

Abstract
We identified YDR499W as a Saccharomyces cerevisiae open reading frame with homology to several checkpoint proteins, including S.cerevisiae Rfc5p and Schizosaccharomyces pombe Rad26. Disruption of YDR499W (termed LCD1) results in lethality that is rescued by increasing cellular deoxyribonucleotide levels. Cells lacking LCD1 are very sensitive to a range of DNA-damaging agents, including UV irradiation, and to the inhibition of DNA replication. LCD1 is necessary for the phosphorylation and activation of Rad53p in response to DNA damage or DNA replication blocks, and for Chk1p activation in response to DNA damage. LCD1 is also required for efficient DNA damage-induced phosphorylation of Rad9p and for the association of Rad9p with the FHA2 domain of Rad53p after DNA damage. In addition, cells lacking LCD1 are completely defective in the G1/S and G2/M DNA damage checkpoints. Finally, we reveal that endogenous Mec1p co-immunoprecipitates with Lcd1p both before and after treatment with DNA-damaging agents. These results indicate that Lcd1p is a pivotal checkpoint regulator, involved in both the essential and checkpoint functions of the Mec1p pathway.
Keywords: checkpoint/DNA damage/MEC1/protein phosphorylation/RAD53
Introduction
DNA damage can have deleterious consequences for survival and cells have adopted multiple strategies for tolerating damage to their genetic material. Several different, well conserved repair systems can physically remove or bypass specific types of DNA lesion (Friedberg et al., 1995). Also, in response to DNA damage, cells can slow down their progression through different cell cycle phases, to provide time for repair to occur and to prevent mutations from being propagated. These cell cycle delays, termed ‘checkpoints’ (Weinert and Hartwell, 1988), prevent replication of damaged DNA (G1/S and intra-S checkpoints) or segregation of damaged chromosomes (the G2/M checkpoint). In addition, the DNA replication checkpoint ensures that the cell has the appropriate DNA content before entering mitosis.
Much attention has focused on the identification of genes involved in DNA repair and checkpoint control, and the budding yeast Saccharomyces cerevisiae has been instrumental in this regard. The first yeast checkpoint gene was identified by Weinert and Hartwell (1988), who showed that the RAD9 gene is required for the G2/M checkpoint in response to DNA damage. Subsequent work from many laboratories has shown that RAD9 is part of the MEC1 pathway—a complex protein phosphorylation cascade that is activated in response to DNA damage and inhibition of DNA replication (reviewed in Lowndes and Murguia, 2000). Mec1p is a member of a family of large protein kinases, termed PIKKs, which have sequence similarity in their catalytic domain to phosphatidylinositol 3-kinase. In some circumstances, Mec1p functions redundantly with Tel1p (Greenwell et al., 1995; Morrow et al., 1995; Sanchez et al., 1996; Vialard et al., 1998), another PIKK and the closest S.cerevisiae relative of Mec1p. PIKKs are conserved throughout evolution, with homologues of Mec1p and Tel1p being found in Schizosaccharomyces pombe (termed Rad3 and Tel1, respectively) and in humans (ATR and ATM). In S.pombe, Rad3 appears to be constitutively bound to the Rad26 protein (Lindsay et al., 1998; Martinho et al., 1998), which is the only component of the Rad3 pathway that, when mutated, leads to a phenotype as severe as that caused by mutations in Rad3 (Al-Khodairy et al., 1994; Lindsay et al., 1998). This indicates that Rad26 plays a crucial role in the Rad3 pathway.
Schizosaccharomyces pombe Rad3 is not essential for cell viability, whereas disruption of the S.cerevisiae MEC1 gene causes lethality (Kato and Ogawa, 1994; Weinert et al., 1994). However, this lethality can be suppressed by increasing the intracellular concentration of deoxyribonucleotides (dNTPs), either by overexpression of the catalytic subunits (Rnr1p, Rnr3p) of the ribonucleotide reductase (RNR) tetramer (Desany et al., 1998), which regulates the rate-limiting step of dNTP synthesis, or by disrupting the gene encoding Sml1p (Zhao et al., 1998), which directly binds to and inhibits Rnr1p (Chabes et al., 1999). At present, the molecular basis for the essential function of MEC1 during the normal cell cycle is unclear. In addition, as is the case for S.pombe cells lacking Rad3 (Lindsay et al., 1998), S.cerevisiae cells disrupted for MEC1 function are exquisitely sensitive to the DNA replication inhibitor hydroxyurea (HU) and to a wide range of DNA-damaging agents (Weinert et al., 1994; Sanchez et al., 1996; Desany et al., 1998). Indeed, an intact MEC1 gene is required for the DNA replication checkpoint and for DNA damage checkpoint responses at all cell cycle stages (Weinert et al., 1994; Paulovich and Hartwell, 1995). Although increasing intracellular dNTP levels overcomes the lethality of cells lacking MEC1, the checkpoint defects and sensitivity to genotoxic insults of these cells, or of cells containing hypomorphic alleles of MEC1, are not affected by RNR overexpression or by deletion of SML1 (Desany et al., 1998; Zhao et al., 1998).
It has not yet been demonstrated that Mec1p has intrinsic protein kinase activity, but it has been shown that several key regulators of the Mec1p-dependent signalling pathway become phosphorylated in a MEC1-dependent manner in response to DNA damage or if DNA replication is blocked. For example, the phosphorylation state and protein kinase activity of Rad53p increase in response to DNA-damaging agents or HU in a MEC1-dependent fashion (Allen et al., 1994; Sun et al., 1996), and in the presence of these agents, cells harbouring mutations in RAD53 rapidly lose viability (Allen et al., 1994; Sun et al., 1996; Fay et al., 1997). Rad53p thus acts as a downstream effector of the Mec1p-dependent signalling pathway. Like MEC1, RAD53 is an essential gene, and overexpression of RNR subunits or deletion of SML1 can suppress the lethal phenotype of null mutations within RAD53 (Allen et al., 1994; Desany et al., 1998; Zhao et al., 1998). The Chk1p protein kinase also lies downstream of Mec1p on a branch of the pathway that is at least partly distinct from the branch that involves Rad53p (Sanchez et al., 1999; J.Rouse and S.P.Jackson, unpublished data). Signifi cantly, work from several laboratories has shown that phosphorylation of Rad53p and Chk1p in response to DNA damage requires an intact RAD9 gene, and that exposure of cells to DNA damage results in rapid and sustained hyperphosphorylation of Rad9p, in a manner that depends on MEC1 (de la Torre-Ruiz et al., 1998; Emili, 1998; Vialard et al., 1998; Sanchez et al., 1999). In this regard then, Rad9p functions downstream of Mec1p, but upstream of the effector kinases Chk1p and Rad53p.
The RAD24 epistasis group (RAD24, RAD17, MEC3 and DDC1) functions additively with RAD9, but on a separate branch of the pathway, in mediating phosphorylation and activation of Rad53p in response to DNA damage (Paulovich et al., 1997; de la Torre-Ruiz et al., 1998). Mutations in any of these genes result in sensitivity to DNA damage, but not HU, and cause defects in both the G1/S and G2/M DNA damage checkpoints (Lydall and Weinert, 1995; Paulovich and Hartwell, 1995; Longhese et al., 1997). Notably, Rad24p has sequence similarity to the small subunits of replication factor C (RFC; Griffiths et al., 1995). The RFC complex, which comprises a large subunit (Rfc1p) and four small subunits (Rfc2p–5p; Cullmann et al., 1995), is a key regulator of initiation of DNA synthesis. Two groups have shown that Rad24p forms a distinct complex with the small RFC subunits, but not with Rfc1p (Shimomura et al., 1998; Green et al., 2000). In addition, cells harbouring temperature-sensitive alleles of either RFC2 (Noskov et al., 1998) or RFC5 (Sugimoto et al., 1996, 1997) have defective DNA damage and replication checkpoints, and fail to activate Rad53p in response to genotoxic agents.
From several database searches, we identified S.cerevisiae YDR499W as a previously uncharacterized open reading frame (ORF) whose product has amino acid sequence homology to several DNA repair and DNA damage checkpoint proteins, including budding yeast Rfc5p and Rad50p, and S.pombe Rad26. Here, we show that disruption of this gene (termed LCD1 for lethal, checkpoint-defective, DNA damage sensitive) results in lethality that can be rescued by increasing cellular dNTP levels. Cells lacking LCD1 are extremely sensitive to DNA damage and to inhibition of DNA replication, and have major cell cycle checkpoint defects. Furthermore, we reveal that the activation of key effector molecules of the MEC1 pathway requires the LCD1 gene, and that Lcd1p interacts physically with Mec1p. These results identify Lcd1p as a key checkpoint component and provide insights into the molecular basis for Mec1p-dependent signalling responses.
Results
Saccharomyces cerevisiae LCD1 contains sequence motifs found in several DNA repair and cell cycle checkpoint proteins
When the sequence of S.cerevisiae Rfc5p was used in a BLAST search of the yeast protein database, the best matches corresponded to the small subunits of RFC, Rad24p (Griffiths et al., 1995; Green et al., 2000) and one other ORF that is situated on chromosome IV: Ydr499w (Figure 1A). For reasons discussed below, we hereafter refer to the YDR499W gene as LCD1. Lcd1p has a high proportion of acidic residues (pI of 5.3) and has a predicted molecular mass of 87 kDa. The observed sequence homology (25% identity, 45% similarity; Figure 1B) extends over a region of ∼180 amino acid residues that align with the C-terminal half of Rfc5p. Several of the residues conserved between Rfc5p and Lcd1p were found to be conserved amongst Rfc5 proteins from diverse species (data not shown), suggesing that these residues are likely to be important for biological function. Interest ingly, we also noticed sequence homology between Lcd1p and Rad50p (data not shown), which is involved in DNA double-strand break repair in S.cerevisiae, and between Lcd1p and S.pombe Rad26, a fission yeast checkpoint gene with no obvious homologue in S.cerevisiae (Figure 1A). The degree of homology between Lcd1p and Rad26, however, is low and occurs over three small patches (Figure 1C) with the largest patch showing 23% overall identity. This latter region is predicted to form a coiled-coil structure in both proteins. Rad26 does not have detectable homology, however, to either Rfc5p or Rad50p. Because the most similar sequences to Lcd1p in several organisms are involved in cell cycle checkpoints and/or DNA repair, we investigated the potential roles for this uncharacterized protein in these cellular processes.

Fig. 1. Lcd1p shows sequence homology to several checkpoint proteins and disruption of LCD1 results in lethality that can be rescued by RNR3 overexpression. (A) Schematic representation of regions of sequence homology between Lcd1p and various checkpoint proteins. (B and C) The entire amino acid sequence of S.cerevisiae Ydr499w (B) or the entire S.pombe Rad26 amino sequence (C) was used as query in a WU-BLAST search at the Saccharomyces Genome Database (genome-www.stanford.edu/Saccharomyces/). Sequences were aligned using the ClustalX program and the Boxshade Server (www.ch.embnet.org/). Identical amino acid residues are shaded in black and similar residues are highlighted in grey. (D) Ten-fold serial dilutions of cells containing pLCD1, and either pRNR3-TRP1 or an empty vector (pRS414) that contains the TRP1 gene, were plated onto SC-TRP and SC-TRP containing 5-FOA, and incubated for 4 days at 30°C. LCD1 indicates the parental strain carrying pRS414 and pLCD1.
Disruption of LCD1 causes lethality that can be suppressed by increasing cellular dNTP pools
To investigate the functions of Lcd1p, we analysed the consequences of ablating the gene encoding it. Sporulation and subsequent tetrad analysis of diploid cells in which one copy of LCD1 was disrupted showed that a maximum of two of four spores in each tetrad was viable, and in all cases viability failed to segregate with the marker used to disrupt LCD1 (data not shown). This suggests that an intact LCD1 gene is required for cell growth. There are several potential explanations for the lethality of LCD1 disruption. As mentioned earlier, Lcd1p has sequence similarity to both S.cerevisiae Rfc5p and S.pombe Rad26. Since RFC5 has been shown to interact genetically with the MEC1 pathway, and since Rad26 interacts with Rad3 (the S.pombe homologue of Mec1p), we speculated that Lcd1p might also be involved in regulation of the MEC1 pathway. As discussed above, MEC1 plays an essential role during an unperturbed cell cycle, and the lethality of Δmec1 cells can be rescued either by overexpressing the catalytic subunits of RNR (Desany et al., 1998) or by disrupting the SML1 gene (Zhao et al., 1998). Notably, Southern blotting and PCR analyses showed that whereas LCD1 could not be disrupted in wild-type haploid cells, it could be disrupted in haploid cells overexpressing Rnr3p or lacking SML1 (data not shown). This suggested that the lethality of LCD1 disruption might be rescued by increasing cellular dNTP pools.
To investigate the above possibility more thoroughly, LCD1-disrupted cells containing an RNR3-overexpression plasmid (pRNR3) were transformed with pLCD1, a plasmid harbouring the URA3 gene and LCD1 under the control of its own promoter. These cells were next encouraged to lose pRNR3 by growth in non-selective medium, then the resulting cells were tested for the retention or loss of the plasmid markers and for their ability to grow in the presence of 5-fluoro-orotic acid (FOA). This drug is toxic to cells containing functional URA3, and thus cells that cannot lose the pLCD1 plasmid should die in its presence. These studies revealed that cells still retaining pRNR3 grew well on medium containing or lacking 5-FOA, whereas those that did not harbour this plasmid were only able to grow in the absence but not in the presence of 5-FOA (and thus, in the absence of Rnr3p overexpression, require the pLCD1 plasmid to survive; Figure 1D). In parallel studies, we verified that haploid cells disrupted for SML1 are also able to sustain deletion of LCD1 (data not shown). Taken together, these data indicate that increased dNTP pools are necessary for cellular survival in the absence of LCD1.
Cells lacking LCD1 are sensitive to DNA damage and inhibition of DNA replication
Further analyses of LCD1-disrupted cells revealed that they have an apparently normal cell morphology and cell cycle distribution, and grow at 23, 30 or 37°C, albeit more slowly than wild-type cells at each of these temperatures (data not shown). Given the sequence homology between LCD1 and certain DNA damage response genes, and the similarity between LCD1 and MEC1 in regard to their essential function(s), we were prompted to investigate the potential role of LCD1 in responding to DNA damage. Cells lacking LCD1 are extremely sensitive to UV irradiation (Figure 2A) and to the presence of increasing concentrations of the alkylating agent methyl-methane-sulfonate (MMS; Figure 2B)—agents that primarily cause helix-distorting modifications to DNA. Δlcd1 cells are also hypersensitive to ionizing radiation (IR; Figure 2C), an agent that causes DNA double-strand breaks (DSBs), compared with wild-type cells. Introduction of the pLCD1 plasmid, bearing the LCD1 gene under the regulation of its own promoter, fully rescued the sensitivity of Δlcd1Δsml1 cells to all types of DNA damage examined (data not shown). Significantly, cells lacking LCD1 were at least an order of magnitude more sensitive to MMS or UV than the Δmec1Δsml1 strain (Figure 2A and B). Furthermore, cells lacking both LCD1 and MEC1 were not significantly more sensitive to DNA damage than cells lacking LCD1 alone, indicating that these genes are at least partially epistatic (Figure 2A–C).

Fig. 2. LCD1 disruption results in hypersensitivity to DNA damage and hydroxyurea. Δsml1, Δlcd1Δsml1, Δmec1Δsml1 and Δlcd1Δmec1Δsml1 cells were grown to mid-log phase (OD600 of 0.6) in liquid culture. At this point, in the case of MMS (B) or HU (D), the relevant drug was added and the cells grown at 30°C for a further 2 h. Cell suspensions were diluted 100-fold, plated onto YPD agar and incubated at 30°C for 3 days. In the case of UV irradiation (A), cells were diluted, spread onto YPD agar and plates were placed under a germicidal UV lamp and irradiated at 254 nm, at a delivery rate of 3 J/m2/s. For experiments involving IR (C), diluted cells were irradiated with a Torrex X-ray machine, at a delivery rate of 3.3 Gy/min, before spreading onto YPD agar.
To see whether LCD1 is involved in cellular responses to inhibition of DNA replication, we examined the effect of exposing Δlcd1 mutant cells to HU. This drug inhibits DNA replication, so cells defective in the DNA replication checkpoint (e.g. Δmec1Δsml1 cells) cannot survive in the presence of HU. As shown in Figure 2D, cells lacking LCD1 are extremely sensitive to the presence of HU, even more so than Δmec1Δsm1 mutant cells. These data imply that LCD1 plays a crucial role in allowing cells to survive when DNA replication is hindered.
LCD1 is required for Rad53p activation in response to DNA damage or stalled DNA replication forks and activation of Chk1p in response to DNA damage
Given the phenotypic similarities between cells lacking MEC1 and cells lacking LCD1, we examined the effect of disrupting LCD1 on the function of individual effector molecules of the MEC1 signalling pathway. When normal cells are exposed to DNA damage or cannot complete DNA replication, the phosphorylation state and kinase activity of Rad53p increase in a MEC1-dependent manner (Allen et al., 1994; Sun et al., 1996). This increase in Rad53p activity is thought to elict cell cycle checkpoints (Allen et al., 1994; Sun et al., 1996; Fay et al., 1997) and perhaps to orchestrate DNA repair (see Discussion). To see whether activation of Rad53p depends on LCD1, Rad53p kinase activity was determined after treating cells with different agents, by measuring the ability of Rad53p to autophosphorylate, as described previously (Pellicioli et al., 1999). Rad53p kinase activity is low in untreated cells (U; Figure 3A, top panel), whereas exposure of wild-type cells to MMS (M) or the UV-mimetic drug 4-nitroquinoline oxide (4-NQO; N) stimulates its kinase activity 20- to 25-fold. Similarly, addition of phleomycin (P; Figure 3A) or induction of the restriction endonuclease EcoRI (data not shown), both of which induce DNA DSBs, also leads to activation of Rad53p, albeit to a lower extent.

Fig. 3. LCD1 is required for activation of Rad53p in response to HU or DNA-damaging agents and for activation of Chk1p in response to DNA damage. Cells (all carrying the pRNR3 plasmid) were grown to mid-log phase and incubated in the presence of MMS (0.02%; M), 4-NQO (5 µg/ml; N), phleomycin (5 µg/ml; P) (A and C) or HU (0.1 M; B), or without any drug addition (U), for 2 h at 30°C. After lysis in TCA, extracts were subjected to in situ analysis of Rad53p activity (A and B, top panels) or western blot analysis to assess Rad53p protein (bottom panels; 12% SDS–polyacrylamide gels), using gel conditions that compacted the phosphoforms of this protein. Rad53p autophosphorylation was quantitated using a phosphoimager (middle panels). Phosphorylation of HA-tagged Chk1p was analysed by electrophoresis of extracts from cells transformed with pHA-CHK1 on 10% SDS–polyacrylamide gels (C). Molecular weight markers (kilodaltons) are shown on the right-hand side of the upper panel in each case.
Strikingly, activation of Rad53p in response to any of the above agents is severely reduced in cells lacking LCD1 (Figure 3A), even at the highest concentrations of drugs used (data not shown). In control experiments, the DNA damage-induced autophosphorylating band at 90 kDa (the apparent mass of Rad53p by western blot analysis) was absent in strains disrupted for RAD53, whereas the non-specific bands of lower molecular weight were still present in such strains (data not shown and Pellicioli et al., 1999). Consistent with the above data, the DNA damage-induced hyperphosphorylation of Rad53p that occurs in wild-type strains was found to be essentially abolished in LCD1-disrupted cells (data not shown). Furthermore, the induction of Rad53p kinase activity in response to HU was also abrogated in Δlcd1 mutant cells (Figure 3B, top panel). Importantly, in each case the Rad53p activation defect of the LCD1-disrupted strain was restored by the introduction of pLCD1 (Figure 3A and B), which expresses Lcd1p at endogenous levels, and western blot analysis revealed that the levels of Rad53p protein remained essentially constant in the various genetic backgrounds and conditions employed (Figure 3A and B, lower panels; electrophoresis conditions that compacted the phospho forms of Rad53p were used to aid comparison of protein levels). Taken together, these results reveal that LCD1 plays a crucial role in regulating Rad53p activity in response to a number of genotoxic agents.
It was recently shown that the Chk1p protein kinase also becomes phosphorylated in a MEC1-dependent manner in response to DNA damage (Sanchez et al., 1999; J.Rouse and S.P.Jackson, unpublished data), and it is thought that Chk1p catalyses DNA damage-induced phosphorylation and stabilization of the anaphase inhibitor Pds1p, thereby arresting cells in mitosis (Cohen-Fix and Koshland, 1997; Sanchez et al., 1999). A potential role for LCD1 in phosphorylation of haemagglutinin (HA)-tagged Chk1p (Sanchez et al., 1999) was examined by western blot analysis. Treatment of wild-type cells with MMS or 4-NQO, and to a lesser extent phleomycin, induced an electrophoretic mobility shift of HA-Chk1p (Figure 3C) compared with HA-Chk1p from untreated cells. The band that cross-reacted with the HA antibody was not observed in cells that do not express this HA-Chk1p, and the electrophoretic mobility shift observed is indicative of protein phosphorylation, as treatment of cell extracts with phage λ phosphatase reverses this shift (Sanchez et al., 1999; data not shown). In contrast, Chk1p did not become phosphorylated in response to any of the DNA-damaging agents examined in cells lacking LCD1, and introduction of a plasmid expressing Lcd1p at endogenous levels restored the DNA damage-induced phosphorylation of Chk1p observed in wild-type cells (Figure 3C). Thus, an intact LCD1 gene is required for DNA damage-induced activation of Chk1p.
LCD1 is required for DNA damage-induced phosphorylation of Rad9p
Previous work has established that DNA damage results in the generation of a series of hyperphosphorylated forms of S.cerevisiae Rad9p that migrate more slowly on SDS–polyacrylamide gels than the unmodified protein (Emili et al., 1998; Vialard et al., 1998). Furthermore, in the G1 phase of the cell cycle, DNA damage-induced phosphorylation of Rad9p is dependent on MEC1 and on the RAD24 epistasis group (Emili et al., 1998; Vialard et al., 1998). In contrast, in S- or M-phase, MEC1 and TEL1 function redundantly in this regard and the RAD24 epistasis group is not required (de la Torre-Ruiz et al., 1998). We therefore investigated the potential role of LCD1 in Rad9p phosphorylation by western immunoblot analysis. As shown in Figure 4A, Rad9p becomes hyperphosphorylated in G1-arrested wild-type cells after they have been exposed to 4-NQO (the diminished overall Rad9p signal after DNA damage is because of multiple phosphorylation variants and not a result of changes in Rad9p levels; see the legend to Figure 4). Notably, this induction of Rad9p phosphorylation is markedly reduced in strains disrupted for LCD1 and is also reduced, albeit to a lesser extent, in cells lacking MEC1 (Figure 4A). By contrast, and consistent with previous work (de la Torre-Ruiz, 1998), for asynchronous cultures of wild-type, Δlcd1 mutant or Δmec1 mutant cells, there was little difference in gross phosphorylation of Rad9p in response to either MMS or 4-NQO (data not shown). These data therefore reveal parallels between LCD1 and MEC1 in terms of their effects on Rad9p phosphorylation after DNA damage.

Fig. 4. LCD1 is required for phosphorylation of Rad9p in response to DNA damage and for the association of Rad53p-FHA2 with phosphorylated Rad9p. (A) Cells (all carrying the pRNR3 plasmid) were grown to mid-log phase and incubated in the presence or absence of 4-NQO (5 µg/ml) for 2 h at 30°C, after arresting cells in G1 by addition of α-factor for 2 h. Cells were lysed in TCA, and extracts were subjected to western blot analysis with anti-Rad9p polyclonal antibodies, after electrophoresis on 6.5% SDS–polyacrylamide gels. (B) Cells were treated as described in (A) and native cell extracts were prepared (see Materials and methods). Association of the FHA2 domain from Rad53p fused to GST (upper panel) was analysed as described previously (Durocher et al., 1999). Alternatively, aliquots of native cell extracts were treated with phage λ phosphatase (10 U) for 30 min at 30°C in the presence of 2 mM MnCl2, to dephosphorylate Rad9p. After addition of SDS–PAGE sample buffer, samples were subjected to western blotting with anti-Rad9p antibodies, after electrophoresis on 6.5% SDS–polyacrylamide gels (lower panel). Molecular weight markers (kilodaltons) are shown on the right-hand side of each panel.
After DNA damage, Rad53p interacts with the phosphorylated forms of Rad9p, and this is mediated by each of the two FHA domains of Rad53p (Sun et al., 1998; Durocher et al., 1999). To see whether this interaction is dependent on LCD1, we took cultures of wild-type, Δlcd1 or Δlcd1 cells that were complemented by pLCD1, grew these in the presence or absence of 4-NQO for 2 h, and then generated extracts from them. The extracts were incubated with glutathione–agarose beads containing glutathione S-transferase (GST) alone or a GST fusion of the C-terminal FHA domain (FHA2) of Rad53p. Finally, after washing the beads, we tested for Rad9p association by western immunoblot analysis. Consistent with previous findings (Durocher et al., 1999), in the case of wild-type cells Rad9p binds efficiently to Rad53p-FHA2 following exposure to the DNA-damaging agent (Figure 4B, upper panel). Significantly, disruption of LCD1 leads to a dramatic decrease in binding, and this defect is complemented by pLCD1 (Figure 4B, upper panel; as shown in the lower panel, treatment of extracts with λ protein phosphatase before electrophoresis led to dephosphorylation of Rad9p and allowed the demonstration that overall levels of Rad9p were essentially equal in all samples). Taken together, these results indicate that LCD1 is required for the efficient DNA damage-induced phosphorylation of Rad9p and that, in the absence of LCD1 function, Rad9p is no longer able to bind to the FHA2 domain of Rad53p after exposure to DNA damage.
LCD1 is required for the DNA damage checkpoint in the G1 and G2/M phases of the cell cycle
Work from several laboratories has led to the conclusion that an intact RAD9 gene and activation of Rad53p are required for the DNA damage checkpoint. The defect in DNA damage-induced Rad53p activation and Rad9p phosphorylation seen in Δlcd1 [pRNR3] cells suggested that LCD1 may play a role in checkpoint control. To test this we employed an assay developed by Garvik et al. (1995) that uses a temperature-sensitive allele of CDC13. Cdc13p binds to telomeres, and defects in its gene lead to the generation of single-stranded telomeric DNA that causes prolonged metaphase arrest (Garvik et al., 1995; Lydall and Weinert, 1995; Gardner et al., 1999). In this assay, cells synchronized in G1 by α-factor are released at a temperature that is restrictive for the cdc13-1 allele and, during the following S phase, DNA damage is generated. Checkpoint-proficient cells arrest in metaphase as large budded cells, with a single nucleus positioned at the bud neck. Checkpoint-deficient cells, on the other hand, proceed past metaphase and, to aid detection of these cells, a second temperature-sensitive mutation, cdc15-2, is used to prevent entry into the next cell cycle by arresting cells late in mitosis as large budded cells with a partitioned nucleus (Lydall and Weinert, 1995). For cultures of cdc13-1 cdc15-2 cells containing or lacking pRNR3, there is an almost total metaphase arrest at the restrictive temperature, indicating efficient checkpoint function (Figure 5A). In contrast, cdc13-1 cdc15-2 cells lacking LCD1 fail to mediate checkpoint arrest and instead proceed to the cdc15 arrest point. Indeed, Δlcd cells show the same degree of checkpoint defect in this assay as cells lacking the RAD9 gene (Figure 5A). Consistent with the above data, after release from α-factor arrest, incubation of cells at the restrictive temperature was found to result in the activation of Rad53p in wild-type cells, but not in either Δlcd1 [pRNR3] cells or Δrad9 [pRNR3] cells (Figure 5B). Importantly, the introduction of pLCD1 into the Δlcd1 strain restored both the cdc13-1-induced DNA damage checkpoint (data not shown) and Rad53p activation (Figure 5B). Thus, cells lacking LCD1 show a major defect in the G2/M DNA damage checkpoint, and this correlates with an inability to activate Rad53p. Cells lacking LCD1 are not significantly sensitive to the microtubule poison, nocodazole, compared with cells lacking the spindle checkpoint gene BUB3 (data not shown), and it is therefore unlikely that LCD1 plays a role in the cell cycle checkpoint that monitors spindle integrity and prevents mitosis until each kinetochore has the correct number of microtubles attached.

Fig. 5. LCD1 is required for the G1 and G2/M DNA damage checkpoints. Cells harbouring cdc13-1 and cdc15-2 temperature-sensitive alleles were grown to early log phase at 23°C, arrested in G1 by addition of α-factor (5 µg/ml) for 120 min, and released into YPD at 37°C. (A) Samples were taken every 60 min, fixed and stained with DAPI. The percentage of budded cells with one nucleus in each bud (post-M phase cells) was counted in a population of ∼200 cells by fluorescence microscopy. (B) After 180 min, cells were lysed in TCA and subjected to in situ analysis of Rad53p kinase activity (upper panel). Rad53p autophosphorylation was quantitated using a phosphoimager (lower panel). (C) Cells were arrested in G1 by addition of α-factor for 120 min, washed and released into YPD at 30°C. Samples were taken at 15 min intervals and the proportion of cells with buds was counted in a population of ∼150 cells.
A role for LCD1 in the G1 DNA damage checkpoint was also investigated. To do this, cells were arrested in G1 with α-factor and the culture was split in two; one half was UV irradiated and the other half left untreated. After release from arrest, the effect of UV irradiation on the ability of these cells to re-enter the cell cycle was examined. As shown in Figure 5C, treatment of wild-type cells with UV causes a marked delay in cell cycle re-entry, as judged by the appearance of buds. The time taken for 50% of wild-type cells to start budding after release from G1 increased from ∼40 min without UV irradiation to ∼110 min following UV irradiation. In contrast, for cells disrupted for RAD9, this delay was less pronounced, with ∼50% of cells having budded after ∼70 min. Strikingly, when exposed to UV light, cells lacking LCD1 completely did not exhibit any delay in budding after release from G1 (Figure 5C), suggesting that these cells are completely defective in the G1 DNA damage checkpoint. Taken together with the previous series of experiments, this indicates that LCD1 plays an absolutely crucial role in DNA damage checkpoint response at various stages of the cell cycle.
Lcd1p interacts with Mec1p
The phenotypic similarities of cells defective in LCD1 or MEC1 suggested that the products of these genes might interact. To see whether this is the case, we generated an LCD1 expression plasmid in which LCD1 is under the control of its own promoter and yields an Lcd1p product that contains an HA epitope tag at its N-terminus. Importantly, this plasmid (pHA-LCD1) was found to fully complement the DNA damage hypersensitivity phenotype of Δlcd1 [pRNR3] cells and to complement the lethal phenotype of the chromosomal LCD1 disruption (i.e. pRNR3 could be lost from Δlcd1 cells containing pHA-LCD1 without loss of viability; data not shown). Western blot analysis revealed a band of ∼90 kDa that cross-reacts with anti-HA antibodies in extracts from cells transformed with pHA-LCD1 (Figure 6A, lane 2), but not in extracts from cells transformed with pLCD1 (Figure 6A, lane 1). Next, we introduced pHA-LCD1 or pLCD1 into cells in which the genomic copy of MEC1 had an N-terminal FLAG tag. The presence of this epitope tag did not disrupt any of the known functions of the MEC1 gene (D.Durocher, unpublished data). Extracts from these cells (Figure 6A, lane 4), but not from cells with untagged MEC1 (Figure 6A, lane 3), showed a band of ∼260 kDa that cross-reacts with anti-FLAG antibodies in western blot analysis.
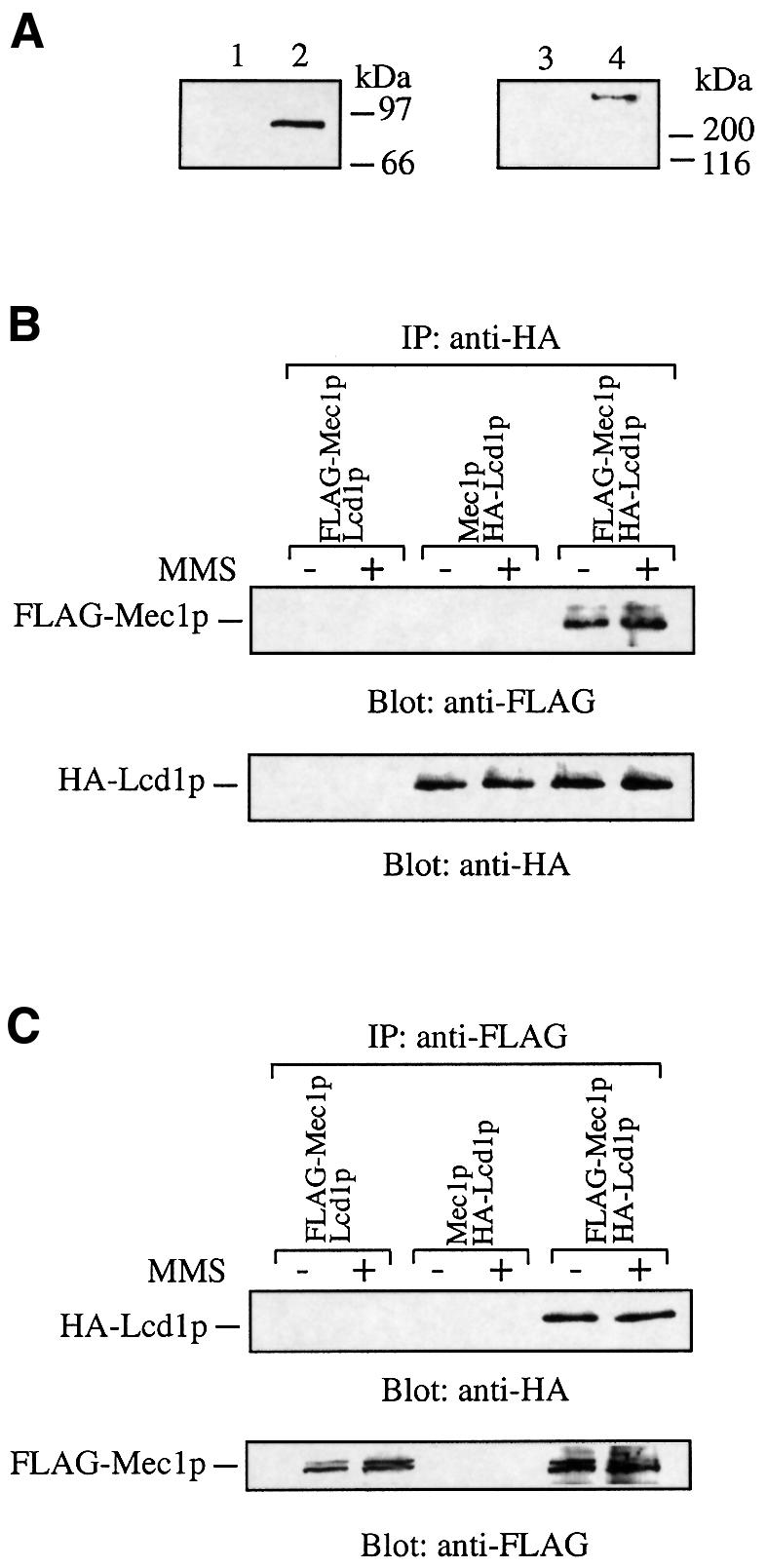
Fig. 6. Lcd1p interacts with Mec1p. (A) Extracts (150 µg) from Δlcd1 cells transformed with either pLCD1 (lane 1) or pHA-LCD1 (lane 2) were electrophoresed on a 7.5% SDS–polyacrylamide gel and subjected to western blot analysis with anti-HA antibodies (left panel). Extracts (150 µg) from cells in which the endogenous copy of the MEC1 gene did (lane 4) or did not (lane 3) bear an N-terminal FLAG–histidine tag were electrophoresed on a 6.5% SDS–polyacrylamide gel and subjected to western blot analysis with anti-FLAG antibodies (right panel). (B and C) Cells containing pHA-LCD1, in which the endogenous copy of the MEC1 gene bore an N-terminal FLAG–histidine tag, were incubated in the presence or absence of MMS (0.02%) for 2 h at 30°C. Native extracts were prepared, and aliquots (2 mg) were incubated with anti-HA antibodies (B) or anti-FLAG antibodies (C) conjugated to protein G–Sepharose. After shaking for 2 h at 4°C, the beads were washed and subjected to western blot analysis with the indicated antibody. As a control, extracts from cells with FLAG-tagged MEC1 gene bearing a pLCD1 or cells with an untagged genomic copy of MEC1, containing pHA-LCD1 or parental cells (data not shown), were subjected to the same analysis.
To see whether Lcd1p can interact with Mec1p, we generated extracts from these strains, immunoprecipitated HA-Lcd1p or FLAG-Mec1p and tested the precipitates for the presence of either protein by western immunoblot analysis. As shown in Figure 6B, FLAG-tagged Mec1p was present in anti-HA immunoprecipitates, using extracts from cells expressing HA-Lcd1p, but not from cells expressing untagged Lcd1p. In the reciprocal experiment, HA-Lcd1p was observed in anti-FLAG immunoprecipitates from extracts of cells expressing FLAG-tagged genomic Mec1p, but not when cells expressing untagged Mec1p were used (Figure 6C). No anti-HA or anti-FLAG cross-reacting bands were detected when immunoprecipitations were performed from extracts of the parental strain expressing Lcd1p and Mec1p in their untagged forms (data not shown). In Figure 6B and C, Mec1p appears as a doublet at ∼260 kDa after western blotting. It is unlikely that these forms of Mec1p are phosphorylated variants, as treatment of cell extracts with protein phosphatases before immunoprecipitation has no effect on this doublet (data not shown). Notably, there was little detectable difference in the levels of Mec1p in wild-type cells compared with cells lacking Lcd1p, suggesting that the interaction of Lcd1p and Mec1p is not required to stabilize the Mec1p protein. In addition, the interaction of Lcd1p with Mec1p does not appear to be affected by treatment of cells with MMS (Figure 6B and C) or 4-NQO (data not shown) and is not affected by washing the immunoprecipitates with 1 M sodium chloride (data not shown). These results indicate that Lcd1p forms a tight and apparently constitutive complex with Mec1p in vivo.
Discussion
Lcd1p has homology to S.cerevisiae Rfc5p and S.pombe Rad26
In our searches of the protein sequence databases, LCD1 consistently emerged as showing homology to Rfc5p, with ∼25% overall identity over a stretch of 180 amino acid residues (Figure 1A and B). Although the region of Rfc5p that is similar to Lcd1p has not yet been characterized, this domain is likely to be functionally important, as some of these residues are conserved between Rfc5 family members from different species. The RFC subunits form a multiprotein complex in vivo but the region(s) of each subunit required for these interactions has not been defined. It will be interesting to examine whether Lcd1p can associate with RFC or with the RFC complex that contains Rad24p (Shimomura et al., 1998; Green et al., 2000) and, if so, what the functional effects of this might be.
We also found that LCD1 is the most similar protein in the S.cerevisiae genome to S.pombe Rad26, although the homology is low and only extends over short stretches of amino acid sequence (Figure 1A and C). To date, S.cerevisiae orthologues of all six S.pombe checkpoint Rad genes have been identified, with the notable exception of Rad26. Since Rad26 is one of the most crucial components of the Rad3 pathway (Al-Khodairy et al., 1994; Lindsay et al., 1998; Edwards et al., 1999), it seems likely that other organisms will possess an orthologue of this protein. To date, however, the Aspergillus nidulans uvsD gene is the only Rad26 homologue that has been unambiguously identified (De Souza et al., 1999), with sequence homology extending over most of the ORF. Notably, the phenotype of cells lacking LCD1 is in some ways similar to that of S.pombe rad26.d mutants, raising the possibility that LCD1 and Rad26 are functionally equivalent despite the fact that they only possess weak homology to one another. Whatever the case, it is clear that, like S.pombe Rad26, S.cerevisiae LCD1 plays a crucial role in DNA damage signalling responses (Figure 7). Consistent with this, while this manuscript was under revision, YDR499W was identified by Paciotti et al. (2000; referred to therein as DDC2) as a key regulator of cell cycle checkpoint responses.

Fig. 7. The Lcd1p/Mec1p signalling pathway. Lcd1p interacts with Mec1p and controls activation of key effector molecules such as Rad53p, in response to DNA damage or inhibition of DNA replication. An intact RAD9 gene is required for activation of both Rad53p and Chk1p in response to DNA damage, and the RAD17/RAD24 epistasis group is also required for DNA damage-induced activation of Rad53p. Black boxes represent discrete complexes involved in the DNA damage response. HU-induced activation of Rad53p involves a different group of genes, e.g. POL2 (right-hand side). Chk1p mediates arrest in mitosis, but is not involved in DNA repair, whereas Rad53p and its effector molecules are involved in both lesion repair and checkpoint regulation.
The essential function of LCD1
As is the case for MEC1 and RAD53, disruption of LCD1 causes lethality that can be suppressed by elevation of dNTP levels. The molecular basis for this suppression is at present unclear, but there are several possible explanations. It has been shown that the firing of late replication origins is advanced in cells lacking RAD53 or MEC1 (Santocanale and Diffley, 1998; Shirahige et al., 1998). Activation of replication complexes and nucleotide supply must be co-ordinated to prevent nucleotide depletion, and it may be that LCD1, MEC1 and RAD53 function in a pathway which ensures that this occurs. Consistent with this idea, temperature-sensitive mutations in DBF4, which slow origin firing, can suppress the lethality of cells lacking MEC1 (Desany et al., 1998). Another explanation for the essential roles of LCD1, MEC1 and RAD53, which is not mutually exclusive, is that these genes act in a pathway that up-regulates dNTP biosynthesis during normal S phase to allow efficient DNA replication—perhaps by disrupting or modulating the Sml1p–Rnr1p interaction (Zhao et al., 1998; Chabes et al., 1999).
The role of Lcd1p in cellular responses to DNA damage and blocked DNA replication
Cells lacking LCD1 are extremely sensitive to UV, IR and MMS, suggesting a major defect in responding to the types of DNA lesions that are generated by these agents. Strikingly, Δlcd1 cells are significantly more sensitive to MMS or UV than Δmec1 cells. Furthermore, the absence of an intact MEC1 gene does not further hypersensitize Δlcd1 cells to DNA damage (Figure 2A–C), suggesting that in terms of MEC1 function these genes are epistatic. Although other possibilities exist and, as discussed below, this raises the possibility that in addition to its role in regulating the Mec1p pathway, Lcd1p functions in other ways to potentiate cellular survival in the presence of DNA damage. Regulation of other signalling and DNA repair pathways other than the MEC1 pathway, by LCD1, is currently under investigation in this laboratory.
Previous work has established that the FHA domains of Rad53p bind specifically to the phosphorylated forms of Rad9p that arise after DNA damage, and that this is required for Rad53p activation (Sun et al., 1998; Durocher et al., 1999). In line with this, the ability of FHA2 to interact with Rad9p in extracts from cells exposed to DNA damage is severely reduced in cells lacking LCD1, and this correlates with lack of activation of Rad53p and Chk1p induced by DNA damage. DNA damage-induced Rad9p phosphorylation in G1-arrested cells is also reduced when LCD1 is absent and this reduction is at least as great as that in cells lacking MEC1. The ability of Rad53p-FHA2 to bind to Rad9p in extracts from asynchronous cultures of Δlcd1 cells is severely decreased compared with wild-type cells. We found that DNA damage-induced phosphorylation of Chk1p also requires an intact LCD1 gene, which is probably also a reflection of the defect in Rad9p phosphorylation seen in these cells, as Chk1p phosphorylation is dependent on RAD9 (Sanchez et al., 1999). Taken together, these results show that LCD1 is required for changing the activity of several key signal transducers that operate in the MEC1-dependent DNA damage signalling pathway (Figure 7).
Cells lacking LCD1 also display a high degree of sensitivity to even low concentrations of HU, indicating a role for Lcd1p in responding to stalled DNA replication forks. Also, activation of Rad53p by DNA damage or replication blocks is severely reduced in cells lacking LCD1. At present, the molecular mechanism(s) whereby the Mec1p pathway ‘senses’ stalled replication forks remains unclear. Proteins such as DNA polymerase ε (Pol2p) and the RFC subunits, which have roles in DNA synthesis, also function in checkpoint responses (Navas et al., 1995; Sugimoto et al., 1996; Shimomura et al., 1998), and the Sgs1p DNA helicase that is needed for faithful DNA replication is also required for activation of Rad53p in response to stalled replication forks (Frei and Gasser, 2000). It will clearly be of interest to see whether Lcd1p interacts with or regulates the functions of any of these DNA replication-associated proteins.
LCD1 is a key regulator of cell cycle checkpoints
The hypersensitivity of Δlcd1 cells to DNA damage and DNA replication blocks raises the possibility that Lcd1p plays a role in slowing cell cycle progression under these circumstances to prevent cell cycle catastrophe. Indeed, when DNA damage is induced using a temperature-sensitive cdc13 allele, Δlcd1 cells fail to arrest in metaphase and proceed through the cell cycle with the same kinetics as cells lacking RAD9. This failure of Δlcd1 cells to arrest in metaphase in response to cdc13-1-induced DNA damage correlates with lack of Rad53p activation. Notably, we have found that in G1 or G2/M these defects are more severe than those exhibited by cells lacking Rad9p, suggesting that LCD1 acts upstream of Rad9p and/or functions in other checkpoint pathways in addition to those that involve Rad9p (Figure 7).
It was shown recently that although disruption of the S.cerevisiae CHK1 gene results in a defective G2/M DNA damage checkpoint, Δchk1 cells are not hypersensitive to DNA-damaging agents (Sanchez et al., 1999; J.Rouse and S.P.Jackson, unpublished data). Also, arresting Δrad9 cells in metaphase has been shown to have little or no effect on their viability after exposure to IR (Aboussekhra et al., 1996). These observations imply that the sensitivity of cells lacking LCD1, for example, is not due primarily to a checkpoint defect, and instead may be the result of an inability to mediate the efficient repair of DNA lesions. Results from several recent studies are consistent with this notion. For example, it has been demonstrated that genes in the RAD24 epistasis group are required for efficient non-homologous end-joining of DSBs, and artificially imposing cell cycle delays only partially rescues this defect (de la Torre-Ruiz and Lowndes, 2000). Also, the Sir and Ku DSB repair proteins re-localize from discrete nuclear foci to sites of DNA damage in a MEC1-dependent manner (Martin et al., 1999; McAinsh et al., 1999; Mills et al., 1999), although none of these proteins have checkpoint defects. Finally, in this light, Rad55p, a protein involved in recombinational repair (Kanaar et al., 1998) but not in checkpoint regulation, was found to become phosphorylated in a Mec1p pathway-dependent manner (Bashkirov et al., 2000). In light of these points, we are currently investigating the potential role of Lcd1p in facilitating the repair of various forms of DNA damage.
Physical and genetic interactions between Lcd1p and Mec1p
The phenotypic similarities between cells lacking LCD1 or MEC1 suggested that the products of these genes might interact. Indeed, we find that Lcd1p and Mec1p can be efficiently immunoprecipitated with one another, even under stringent conditions. Furthermore, exposure of cells to DNA damage has no detectable effect on the extent or stability of the Lcd1p–Mec1p interaction. Taken together with the functional data, the observation that Mec1p interacts with Lcd1p indicates that the actions of these two proteins in the cell are likely to be closely interlinked. It will be of great interest to determine whether the interaction is direct and to investigate whether it is required for the checkpoint and/or the essential functions of these proteins. It will also be interesting to determine what proportion of total cellular Lcd1p exists in complex with Mec1p. The observation that cells lacking LCD1 are more sensitive to MMS or UV than Δmec1 cells suggests that Lcd1p can function, at least partially, independently of MEC1, perhaps by regulating other pathways.
Materials and methods
Plasmids and antibodies
pBAD79, a TRP1 plasmid bearing the RNR3 gene under the control of the constitutive glyceraldehyde-3-phosphate dehydrogenase (GAP) promoter, and pRS415-HA-CHK1 were kind gifts from Dr Steve Elledge and are referred to as pRNR3 and pHA-CHK1, respectively. pLCD1 was constructed by gap repair as follows. The YDR499W ORF and 500 bp upstream of the start ATG were amplified (primers 499C and 499D) with 45 bp of homology to bases 1939–1984 and 2101–2146 around the multiple cloning site of the centromeric plasmid pRS416 (which contains the URA3 marker), respectively. Cells were transformed with BamHI– HindIII-digested pRS416 and the 499C/499D PCR product (5 µg), and transformants were selected by growth on medium lacking uracil. Plasmids isolated from positive colonies were transformed into Escherichia coli, and the presence of the YDR499W gene verified by restriction digestion and sequencing. Polyclonal anti-Rad53p and anti-Rad9p antibodies were kind gifts from Dr Noel Lowndes. pHA-LCD1 was constructed by mutating the start codon of YDR499W in pLCD1 to an NcoI restriction site by using the QuikChange site-directed mutagenesis kit (Stratagene). Oligonucleotides encoding the tag were ligated into the NcoI site of the vector. Mouse monoclonal anti-FLAG antibodies (clone M2) were from Sigma, anti-HA antibodies (12CA5) were from Boehringer and rabbit polyclonal anti-histidine tag antibodies were from Santa Cruz Biotechnology.
Yeast strains
Standard genetic techniques were used for manipulating yeast strains (Adams et al., 1997), and media and growth of yeast strains were as described previously (Rose et al., 1990). Δmec1Δsml1 cells were a kind gift from Dr Rodney Rothstein. cdc13-1 cdc15-2 cells and cells bearing an N-terminally FLAG-His6-tagged genomic copy of the MEC1 gene were a kind gift from Dr Daniel Durocher in this laboratory. To generate cells lacking LCD1, a fragment of this gene, spanning 700 bp upstream of the start ATG and the first 1040 bp of the ORF, was amplified and cloned into pGEM-T. The resulting vector, pGEM499, was digested with XhoI and HindIII to excise 350 bp upstream of LCD1 and the first 688 bp of coding sequence. The LEU2 gene and 1.2 kb upstream of the start ATG were amplified and cloned into pCR-Script. An XhoI–HindIII fragment containing the LEU2 ORF and 1 kb of 5′ non-coding sequence was inserted into pGEM499 to create pGEM499::LEU2. The NcoI–NotI fragment (5 µg) of this vector was transformed into cells containing pRNR3. Disruption of LCD1 was checked by genomic PCR and confirmed by Southern blot analysis.
To generate Δlcd1Δsml1 and Δlcd1Δmec1Δsml1 cells, the Δlcd1 [pRNR3] strain was crossed with Δmec1Δsml1 cells. Diploids were sporulated and, after tetrad dissection, strains of interest were isolated by following segregation of sensitivity to HU and the relevant markers. Loss of the RNR3 plasmid was encouraged by growth in the presence of tryptophan and verifed by loss of ability to grow in the absence of this amino acid.
In situ kinase assay for Rad53p
Briefly, cells were grown to early log phase, at which point the relevant genotoxic agent was added at the appropriate concentration for the indicated time. Cells were collected by centrifugation and resuspended in 4 vols of trichloroacetic acid (TCA) (20%), and after addition of 1 vol. of glass beads, cells were lysed using a mini bead beater. The resulting lysate was centrifuged, the supernatant discarded and 2 vols of SDS–PAGE sample buffer, adjusted to 0.2 M Tris–HCl pH 8.8, were added to the pellet and mixed thoroughly. After boiling, samples were centrifuged and the supernatants stored at –20°C until required. Samples were run on 10% SDS–polyacrylamide gels and transferred to Immobilon-P in the absence of methanol, and the resulting filters were subjected to in situ analysis of Rad53p kinase activity as described previously (Pellicioli et al., 1999).
Analysis of the G1 DNA damage checkpoint
Cells were grown to early log phase and arrested in G1 by the addition of α-factor (5 µg/ml) for 60–90 min, at which point arrest was verified by the absence of budded cells (<1%). For UV irradiation, cells were collected by centrifugation, spread on YPD plates and irradiated with a germicidal lamp at 254 nm at the indicated dose, at a rate of 3 J/m2/s. Cells were scraped into YPD and released from G1 arrest by extensive washing through a filter apparatus with YPD, and incubated at 30°C. Samples were taken at 15 min intervals and the proportion of budded cells was counted by light microscopy.
Analysis of the G2/M DNA damage checkpoint
Cell cycle arrest at G2/M phase, in response to cdc13-induced DNA damage, was assayed as previously described (Garvik et al., 1995). Briefly, cells were grown to early log phase in YPD at 23°C (permissive temperature for cdc13-1 and cdc15-2), at which point cells were synchronized in G1 by the addition of α-factor (5 µg/ml) for ∼120 min. When budded cells accounted for >1% of the population, cells were released from arrest by washing five times in YPD before incubation in pre-warmed YPD at the restrictive temperature (36°C). At the times indicated, aliquots of cells were removed, fixed in 70% ethanol and stained with 4′,6-diamidino-2-phenylindole (DAPI). The percentage of budded cells with one nucleus in each bud (post-M phase cells) was counted in a population of ∼200 cells by fluorescence microscopy.
Preparation of native cell extracts
To prepare native extracts, cells were grown to early log phase and incubated in the presence or absence of DNA-damaging agents for 2 h at 30°C. After centrifugation, cells were washed twice in water and once in 1 vol. of 2× extraction buffer (100 mM HEPES pH 7.4, 400 mM sodium acetate, 100 mM magnesium acetate, 1 mM EDTA, 20% glycerol, 1 mM sodium orthovanadate, 1 µM microcystin-LR, 0.1% 2-mercaptoethanol and a cocktail of protease inhibitors). After packing cells into a syringe sealed at the tip, a hole was pierced in the tip, through which the cells were extruded in a thin stream into liquid nitrogen. The frozen material was then placed in ground dry ice and ground in a coffee grinder for ∼3 min. This mixture was placed on ice, and after the dry ice had sublimed, the lysed cells were resuspended in 1 vol. of cold 2× extraction buffer, on ice. The lysate was centrifuged at 13 500 r.p.m. at 4°C and the supernatant was aliquoted and stored at –80°C until use.
Western blotting and immunoprecipitation
Rad53p and Rad9p western immunoblots were performed as described previously (Vialard et al., 1998). In co-immunoprecipitation experiments, aliquots of native extract (2 mg protein) from cells expressing either HA-Lcd1p or FLAG-Mec1p, or both, were incubated with protein G– Sepharose beads conjugated to mouse monoclonal anti-FLAG antibodies (1 µg) or anti-HA monoclonal antibodies (1 µg) on a shaking platform at 4°C for 2 h. After centrifugation, the beads were washed three times in WB buffer (50 mM HEPES pH 7.4, 0.2% NP-40 protease inhibitors and 0.1% 2-mercaptoethanol) containing 0.75 M sodium chloride and twice in WB buffer without salt. The beads were boiled for 5 min in SDS sample buffer and electrophoresed on 6.5% polyacrylamide gels, and subjected to western blot analysis with the appropriate antibody.
Acknowledgments
Acknowledgements
We thank all members of the Jackson laboratory, especially Daniel Durocher, Brandi Williams and Andrew McAinsh, for helpful discussions. We are grateful to Steve Elledge, Ted Weinert and Michael Resnick for providing yeast strains and plasmids, to Noel Lowndes for providing antibodies, and to Daniel Durocher for providing reagents before publication. This work was funded by the Leukaemia Research Fund and the Association for International Cancer Research.
References
- Aboussekhra A., Vialard,J.E., Morrison,D.E., de la Torre-Ruiz,M.A., Cernakova,L., Fabre,F. and Lowndes,N.F. (1996) A novel role for the budding yeast RAD9 checkpoint gene in DNA damage-dependent transcription. EMBO J., 15, 3912–3922. [PMC free article] [PubMed] [Google Scholar]
- Adams A., Gottschling,D.E., Kaiser,C.A. and Stearns,T. (1997) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Al-Khodairy F., Fotou,E., Sheldrick,K.S., Griffiths,D.J.F., Lehmann,A.R. and Carr,A.M. (1994) Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol. Biol. Cell, 5, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J.B., Zhou,Z., Siede,W., Friedberg,E.C. and Elledge,S.J. (1994) The Sad1/Rad53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev., 8, 2401–2415. [DOI] [PubMed] [Google Scholar]
- Bashkirov V.I., King,J.S., Bashkirova,E.V., Schmuckli-Maurer,J. and Heyer,W.-D. (2000) DNA repair protein Rad55 is a terminal substrate of DNA damage checkpoints. Mol. Cell. Biol., 20, 4393–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A., Domkin,V. and Thelander,L. (1999) Yeast Sml1, a protein inhibitor of ribonucleotide reductase. J. Biol. Chem., 274, 36679–36683. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O. and Koshland,D. (1997) The anaphase inhibitor of Saccharomyces cerevisiae, Pds1p, is a target of the DNA damage checkpoint pathway. Proc. Natl Acad. Sci. USA, 94, 14361–14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullmann G., Fien,K., Kobayashi,R. and Stillman,B. (1995) Characterisation of the five replication factor-C genes of Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 4661–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ruiz M.A. and Lowndes,N.F. (2000) The Saccharomyces cerevisiae DNA damage checkpoint is required for efficient repair of double strand breaks by non-homologous end joining. FEBS Lett., 467, 311–315. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ruiz M.A., Green,C.M. and Lowndes,N.F. (1998) RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J., 17, 2687–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desany B.A., Alcasabas,A.A., Bachant,J.B. and Elledge,S.J. (1998) Recovery from DNA replicational stress is the essential function of the S phase checkpoint pathway. Genes Dev., 12, 2956–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza C.P.C., Ye,X.S. and Osmani,S.A. (1999) Checkpoint defects leading to premature mitosis also cause endoreplication of DNA in Aspergillus nidulans. Mol. Biol. Cell, 10, 3661–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher D., Henckel,J., Fersht,A.R. and Jackson,S.P. (1999) The FHA domain is a modular phosphopeptide recognition motif. Mol. Cell, 4, 387–394. [DOI] [PubMed] [Google Scholar]
- Edwards R.J., Bentley,N.J. and Carr,A.M. (1999) A Rad3–Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nature Cell Biol., 1, 393–398. [DOI] [PubMed] [Google Scholar]
- Emili A. (1998) MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol. Cell, 2, 183–189. [DOI] [PubMed] [Google Scholar]
- Fay D.S., Sun,Z.X. and Stern,D.F. (1997) Mutations in SPK1/RAD53 that specifically abolish checkpoint- but not growth-related functions. Curr. Genet., 31, 97–105. [DOI] [PubMed] [Google Scholar]
- Frei C. and Gasser,S.M. (2000) The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev., 14, 81–96. [PMC free article] [PubMed] [Google Scholar]
- Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC. [Google Scholar]
- Gardner R., Putnam,C.W. and Weinert,T. (1999) RAD53, DUN1 and PDS1 define two parallel G2/M checkpoint pathways in budding yeast. EMBO J., 18, 3173–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvik B., Carson,M. and Hartwell,L. (1995) Single-stranded DNA arising telomeres in CDC13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol., 15, 6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green C.M., Erdjument-Bromage,H., Tempst,P. and Lowndes,N.F. (2000) A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol., 10, 39–42. [DOI] [PubMed] [Google Scholar]
- Greenwell P.W., Kronmal,S.L., Porter,S.E., Gassenhuber,J., Obermaier,B. and Petes,T.D. (1995) TEL1, a gene involved in controlling telomere length in Saccharomyces cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell, 82, 823–829. [DOI] [PubMed] [Google Scholar]
- Griffiths D.J.F., Barbet,N.C., McCready,S., Lehmann,A.R. and Carr,A.M. (1995) Fission yeast Rad17: a homolog of budding yeast Rad24 that shares regions of sequence similarity with DNA polymerase accessory proteins. EMBO J., 14, 5812–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaar R., Hoeijmakers,J.H.J. and van Gent,D.C. (1998) Molecular mechanisms of DNA double strand break repair. Trends Cell Biol., 8, 483–489. [DOI] [PubMed] [Google Scholar]
- Kato R. and Ogawa,H. (1994) An essential gene, ESR1, is required for mitotic cell growth, DNA repair and meiotic recombination in Saccharomyces cerevisiae. Nucleic Acids Res., 22, 3104–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H.D., Griffiths,D.J.F., Edwards,R.J., Christensen,P.U., Murray,J.M., Osman,F., Walworth,N. and Carr,A.M. (1998) S phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev., 12, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese M.P., Paciotti,V., Fraschini,R., Zaccarini,R., Plevani,P. and Lucchini,G. (1997) The novel DNA damage checkpoint protein Ddc1p is phosphorylated periodically during the cell cycle and in response to DNA damage in budding yeast. EMBO J., 16, 5216–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes N.F. and Murguia,J.R. (2000) Sensing and responding to DNA damage. Curr. Opin. Genet. Dev., 10, 17–25. [DOI] [PubMed] [Google Scholar]
- Lydall D. and Weinert,T. (1995) Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science, 270, 1488–1491. [DOI] [PubMed] [Google Scholar]
- Martin S.G., Laroche,T., Suka,N., Grunstein,M. and Gasser,S.M. (1999) Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell, 97, 621–633. [DOI] [PubMed] [Google Scholar]
- Martinho R.G., Lindsay,H.D., Flaggs,G., DeMaggio,A.J., Hoekstra,M.F., Carr,A.M. and Bentley,N.J. (1998) Analysis of Rad3 and Chk1 protein kinases defines different checkpoint responses. EMBO J., 17, 7239–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAinsh A.D., Scott-Drew,S., Murray,J. and Jackson,S.P. (1999) DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Curr. Biol., 9, 963–966. [DOI] [PubMed] [Google Scholar]
- Mills K.D., Sinclair,D.A and Guarente,L. (1999) MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to double strand breaks. Cell, 97, 609–620. [DOI] [PubMed] [Google Scholar]
- Morrow D.M., Morrow,M., Tagle,D.A., Shiloh,Y., Collins,F.S. and Hieter,P. (1995) TEL1, a Saccharomyces cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell, 82, 831–840. [DOI] [PubMed] [Google Scholar]
- Navas T.A., Zhou,Z. and Elledge,S.J. (1995) DNA polymerase ε links the DNA replication machinery to the S phase checkpoint. Cell, 80, 29–39. [DOI] [PubMed] [Google Scholar]
- Noskov V.N., Araki,H. and Sugino,A. (1998) The RFC2 gene, encoding the third-largest subunit of the replication factor C complex, is required for an S-phase checkpoint in Saccharomyces cerevisiae. Mol. Cell. Biol., 18, 4914–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciotti V., Clerici,M., Lucchini,G. and Longhese,M.P. (2000) The checkpoint Ddc2, functionally related to S.pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev., 14, 2046–2059. [PMC free article] [PubMed] [Google Scholar]
- Paulovich A.G. and Hartwell,L.H. (1995) A checkpoint regulates the rate of progression through S phase in Saccharomyces cerevisiae in response to DNA damage. Cell, 82, 841–847. [DOI] [PubMed] [Google Scholar]
- Paulovich A.G., Margulies,R.U. and Garvik,B.M. (1997) RAD9, RAD17 and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics, 145, 45–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A., Lucca,C., Liberi,G., Marini,F., Lopes,M., Plevani,P., Romano,A., Di Fiore,P.P. and Foiani,M. (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J., 18, 6561–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M.D., Meluh,P.B. and Hieter,P. (1990) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sanchez Y., Desany,B.A., Jones,W.J., Liu,Q.H., Wang,B. and Elledge,S.J. (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science, 271, 357–360. [DOI] [PubMed] [Google Scholar]
- Sanchez Y., Bachant,J., Wang,H., Hu,F.H., Liu,D., Tetzlaff,M. and Elledge,S.J. (1999) Control of the DNA damage checkpoint by Chk1 and Rad53 protein kinases through distinct mechanisms. Science, 286, 1166–1171. [DOI] [PubMed] [Google Scholar]
- Santocanale C. and Diffley,J.F.X. (1998) A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature, 395, 615–618. [DOI] [PubMed] [Google Scholar]
- Shimomura T., Ando,S., Matsumoto,K. and Sugimoto,K. (1998) Functional and physical interaction between Rad24 and Rfc5 in the yeast checkpoint pathways. Mol. Cell. Biol., 18, 5485–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahige K., Hori,Y., Shiraishi,K., Yamashita,M., Takahashi,K., Obuse,C., Tsurimoto,T. and Yoshikawa,H. (1998) Regulation of DNA replication origins during cell cycle progression. Nature, 395, 618–621. [DOI] [PubMed] [Google Scholar]
- Sugimoto K., Shimomura,T., Hashimoto,K., Araki,H., Sugino,A. and Matsumoto,K. (1996) Rfc5, a small subunit of replication factor C complex, couples DNA replication and mitosis in budding yeast. Proc. Natl Acad. Sci. USA, 93, 7048–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K., Ando,S., Shimomura,T. and Matsumoto,K. (1997) Rfc5, a replication factor C component, is required for regulation of Rad53 protein kinase in the yeast checkpoint pathway. Mol. Cell. Biol., 17, 5905–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.X., Fay,D.S., Marini,F., Foiani,M. and Stern,D.F. (1996) Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev., 10, 395–406. [DOI] [PubMed] [Google Scholar]
- Sun Z.X., Hsiao,J., Fay,D.S. and Stern,D.F. (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science, 281, 272–274. [DOI] [PubMed] [Google Scholar]
- Vialard J.E., Gilbert,C.S., Green,C.M. and Lowndes,N.F. (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J., 17, 5679–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T.A. and Hartwell,L.H. (1988) The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science, 241, 317–322. [DOI] [PubMed] [Google Scholar]
- Weinert T.A., Kiser,G.L. and Hartwell,L.H. (1994) Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev., 8, 652–665. [DOI] [PubMed] [Google Scholar]
- Zhao X.L., Muller,E.G.D. and Rothstein,R. (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell, 2, 329–340. [DOI] [PubMed] [Google Scholar]