNAcα1,2Galβ1pMP, GlcNAcα1,3Galβ1pMP, GlcNAcα1,6Galβ1pMP, and GlcNAcα1,4GlcAβ1pMP, incubated with AgnC (solid lines) or heat-inactivated AgnC (gray lines).
Next, to investigate the specificity of the enzyme for glycoside linkage between GlcNAc and Gal, we further synthesized GlcNAcα1,2Galβ1pMP, GlcNAcα1,3Galβ1pMP, and GlcNAcα1–6Galβ1pMP. AgnC was incubated with one of the three disaccharide substrates and then analyzed by HPLC (Fig. 2, C–E). Among the four substrates, GlcNAcα1,4Galβ1pMP was hydrolyzed to Galβ1pMP, whereas the other three substrates containing α1,2-, α1,3-, and α1,6-linked GlcNAc were not hydrolyzed. This result suggests that this enzyme selectively acts on α1,4-linked GlcNAc. Because α-1,4-GlcNAc exists in heparin and heparan sulfate, we synthesized GlcNAcα1,4GlcAβ1-pMP containing repetitive units of heparin and heparan sulfate and tested whether AgnC hydrolyzed this substrate. Hydrolysis and release of GlcNAc were not observed using this substrate (Fig. 2G). AgnCΔC also showed no activity toward this substrate (data not shown). Taken together, clostridial αGNase is highly specific for the GlcNAcα1,4Gal structure.
Action of αGNase on PGM
PGM contains a GlcNAcα1, 4Galβ1 epitope at the terminus of the core-1 and core-2 type O-glycans. We first tested whether AgnC and AgnCΔC could release GlcNAc from PGM. PGM (2.5%) was incubated with 0.67 milliunits/ml of AgnC or AgnCΔC, and the reaction was analyzed by TLC. AgnC released GlcNAc from PGM, whereas AgnCΔC showed negligible release of GlcNAc under these conditions (Fig. 3A). We next quantified the amount of GlcNAc released from PGM by HPLC. In a 0.2% mucin solution, AgnC released GlcNAc from PGM significantly faster than AgnCΔC (Fig. 3B, left). The initial velocities of GlcNAc released from PGM were calculated to be 9.1 and 0.30 μmol min−1 mg−1 in AgnC and AgnCΔC, respectively. The activity of AgnC toward PGM was ∼45-fold higher than the activity toward GlcNAcα1,4Galβ1pMP (0.20 μmol min−1 mg−1), whereas the activity of AgnCΔC toward PGM was only one-tenth of the activity toward GlcNAcα1,4Galβ1pMP (2.82 μmol min−1 mg−1) (Table 1). Increasing the mucin concentration to 2.5% did not change the initial velocity of GlcNAc release by AgnC; however, the release increased 15-fold when incubation was performed with AgnCΔC. This indicates that AgnCΔC has a much lower affinity for mucin. Incubation of the two proteins with 2.5% PGM for 20 h showed that the amount of released GlcNAc was more than 10-fold higher when the reaction was performed with AgnC than with AgnCΔC (Fig. 3B, right).
FIGURE 3.

Action of AgnC on PGM. A, PGM (7.5 mg) was incubated with 0.2 milliunits of AgnC or AgnCΔC in 300 μl of PBS for 20 h, and the released GlcNAc was analyzed by TLC. Lanes 1–3, standard Fuc, GlcNAcα1,4Gal (Gn-Gal), and GalNAc (GalN); lane 4, PGM treated with AgnCΔC; lane 5, PGM treated with AgnC; lanes 6–8, standard GlcNAc (Gn), Gal, and sialic acid (Sial). B, time course of the amount of GlcNAc released from PGM. PGM (0.6 mg (left) or 7.5 mg (right)) was incubated with 0.2 milliunits of AgnC or AgnCΔC in 300 μl of PBS, and the released GlcNAc was quantified by HPLC. Circles, AgnC; triangles, AgnCΔC. C, the removal of α-GlcNAc epitopes from PGM as determined by the HIK1083 antibody. PGM (0.6 mg) was incubated with AgnC (0.01 or 1.0 milliunits) or AgnCΔC (1.0 or 10.0 milliunits) in 300 μl of PBS for 24 h. The reaction mixtures were analyzed by sandwich ELISA using the HIK1083 antibody. N.C., without PGM; P.C., without enzymes. Experiments were carried out in triplicate, and the results are presented as mean ± S.D. (error bars).
The action of AgnC and AgnCΔC on PGM using a sandwich ELISA and a HIK1083 monoclonal antibody against the α-linked GlcNAc was evaluated (Fig. 3C). PGM was incubated with AgnC or AgnCΔC and subsequently placed into a HIK1083 antibody-coated well. After washing, biotinylated HIK1083 antibody was added to measure the amount of the remaining α-GlcNAc epitope. The treatment with AgnC (both 0.01 and 1.0 units/ml) reduced the reactivity to the level of the control without PGM, indicating the complete removal of the α-GlcNAc epitope from PGM. In contrast, AgnCΔC treatment did not completely remove the α-GlcNAc epitope even when the concentration was 10 units/ml. This result suggests that the C-terminal section of this enzyme containing three CBM32 domains, two FIVAR and a FN3 domain, is functionally important in the recognition of native mucin glycoproteins.
Binding Activities of the CBM Domains toward PGMs
To characterize the crucial function of the C-terminal tandem CBM domains on substrate recognition, several CBM constructs tagged with GST were expressed, and the binding toward mucins was tested. First, the binding toward glycoproteins/proteoglycans was determined by a dot blot overlay assay. Glycoproteins/proteoglycans were blotted onto the membrane, and CBMs were overlaid (Fig. 4A). CBM(C2–C4), CBM(C2–C5), and CBM(C2–C6)-FIVAR showed strong binding for PGM and moderate binding for crude PGM, human gastric mucin, and bovine submaxillary mucin. Strikingly, CBM(C2–C6) showed strong binding for all three mucin samples; for purified PGM, the membrane may be overexposed. In contrast, all CBMs did not bind fetuin and asialofetuin, which have N- and O-glycans. Next, we quantified the amount of bound CBMs toward PGM by ELISA. CBMs were added to the wells coated by PGM using a HIK1083 antibody and quantified using an anti-GST antibody (Fig. 4B). As observed in the dot blot overlay assay, CBM(C2–C6) showed remarkably higher binding activity than the other positive constructs.
FIGURE 4.
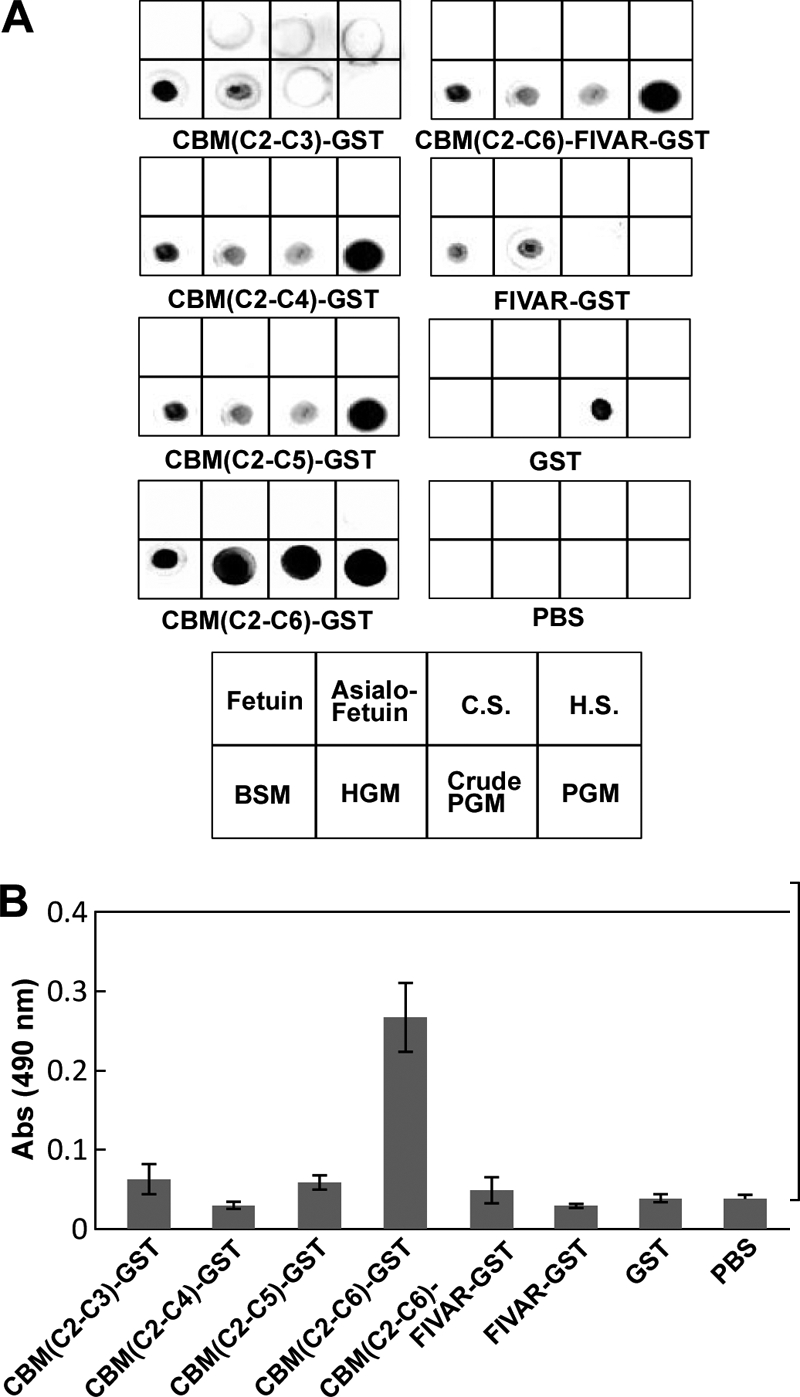
Binding assay of CBM domains toward PGM. A, dot blot overlay assay for the binding of the CBMs to glycoproteins/proteoglycans. 10 μg of fetuin, asialo fetuin, chondroitin sulfate (C.S.), heparin sulfate (H.S.), bovine submaxillary gland mucin, human gastric mucin (HGM), crude PGM, and PGM were dot-blotted onto the membranes. The membranes were incubated with the indicated CBM constructs tagged with GST at the C terminus. Bound proteins were detected by an anti-GST antibody. B, sandwich ELISA for the binding of the CBMs to PGM. CBM constructs were added to PGM-coated wells using the HIK1083 antibody and quantified using an anti-GST antibody. Experiments were carried out in triplicate, and the results are presented as mean ± S.D. (error bars).
Action of αGNase on Cell Surface Mucin
To evaluate the hydrolysis of αGlcNAc expressed on the surface of mammalian cells, we used AGS-α4GnT cells, which stably express GlcNAcα1,4Galβ1 epitopes on the cell surface. Cells previously fixed with formalin were incubated with AgnCΔC and then stained with the HIK1083 antibody. The cells treated with the enzyme stained much weaker than the cells that represented the control group (Fig. 5A). The results were essentially the same when living cells were treated with enzyme before fixation (Fig. 5B). These results clearly showed that AgnCΔC acts on GlcNAcα1,4Galβ1R at the non-reducing end of glycoproteins located on the surface of intact cells. The results were essentially identical when AgnC was tested at the same enzyme concentration (data not shown).
FIGURE 5.

Removal of α-GlcNAc epitopes on mammalian cell surfaces by AgnC. αGlcNAc-containing mucin-expressing AGS-α4GnT cells were treated with AgnCΔC and stained using the HIK1083 antibody followed by FITC-conjugated anti-mouse IgM. Images were analyzed using a fluorescence microscope (bar, 50 μm). A, AGS-α4GnT cells were fixed by formalin and treated with AgnCΔC. B, living AGS-α4GnT cells in the DMEM were treated with AgnCΔC. A-control and B-control, no enzyme; A-1 and B-1, 10.2 milliunits/well; A-2 and B-2, 40.2 milliunits/well.
Stereochemistry of the Hydrolysis Catalyzed by the Recombinant αGNase
The stereochemical course of the hydrolysis of His6-tagged AgnCΔC was determined by 1H NMR using GlcNAcα1pNP as a substrate (Fig. 6). The chemical shifts (anomeric proton of GlcNAcαOH was δ = 5.25 ppm, J = 3.4 Hz; that of GlcNAcβOH was δ = 4.75 ppm, J = 8.3 Hz) of the two anomers of GlcNAc were assigned prior to the experiment. The ratio of GlcNAcαOH/GlcNAcβOH was 65:35 based on the intensities of the signals in the NMR spectrum. After recording a reference spectrum (t = 0 min), the enzyme was added to the reaction mixture. Between 9 and 20 min, the signal derived from the anomeric equatorial proton of GlcNAcα1pNP of the substrate decreased slightly, and a new doublet, which corresponded to the GlcNAcαOH, appeared. Between 20 and 34 min, a second new peak (4.75 ppm) was detected, which corresponded to the axial proton of the GlcNAcβOH. At this time, the α/β anomer ratio of the GlcNAcOH was 86:14. As the reaction proceeded, the signals of the anomeric protons of the hydrolyzed products increased, and the ratio of the α/β anomer of the GlcNAc gradually changed to reach an equilibrium (α/β ratio = 65/35). These results indicate that hydrolysis by AgnCΔC proceeded with retention of the anomeric configuration.
FIGURE 6.

1H NMR spectra showing the hydrolysis of GlcNAcα1pNP after the addition of AgnCΔC. A, reaction scheme and assignment of the anomeric protons. B, the reaction was monitored by 1H NMR at different times, and the regions of the signals for the anomeric protons (4.4–6.1 ppm) are shown. The signals for the equatorial anomeric proton of the GlcNAc residue (Ha′eq; 5.8 ppm, J = 3.4 Hz) appeared after 9 min, and then the signals for the axial anomeric proton of the GlcNAc residue (Ha′ax; 4.75 ppm, J = 8.3 Hz) appeared after 20 min as a consequence of mutarotation. The large signal around δ = 4.65 is the HDO signal.
Physiological Roles of AgnC in the Growth of C. perfringens
The agnC gene of C. perfringens strain 13 was disrupted by inserting, in the sense orientation, a group II intron (∼900 bp) between nucleotides 303 and 304 of the agnC ORF (Fig. 7A (I)). The presence of an intron insertion in the agnC gene was first shown by PCR using agnC-specific primers (Fig. 7A (II)). Sequencing of the agnC gene then confirmed that the group II intron had inserted into the expected site of the agnC ORF. The presence of a single intron insertion in the agnC disruptant was shown by Southern blot analysis (Fig. 7, A (III)). The αGNase assay using GlcNAcα1,4Galβ1pMP as a substrate demonstrated that agnC disruptant completely lost αGNase activity (Fig. 7B, lane 5), indicating that AgnC is the only enzyme that removes the α-GlcNAc cap.
FIGURE 7.

Disruption of the agnC gene in C. perfringens. A, intron-based disruption to create an agnC null mutant. Wild type and intron-inserted agnC genes and the annealing sites of primers for PCR are graphically depicted (I). II, PCR analysis using genomic DNA from wild type strain and agnC disruptant as templates. III, Southern blot analysis of wild type strain and an agnC disruptant. B, αGNase activity of wild type strain or agnC disruptant. GlcNAcα1,4Galβ1pMP (GnG) was incubated with each bacterial culture supernatant at 37 °C for 20 h and analyzed by TLC. Lane 1, standard GlcNAc (Gn); lane 2, GlcNAcα1,4Galβ1pMP; lane 3, GlcNAcα1,4Galβ1pMP incubated with culture supernatant from wild type strain; lane 4, culture supernatant from wild type strain; lane 5, GlcNAcα1,4Galβ1pMP incubated with culture supernatant from agnC disruptant; lane 6, culture supernatant from agnC disruptant. C, growth of wild type strain and agnC disruptant. Wild type strain (filled symbols) and agnC disruptant (open symbols) were cultured at 37 °C under anaerobic conditions in minimal medium supplemented with 0.5% gastric mucin (I); minimal medium supplemented with 10 mm glucose (II); and GAM (diamonds) and minimal (squares) media (III). The assay was performed independently three times, and the results are presented as mean ± S.D. (error bars).
To investigate whether AgnC is involved in the utilization of αGlcNAc-capped mucin, we compared the growth of wild type and agnC disruptant. When the strains were grown in GAM medium, there was no difference of the growth between wild type and agnC disruptant (Fig. 7, C (III)). Both wild type and agnC disruptant were unable to grow on minimal medium in the absence of carbohydrate sources (Fig. 7C (III)). Growth of wild type and agnC disruptant on 10 mm glucose in minimal medium was similar (Fig. 7C (II)); however, on 0.5% gastric mucin, agnC disruptant decreased the growth rate compared with wild type (Fig. 7C (I)). These results suggest that GH89 αGNase (AgnC) of C. perfringens strain 13 plays an important role in the utilization of αGlcNAc-containing class III mucin.
DISCUSSION
The GH89 family is composed exclusively of αGNases and is distributed from bacteria to higher eukaryotes. The mammalian αGNases have been well characterized as lysosomal enzymes that degrade heparin and heparan sulfate. In contrast, αGNases from other organisms have been poorly investigated. We have previously expressed AgnCΔC, a deletion protein of αGNase encoded by CPE0866 in the genome of C. perfringens strain 13 and reported preliminary results on its properties (31). The x-ray crystal structure of αGNase (CpGH89 encoded by CPF_0859) from C. perfringens ATCC 13124 was previously reported as a model protein of human αGNase. Here, a truncated protein construct containing an N-terminal CBM32 domain and a GH89 catalytic domain (aa 23–893) was used for the overexpression and crystallization analysis. The enzyme activity of CpGH89 was confirmed using only synthetic substrates, such as GlcNAcα1pNP (20). Consequently, the natural substrate for CpGH89 was not identified. In this study, we expressed AgnC from C. perfringens strain 13, which shares 97% identity with CpGH89 from C. perfringens ATCC 13124 and found that AgnC hydrolyzed not only GlcNAcα1pNP but also GlcNAcα1,4Gal in O-glycans of gastroduodenal mucin. AgnC was highly specific toward α1,4-linked GlcNAc (i.e. the enzyme showed no catalytic activity toward GlcNAcα1,3Gal or GlcNAcα1,6Gal). In addition, AgnC did not hydrolyze GlcNAcα1,4GlcA, a repetitive unit of heparin and heparan sulfate (Fig. 2). This result indicates that AgnC is not a heparin/heparan sulfate-degrading enzyme. GlcNAcα1,4IdoA also occurs in heparin and heparan sulfate; however, the activity of AgnC toward the disaccharide has not been examined because its chemical synthesis was difficult (21). We also found that heterologously expressed human αGNase acted on GlcNAcα1,4GlcAβ1pMP but not on GlcNAcα1,4Galβ1pMP,4 suggesting that the different substrate specificities may be due to the different structures of the catalytic sites of the two enzymes.
We have successfully expressed full-length AgnC and a truncated version of the protein, AgnCΔC, using the pCold vector system. Both enzymes showed similar kinetic parameters against GlcNAcα1pNP; however, the reactivity of AgnC for PGM was significantly higher than that of AgnCΔC (Fig. 3). Conceivably, AgnC prefers branched and/or clustered O-glycans over small monomeric substrates, such as GlcNAcα1,4Galβ1pMP and GlcNAcα1pNP. The C-terminal region of AgnC may enhance the affinity for PGM. In this region, there are three CBM32 domains (C4–C6), two FIVAR domains, and one FN3 domain. The results of binding assays using various C-terminal protein constructs indicated that the tandem CBM region containing C4–C6 is strongly involved in binding to PGM. Thus, these three CBMs are required for high affinity toward PGM. CBM32 domains are frequently found in microbial glycosidases (32–34). Several CBM32s have been characterized to recognize Gal or GlcNAc (27). The C5 CBM32 domain of AgnC shows 42% homology with the CBM32 of clostridial β-N-acetylhexosaminidase (CpGH84), which was shown to recognize Gal, N-acetyllactosamine (LacNAc), or 2′-fucosyl-LacNAc by crystallographic analyses (27, 28, 35). Because residues interacting with Gal are highly conserved in the CBM32s of AgnC, they may also recognize Gal residues in O-glycans of the class III mucin and thus facilitate the hydrolysis reaction. The FIVAR and FN3 domains are also found in many microbial glycosidases, but the functions of these modules remain unresolved.
C. perfringens represents one of the major intestinal pathogens in humans. This enterobacterium has a series of mucin-degrading enzymes. The most important enzyme is endo-α-N-acetylgalactosaminidase, which hydrolyzes the glycosidic bond between α-GalNAc and Thr/Ser to release both the core-1 disaccharide and core-2 trisaccharide from mucin (36). To expose the core structures, this bacterium secretes several endo- and exoglycosidases. Three unique endo-β-galactosidases have been reported to act on the terminal sugar epitopes: Galα1,3Gal-releasing endo-β-galactosidase C (37), GlcNAcα1,4Gal-releasing endo-β-galactosidase (GngC) (15, 38), and blood group A- and B-trisaccharide-releasing endo-β-galactosidase (39). The exoglycosidases, such as sialidases (29), α-fucosidases (40), and β-N-acetylhexosaminidases (28, 35, 41), are also present in this pathogen. We have previously speculated that the GlcNAcα1,4Gal epitope is first released by GngC, and then the disaccharide is hydrolyzed by αGNase. However, the candidate gene encoding GngC has not been found in the genome of C. perfringens strain 13. Thus, the removal of the α-GlcNAc cap may be mediated by either αGNase or GngC. In fact, C. perfringens strain 13 showed αGNase activity but not GlcNAcα1,4Gal-releasing GngC activity (Fig. 7B). The agnC knock-out C. perfringens strain 13, which is unable to remove α-GlcNAc cap, showed significantly reduced growth in gastric mucin-containing minimal medium that may mimic the carbohydrate-limited environment of the lower intestine (Fig. 7C).
In conclusion, our results suggest that the natural substrate of clostridial αGNases is the α-GlcNAc-containing class III mucin secreted from the stomach and duodenum. The secreted mucin may flow into the intestines, where the enzyme plays an essential role in the utilization of α-GlcNAc-capped O-glycans.
Supplementary Material
Acknowledgments
We thank Dr. T. Shimizu (Kanazawa University) for providing C. perfringens strain 13 and its genomic DNA; M. Mori (The Noguchi Institute) and Drs. K. Haneda and M. Kurihara (Kanagawa Institute of Technology) for providing PGM; and Dr. T. Katayama (Ishikawa Prefectural University) for critically reading the manuscript.
This work was supported in part by a grant from the Kieikai Research Foundation (to H. A.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2.
M. Fujita, A. Tsuchida, K. Goto, K. Osumi, Y. Hirose, and M. Mizuno, unpublished results.
- α4GnT
- α1,4-N-acetylglucosaminyltransferase
- αGNase
- α-N-acetylglucosaminidase
- GH89
- glycoside hydrolase
- HMBC
- 1H-detected heteronuclear multiple-bond correlation
- HMQC
- 1H-detected heteronuclear quantum coherence
- PGM
- porcine gastric mucin
- pMP
- p-methoxyphenyl
- pNP
- p-nitrophenyl
- TF
- trigger factor
- FIVAR
- found in various architectures
- FN3
- fibronectin type 3
- nt
- nucleotides
- aa
- amino acids
- ESI
- electrospray ionization.
REFERENCES
- 1. Parolis H., Parolis L. A., Stanley S. M., Dutton G. G. (1990) Carbohydr. Res. 200, 449–456 [DOI] [PubMed] [Google Scholar]
- 2. Jann B., Shashkov A. A., Kochanowski H., Jann K. (1994) Carbohydr. Res. 264, 305–311 [DOI] [PubMed] [Google Scholar]
- 3. Hsieh T. C., Lester R. L., Laine R. A. (1981) J. Biol. Chem. 256, 7747–7755 [PubMed] [Google Scholar]
- 4. Lloyd K. O., Kabat E. A., Beychok S. (1969) J. Immunol. 102, 1354–1362 [PubMed] [Google Scholar]
- 5. Van Halbeek H., Gerwig G. J., Vliegenthart J. F., Smits H. L., Van Kerkhof P. J., Kramer M. F. (1983) Biochim. Biophys. Acta 747, 107–116 [DOI] [PubMed] [Google Scholar]
- 6. Ishihara K., Kurihara M., Goso Y., Urata T., Ota H., Katsuyama T., Hotta K. (1996) Biochem. J. 318, 409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ota H., Hayama M., Nakayama J., Hidaka H., Honda T., Ishii K., Fukushima M., Uehara T., Kurihara M., Ishihara K., Hotta K., Katsuyama T. (2001) Am. J. Clin. Pathol. 115, 69–79 [DOI] [PubMed] [Google Scholar]
- 8. Mikami Y., Kiyokawa T., Hata S., Fujiwara K., Moriya T., Sasano H., Manabe T., Akahira J., Ito K., Tase T., Yaegashi N., Sato I., Tateno H., Naganuma H. (2004) Mod. Pathol. 17, 962–972 [DOI] [PubMed] [Google Scholar]
- 9. Utsugi K., Hirai Y., Takeshima N., Akiyama F., Sakurai S., Hasumi K. (1999) Gynecol. Oncol. 75, 345–348 [DOI] [PubMed] [Google Scholar]
- 10. Nakayama J., Yeh J. C., Misra A. K., Ito S., Katsuyama T., Fukuda M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 8991–8996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawakubo M., Ito Y., Okimura Y., Kobayashi M., Sakura K., Kasama S., Fukuda M. N., Fukuda M., Katsuyama T., Nakayama J. (2004) Science 305, 1003–1006 [DOI] [PubMed] [Google Scholar]
- 12. Lee H., Kobayashi M., Wang P., Nakayama J., Seeberger P. H., Fukuda M. (2006) Biochem. Biophys. Res. Commun. 349, 1235–1241 [DOI] [PubMed] [Google Scholar]
- 13. Lee H., Wang P., Hoshino H., Ito Y., Kobayashi M., Nakayama J., Seeberger P. H., Fukuda M. (2008) Glycobiology 18, 549–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kobayashi M., Lee H., Nakayama J., Fukuda M. (2009) Curr. Drug. Metab. 10, 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ashida H., Anderson K., Nakayama J., Maskos K., Chou C. W., Cole R. B., Li S. C., Li Y. T. (2001) J. Biol. Chem. 276, 28226–28232 [DOI] [PubMed] [Google Scholar]
- 16. Roseman S., Dorfman A. (1951) J. Biol. Chem. 191, 607–620 [PubMed] [Google Scholar]
- 17. Weber B., Blanch L., Clements P. R., Scott H. S., Hopwood J. J. (1996) Hum. Mol. Genet. 5, 771–777 [DOI] [PubMed] [Google Scholar]
- 18. von Figura K. (1977) Eur. J. Biochem. 80, 535–542 [DOI] [PubMed] [Google Scholar]
- 19. Hopwood J. J., Elliott H. (1982) Clin. Chim. Acta 120, 77–86 [DOI] [PubMed] [Google Scholar]
- 20. Ficko-Blean E., Stubbs K. A., Nemirovsky O., Vocadlo D. J., Boraston A. B. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6560–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmidt R. R., Michel J., Roos M. (1984) Liebigs Ann. Chem. 1343–1357 [Google Scholar]
- 22. Shimizu T., Ohtani K., Hirakawa H., Ohshima K., Yamashita A., Shiba T., Ogasawara N., Hattori M., Kuhara S., Hayashi H. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 996–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anderson K., Li S. C., Li Y. T. (2000) Anal. Biochem. 287, 337–339 [DOI] [PubMed] [Google Scholar]
- 24. Chen Y., McClane B. A., Fisher D. J., Rood J. I., Gupta P. (2005) Appl. Environ. Microbiol. 71, 7542–7547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Homer K. A., Kelley S., Hawkes J., Beighton D., Grootveld M. C. (1996) Microbiology 142, 1221–1230 [DOI] [PubMed] [Google Scholar]
- 26. Homer K. A., Patel R., Beighton D. (1993) Infect. Immun. 61, 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abbott D. W., Eirín-López J. M., Boraston A. B. (2008) Mol. Biol. Evol. 25, 155–167 [DOI] [PubMed] [Google Scholar]
- 28. Ficko-Blean E., Boraston A. B. (2006) J. Biol. Chem. 281, 37748–37757 [DOI] [PubMed] [Google Scholar]
- 29. Boraston A. B., Ficko-Blean E., Healey M. (2007) Biochemistry 46, 11352–11360 [DOI] [PubMed] [Google Scholar]
- 30. Weissmann B., Rowin G., Marshall J., Friederici D. (1967) Biochemistry 6, 207–214 [DOI] [PubMed] [Google Scholar]
- 31. Fujita M., Kobayashi N., Tsuchida A., Goto K., Osumi K., Mizuno M., Yamanoi T., Ashida H., Haneda K., Nakayama J. (2007) Glycoconj. J. 24, 326 [Google Scholar]
- 32. Fujita K., Oura F., Nagamine N., Katayama T., Hiratake J., Sakata K., Kumagai H., Yamamoto K. (2005) J. Biol. Chem. 280, 37415–37422 [DOI] [PubMed] [Google Scholar]
- 33. Ashida H., Miyake A., Kiyohara M., Wada J., Yoshida E., Kumagai H., Katayama T., Yamamoto K. (2009) Glycobiology 19, 1010–1017 [DOI] [PubMed] [Google Scholar]
- 34. Miwa M., Horimoto T., Kiyohara M., Katayama T., Kitaoka M., Ashida H., Yamamoto K. (2010) Glycobiology 20, 1402–1409 [DOI] [PubMed] [Google Scholar]
- 35. Ficko-Blean E., Gregg K. J., Adams J. J., Hehemann J. H., Czjzek M., Smith S. P., Boraston A. B. (2009) J. Biol. Chem. 284, 9876–9884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ashida H., Maki R., Ozawa H., Tani Y., Kiyohara M., Fujita M., Imamura A., Ishida H., Kiso M., Yamamoto K. (2008) Glycobiology 18, 727–734 [DOI] [PubMed] [Google Scholar]
- 37. Ogawa H., Muramatsu H., Kobayashi T., Morozumi K., Yokoyama I., Kurosawa N., Nakao A., Muramatsu T. (2000) J. Biol. Chem. 275, 19368–19374 [DOI] [PubMed] [Google Scholar]
- 38. Ashida H., Maskos K., Li S. C., Li Y. T. (2002) Biochemistry 41, 2388–2395 [DOI] [PubMed] [Google Scholar]
- 39. Anderson K. M., Ashida H., Maskos K., Dell A., Li S. C., Li Y. T. (2005) J. Biol. Chem. 280, 7720–7728 [DOI] [PubMed] [Google Scholar]
- 40. Gregg K. J., Finn R., Abbott D. W., Boraston A. B. (2008) J. Biol. Chem. 283, 12604–12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ficko-Blean E., Boraston A. B. (2009) J. Mol. Biol. 390, 208–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.