

Abstract
BACKGROUND AND PURPOSE
Many G protein-coupled receptors internalize following agonist binding. The studies were designed to identify novel means to effectively quantify this process using the orexin OX1 receptor and the cannabinoid CB1 receptor as exemplars.
EXPERIMENTAL APPROACH
The human OX1 and CB1 receptors were modified to incorporate both epitope tags and variants (SNAP and CLIP) of the enzyme O6-alkylguanine-DNA-alkyltransferase within their extracellular, N-terminal domain. Cells able to regulate expression of differing amounts of these constructs upon addition of an antibiotic were developed and analysed.
KEY RESULTS
Cell surface forms of each receptor construct were detected by both antibody recognition of the epitope tags and covalent binding of fluorophores to the O6-alkylguanine-DNA-alkyltransferase variants. Receptor internalization in response to agonists but not antagonists could be monitored by each approach but sensitivity was up to six- to 10-fold greater than other approaches when employing a novel, time-resolved fluorescence probe for the SNAP tag. Sensitivity was not enhanced, however, for the CLIP tag, possibly due to higher levels of nonspecific binding.
CONCLUSIONS AND IMPLICATIONS
These studies demonstrate that highly sensitive and quantitative assays that monitor cell surface CB1 and OX1 receptors and their internalization by agonists can be developed based on introduction of variants of O6-alkylguanine-DNA-alkyltransferase into the N-terminal domain of the receptor. This should be equally suitable for other G protein-coupled receptors.
Keywords: G protein-coupled receptor, orexin, cannabinoid, receptor trafficking, receptor internalization, insomnia, narcolepsy, epitope tagging
Introduction
The orexin receptors OX1 and OX2 are members of the rhodopsin-like family of G protein-coupled receptors (GPCRs) (Sakurai, 2005). Ligand pairing of these receptors with the peptides orexin A and orexin B, both derived from the precursor prepro-orexin (Sakurai, 2005), instigated a wide range of studies on their biological function that have centred on the regulation of sleep/wakefulness and in the control of feeding and appetite (Bingham et al., 2006; Kroeger and de Lecea, 2009). Based on such studies agonism of orexin receptors has been suggested as a means to treat narcolepsy, whereas orexin receptor antagonists have been promoted as a potential treatment of sleep disorders such as insomnia. Indeed, the combined OX1 and OX2 antagonist almorexant has undergone late stage clinical trials in this area (Neubauer, 2010). Cannabinoid CB1 and CB2 receptors are also members of the GPCR superfamily (Pertwee and Ross, 2002; Pertwee, 2005) and respond to both a range of endo-cannabinoids as well as psychoactive plant-derived ligands (Pertwee and Ross, 2002; Pertwee, 2005). Based on effects of stimulation of the CB1 receptor to promote appetite, CB1 antagonists/inverse agonists were promoted to limit appetite and support weight loss (Kirkham, 2009). Although the inverse agonist SR141716A (rimonabant) was subsequently withdrawn from clinical use (Burch et al., 2009; Lee et al., 2009) it was shown be to efficaceous at this endpoint. CB1 agonists have also attracted attention as potential agents to treat aspects of pain (Walker and Hohmann, 2005; Coman et al., 2008).
The OX1 receptor is known to link predominantly to the elevation of intracellular [Ca2+] via activation of heterotrimeric G proteins of the Gq class (Smart et al., 1999; 2001; Ammoun et al., 2006; Johansson et al., 2007) whilst the CB1 receptor signals predominantly via the Gi/Go class of Pertussis toxin-sensitive G proteins (Dalton et al., 2009).
As with the vast majority of GPCRs, both of these receptors can be rapidly internalized following binding of agonist (Coutts et al., 2001; Milasta et al., 2005; Ellis et al., 2006; Wu et al., 2008). Ligand regulation of GPCR internalization has been incorporated into a number of strategies to identify receptor agonists and antagonists. These have included the tagging of GPCRs with fluorescent proteins to allow monitoring of the cellular location of the engineered construct in real-time imaging studies (Milligan, 1999; Kallal and Benovic, 2002) and also the use of pH-sensitive fluors. These are linked via antibodies to the extracellular N-terminus of a GPCR and fluoresce much more extensively within the acidic endosomes that the receptor enters upon internalization (Adie et al., 2002; 2003;).
Very recently, the development of SNAP- and CLIP-tagging technologies, based on the activity of the enzyme O6-alkylguanine-DNA-alkyltransferase (Tirat et al., 2006; Gautier et al., 2008; Maurel et al., 2008; Alvarez-Curto et al., 2010), has offered means to covalently attach small molecule fluorophores to proteins engineered to contain the SNAP- or CLIP-tag sequences. These can then potentially be monitored in living cells to provide an index of the cellular location of the protein of interest and its regulation. In the current studies we employed both SNAP- and CLIP tagging to examine ligand-regulated internalization and cell surface recovery of stably expressed forms of the human OX1 and CB1 receptors. In concert with the use of Tag-Lite™ (http://www.htrf.com/technology/tag-lite/) reagents this provided significantly greater sensitivity to measure receptor expression as well as markedly higher signal to background ratios for quantification of receptor internalization, particularly for the SNAP tag.
Methods
Drugs, chemicals reagents and other materials
Lipofectamine transfection reagent was from Invitrogen (Paisley, UK). SB334867, SB408124, CP55940, AM251 and O2050 were from Tocris Biosciences (Avonmouth, UK). Orexin A was from Bachem (UK) Ltd. (St Helens, Merseyside UK). Oligonucleotides were from ThermoElectron (Ulm, Germany) and all materials for tissue culture were from Invitrogen. [3H]-SB674042 and [3H]-SR141716A were from Perkin Elmer (Boston, MA, USA). SR141716A (rimonabant) was a gift of GlaxoSmithKline. Antibodies to epitope tags were obtained from Sigma Aldrich Co. Ltd., Gillingham, Dorset, UK (anti-VSV-G), Roche Diagnostics, Mannheim, Germany (anti-HA) and New England BioLabs, Hitchin, UK (anti-SNAP). SNAP- and CLIP-specific labels were supplied by New England Biolabs (Hitchin, UK) and Taglite™ reagents by Cisbio Bioassays (Bagnols-sur-Cèze, France). All other reagents were obtained from Fisher Scientific (Loughborough, UK) or Sigma Aldrich Co. Ltd.
Test systems used
DNA constructs
The plasmids pSEMS1-26 m (SNAP tag) and pCEMS1-CLIP10m (CLIP tag), as supplied by Covalys Biosciences AG (Witterswil, Switzerland), were modified by the addition of a small linker region encoding the metabotropic glutamate receptor 5 (mGluR5) signal sequence (MVLLLILSVLLLKEDVRGSAQS), and an epitope tag (either HA, YPYDVPDYA or VSV-G, YTDIEMNRLGK) between the Cla1 and EcoR1 sites of the multiple cloning site upstream of the SNAP or CLIP tag (MCS1). The linker was made by annealing two complementary primers containing the sequences described earlier with the addition of a Kozak sequence, start codon and appropriate nucleotides to generate Cla1 and EcoR1 ‘sticky’ ends. The primers were annealed by combining 1 nM of each with 1בmulticore’ buffer (Promega Corporation) in a final volume of 50 µL. This was then heated to 100°C in a boiling water bath for 5 min, after which the bath was then turned off and allowed to cool overnight. The annealed fragment was then purified by gel extraction and ligated into the plasmid by standard techniques. The human orexin OX1 and cannabinoid CB1 receptors were PCR amplified using primers designed to add BamH1 and Not1 sites to the fragment termini. These were then ligated into the multiple cloning site downstream of SNAP or CLIP tags (MCS2) of the modified plasmids described earlier.
In order to create constructs that could be used to make Flp-In™ T-REx™ 293 inducible stable cell lines of these constructs (Ward et al., 2011), the entire insert from the Cla1 site to the Not1 site was cut out and ligated into a modified version of pcDNA5/FRT/TO (Invitrogen) with a Cla1 site added to the multiple cloning site using a linker formed from two annealed primers as described earlier.
Generation and maintenance of stable Flp-In™ T-REx™ 293 cells
To generate Flp-In™ T-REx™ 293 cells able to inducibly express the VSV-G-SNAP-OX1/CB1 or HA-CLIP-CB1 constructs, cells were co-transfected with the plasmids pOG44 and pcDNA5/FRT/TO (Invitrogen) containing the desired cDNA, at a ratio of 9:1 using Lipofectamine. After 48 h the medium was supplemented with 200 µg·mL−1 hygromycin to select for stably transfected cells. Pools of cells were established and tested for inducible expression by the addition of 1 µg·mL−1 doxycycline for 48 h followed by screening for VSV-G, HA- or SNAP/CLIP-tagged protein expression by Western blotting.
Cell lysates and Western blotting
Cells were washed once in cold PBS (120 mM NaCl, 25 mM KCl, 10 mM Na2HPO4 and 3 mM KH2PO4, pH7.4) and harvested with ice-cold RIPA buffer (50 mM HEPES, 150 mM NaCl, 1% Triton X-100, and 0.5% sodium deoxycholate, 10 mM NaF, 5 mM EDTA, 10 mM NaH2PO4, 5% ethylene glycol, pH 7.4) supplemented with complete protease inhibitors cocktail (Roche Diagnostics, Mannheim, Germany). Extracts were passed though a 25 gauge needle and incubated for 15 min at 4°C while spinning on a rotating wheel. Cellular extracts were then centrifuged for 30 min at 14 000×g and the supernatant was recovered.
After heating samples at 65°C for 5 min, both cell lysates and pulldowns were subjected to SDS-PAGE analysis using 4 to 12% Bis-Tris gels (NuPAGE; Invitrogen) and MOPS buffer. After separation, the proteins were electrophoretically transferred to nitrocellulose membrane, which was then blocked (5% fat-free milk powder in PBS with 0.1% Tween-20) at 4°C on a rotating shaker overnight. The membrane was incubated for 3 h with appropriate primary antibody (see Figure legends) in 2% fat-free milk powder in PBS-Tween, washed (3 × 10 min PBS-Tween) and then incubated for 3 h with appropriate secondary antibody [horseradish peroxidase-linked donkey anti-rabbit IgG, sheep anti-mouse horseradish peroxidase (HRP) or goat anti-rat HRP, GE Healthcare, Amersham, UK] diluted 1:10 000 in 2% fat-free milk powder in PBS-Tween. After washing, proteins were detected by enhanced chemiluminescence (Pierce Chemical, Rockford, IL, USA) according to the manufacturer's instructions.
Cell membrane preparation
Pellets of cells were frozen at ∼80°C for a minimum of 1 h, thawed and resuspended in ice-cold 10 mM Tris, 0.1 mM EDTA, pH 7.4 (TE buffer) supplemented with complete protease inhibitors cocktail (Roche Diagnostics, Mannheim, Germany). Cells were homogenized on ice by 40 strokes of a Teflon-glass homogenizer followed by centrifugation at 1000×g for 5 min at 4°C to remove unbroken cells and nuclei. The supernatant fraction was removed and passed through a 25 gauge needle 10 times before being transferred to ultracentrifuge tubes and subjected to centrifugation at 50 000×g for 30 min at 4°C. The resulting pellets were resuspended in ice-cold TE buffer. Protein concentration was assessed and membranes were stored at ∼80°C until required.
Measurements made
[3H]-SB674042 binding assays
Saturation binding curves were established by the addition of 5 µg of membrane protein to assay buffer (25 mM HEPES, 500 mM NaCl, and 2.5 mM MgCl2, pH 7.4, supplemented with 0.3% BSA) containing varying concentrations of [3H]-SB674042 (Langmead et al., 2004) (0.4–20 nM). Non-specific binding was determined in the presence of 3 µM SB408124. Reactions were incubated for 90 min at 25°C, and bound ligand was separated from free by vacuum filtration through GF/C filters (Brandel Inc., Gaithersburg, MD, USA). The filters were washed twice with cold 1× PBS (120 mM NaCl, 25 mM KCl, 10 mM Na2HPO4, 3 mM KH2PO4 pH7.4) and bound ligand was estimated by liquid scintillation spectrometry.
[3H]-SR141716A binding assays
Saturation binding curves for [3H]-SR141716A were determined as in the previous section but with the following detailed differences; 30 µg of membrane protein was added to assay buffer composed of 50 mM TrisHCl, 3 mM MgCl2, 1 mM EDTA and 0.3% BSA pH7.4, containing varying concentrations of [3H]-SR141716A (0.5–12 nM). Non-specific binding was determined by the addition of 10 µM AM251 and unbound ligand was separated by washing with cold 1xPBS supplemented with 0.1% (poly)ethyleneimine.
Calcium mobilization assays
Flp-In™ T-REx™ 293 cells able to express VSV-G-SNAP-OX1 in an inducible manner were grown in poly-D-Lysine coated, black, clear bottom 96-well microtitre plates. Then 24 h after induction with doxycycline, the cells were loaded with the calcium-sensitive dye Fura-2, by changing the media for DMEM containing 3 µM Fura-2. The plates were incubated in the dark for 45 min at 37°C and then washed with 2 × 100 µL per well of HEPES buffer (130 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, and 10 mM d-glucose, pH7.4). Then 100 µL per well HEPES buffer was added and the plate incubated at room temperature for 30 min in the dark. The effect of ligands was then assessed by measuring the calcium response using a FLEX-Station (Molecular Devices, Sunnydale, CA, USA).
ERK1/2 MAP kinase phosphorylation
Flp-In™ T-REx™ 293 cells able to express HA-CLIP-CB1 were plated at a density of 40 000 cells per well in a poly-D-lysine coated 96 well plate. Expression of the construct was simultaneously induced by the addition of 100 ng·mL−1 doxycycline. The cells were allowed to grow overnight and then deprived of serum for 4 h prior to lysis and processing. The cells were lysed using the lysis buffer provided (SureFire AlphaScreen kit, Perkin Elmer) and then processed according to the manufacturer's instructions.
Enzyme-linked immunosorbent assay for cell surface receptor expression
Flp-In™ T-REx™ 293 cells able to express VSV-G-SNAP-OX1/CB1 or HA-CLIP-CB1 in an inducible fashion were seeded into poly-D-Lysine coated, clear, 96 well tissue culture plates at a density of 50 000 cells per well. After incubation for 24 h the media was removed and replaced with 100 µL per well, warmed, primary antibody solution (normal media with 1:1000 dilution of anti-VSV-G, anti-HA or anti-SNAP/CLIP antibody) and incubated at 37°C for 30 min. The primary antibody solution was removed and the wells washed with 100 µL per well warm DMEM HEPES (13.4 g·L−1 Dulbecco's modified Eagle's medium – high glucose, 20 mM HEPES, pH 7.4, filter sterilized). Then 100 µL per well, warmed, secondary antibody solution was added (1:2000 dilution appropriate secondary antibody linked to HRP, 1:1000 dilution Hoechst stain (Hoechst 33342 trihydrochloride trihydrate, Molecular Probes, OR, USA). Following incubation at 37°C for 30 min, the secondary antibody solution was removed and the wells washed with 2 × 100 µL per well warm PBS. During the second PBS wash, the Hoechst staining was measured at 460 nm using a Victor 2 1420 multi-label counter [Perkin Elmer LAS (UK) Ltd. Beaconsfield, UK]. The PBS was completely removed and replaced with 100 µL per well, warmed, TMB substrate (SureBlue Reserve™ TMB peroxidise substrate, Insight Biotechnology, Wembley, UK). After 5 min incubation in the dark at room temperature, the absorption at 620 nm was measured and these values were then corrected for cell number using the Hoechst staining values.
Fluorescence imaging of SNAP-tag proteins in live cells
Cells stably expressing the human OX1 receptor N terminally tagged with SNAP or the human CB1 receptor N-terminally tagged with CLIP were grown on coverslips that had been cleaned with alcohol and treated with 0.1 mg·mL−1 poly-D-lysine. SNAP and CLIP-tag specific dye substrates (SNAP Cell 505, SNAP Surface 549 and CLIP-505) were diluted in complete DMEM medium from a 1 mM stock solution to give a labelling solution of 5 µM with respect to the SNAP/CLIP dye substrate. The cell medium was replaced with labelling solution and incubated at 37°C, 5% CO2 for 30 min (as required cells were pre-treated with SNAP blocking reagent (bromothenylpteridine), diluted as described earlier, for 30 min prior to replacement with SNAP-dye substrate). Cells were washed three times with complete DMEM and once with HEPES physiological saline solution (130 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, and 10 mM d-glucose, pH7.4). The fluorescent samples were then transferred to a microscope chamber where they were imaged using a Zeiss Pascal Exciter inverted confocal microscope (Zeiss, Jena, Germany), equipped with a 63× (N A = 1.4) oil-immersion Plan Apochromat lens. SNAP/CLIP-505 and 549 were excited using either the 488 or 543 laser line with the pinhole set to 1 Airy unit. Frame averaging was set to 4 or 8, and all images were acquired in 12 bit format. Images were exported into Metamorph (version 7.7), for final image processing.
SNAP/CLIP Lumi 4Tb binding studies
Cells expressing the OX1 receptor tagged with the SNAP tag or the CB1 receptor tagged with CLIP were seeded at 100 000 cells per well in solid black 96 well plates (Greiner BioOne, Stonehouse, Gloucestershire, UK), which had been treated with 0.1 mg·mL−1 poly-D-lysine. Following overnight growth the cells were subjected to the required ligand treatments. The growth medium was replaced with 50 µL of either 10 nM Tag-lite SNAP-Lumi4Tb or 20 nM CLIP-Lumi4Tb in 1× labelling medium (all Cisbio Bioassays, Bagnols-sur-Cèze, France). Plates were incubated for 1 h (or 30 min in the case of post-ligand treatment, to allow comparison with the elisa measurements) at 37°C, 5% CO2 in a humidified atmosphere. The plates were subsequently washed four times in 100 µL per well labelling medium and a final 100 µL per well labelling medium added. After excitation at 337 nm, emission at 620 nm was determined using a PheraStar FS homogeneous time-resolved fluorescence (HTRF) compatible reader (BMG Labtechnologies, Offenburg, Germany).
Results
The human orexin OX1 and cannabinoid CB1 receptors were modified at the N-terminus by addition of an N-terminal leader sequence, derived from the metabotropic glutamate 5 receptor, linked in-frame to the VSV-G epitope tag sequence and the 20 kDa SNAP tag protein that is based on mammalian O6-alkylguanine-DNA-alkyltransferase (Figure 1A). A second construct was generated for the human CB1 receptor, which was similar except that the HA epitope tag replaced the VSV-G tag and the SNAP tag was replaced with the CLIP sequence (Figure 1A). The SNAP/CLIP tags can be labelled covalently to incorporate a variety of fluorophores including a terbium cryptate (Lumi-4Tb) with long-lived fluorescence properties (Maurel et al., 2008; Alvarez-Curto et al., 2010). Each of these constructs (VSV-G-SNAP-OX1/CB1 and HA-CLIP-CB1) was cloned into the Flp-In™ locus of Flp-In™ T-REx™ 293 cells and populations of cells harbouring these constructs isolated. Flp-In™ T-REx™ 293 cells allow the synthesis of protein from DNA located at the Flp-In™ locus only upon addition of the antibiotic tetracycline or its analogue doxycycline (Ellis et al., 2006; Canals and Milligan, 2008; Smith et al., 2009). Such cells were either untreated or incubated with doxycycline (10 ng·mL−1, 24 h) prior to harvest and membrane preparation. Separation of membrane proteins by SDS-PAGE and subsequent immunoblotting with anti-VSV-G, anti-HA and anti-SNAP/CLIP antibodies confirmed the lack of expression of the receptor constructs in the absence of antibiotic and the presence of polypeptides of some 83 kDa (OX1 expressing cells) or 85 kDa (CB1 expressing cells) molecular mass following treatment of the cells with doxycycline (Figure 1B). The CB1 receptor was usually detected as a pair of bands (85/82-3 kDa) suggesting either a degree of breakdown or differential post-translational modification of the receptor. Noticeably, immunoblotting with both anti-VSV-G and anti-SNAP/CLIP also revealed a higher molecular mass complex containing VSV-G-SNAP-OX1 (Figure 1B) that is at least consistent with the presence of an SDS-resistant complex containing at least one copy of this receptor. An equivalent complex containing either VSV-G-SNAP-CB1 or HA-CLIP-CB1 was not observed (Figure 1B). The anti-SNAP/CLIP immunoblots also suggested that expression levels of VSV-G-SNAP-OX1 were substantially higher than those of VSV-G-SNAP-CB1 or HA-CLIP-CB1. To explore this ligand-binding studies were performed. When the OX1 antagonist [3H]-SB674042 was employed (Langmead et al., 2004) no substantial specific binding of the ligand was detected without pre-treatment of the cells with doxycycline (Figure 2A i). By contrast, induction of expression of the VSV-G-SNAP-OX1 construct with a maximally effective concentration of doxycycline resulted in a high level of expression of VSV-G-SNAP-OX1 (range 10.7–14.7 pmol·mg−1 protein in individual experiments) (Figure 2A i). Equivalent binding studies employing the CB1 receptor antagonist [3H]-SR141716A indicated HA-CLIP-CB1 expression was also only observed following treatment with doxycycline (Figure 2A ii) and, indeed, that maximum expression levels were substantially lower (range 0.66–1.24 pmol·mg−1 protein) than for VSV-G-SNAP-OX1. Binding studies were also performed using membranes from cells expressing VSV-G-OX1-eYFP and CB1-CFP constructs (Ellis et al., 2006) (Figure 2A iii and iv). These receptors are tagged with autofluorescent proteins at the carboxy terminal and with only an epitope tag or nothing at all at the amino terminal. The Kd values obtained from these and the amino terminal SNAP- or CLIP- tagged receptors were comparable (VSV-G-SNAP-OX1= 1.11 ± 0.09 nM, VSV-G-OX1-eYFP = 1.10 ± 0.42 nM, HA-CLIP-CB1= 0.43 ± 0.19 nM and CB1-CFP = 0.47 ± 0.26 nM) and in line with previously published results (Langmead et al., 2004; Ellis et al., 2006).
Figure 1.
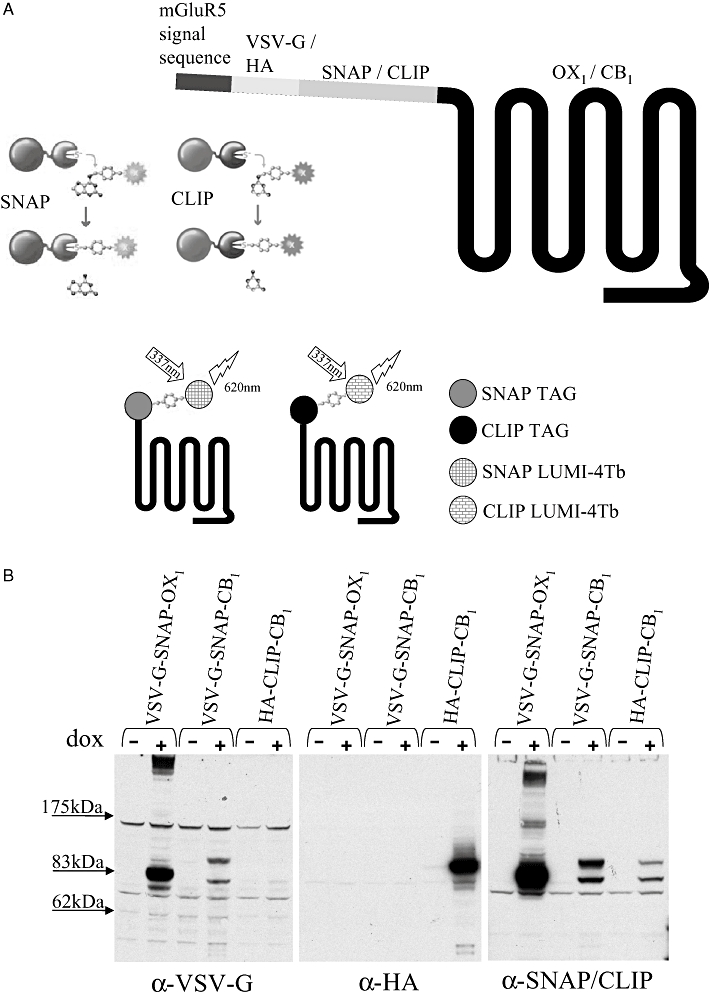
Construction and expression of SNAP- and CLIP-tagged forms of the human OX1 and CB1 receptors. (A) Upper panel: cartoon representation of the signal/leader sequence, an epitope tag and the SNAP or CLIP sequence within the N-terminal extracellular domain of a 7 transmembrane domain GPCR. A schematic cartoon of the basis of selective covalent modification of proteins that incorporate SNAP or CLIP tags is also shown. The fluorophore or other label becomes linked permanently via the O6-alkylguanine-DNA-alkyltransferase activity of the SNAP (SNAP-tag substrates consist of dye conjugated to guanine or chloropyrimidine leaving groups via a benzyl linker)/CLIP (CLIP-tag substrates consist of dye conjugated to a cytosine leaving group via a benzyl linker) proteins. Lower panel: Cartoon representation of the use of SNAP- or CLIP-Lumi4Tb to label cell surface SNAP/CLIP-tagged GPCRs. Excitation with light of 337 nm results in long-lived fluorescence output at 620 nm that can be harnessed to a variety of homogeneous time-resolved fluorescence assays. (B) Lysates prepared from uninduced (− dox) or construct-induced (+ dox) cells that harbour VSV-G-SNAP-OX1, VSV-G-SNAP-CB1 or HA-CLIP-CB1 were resolved by SDS-PAGE and subsequently immunoblotted with anti-VSV-G, anti-HA or anti-SNAP/CLIP (which recognizes both sequences).
Figure 2.
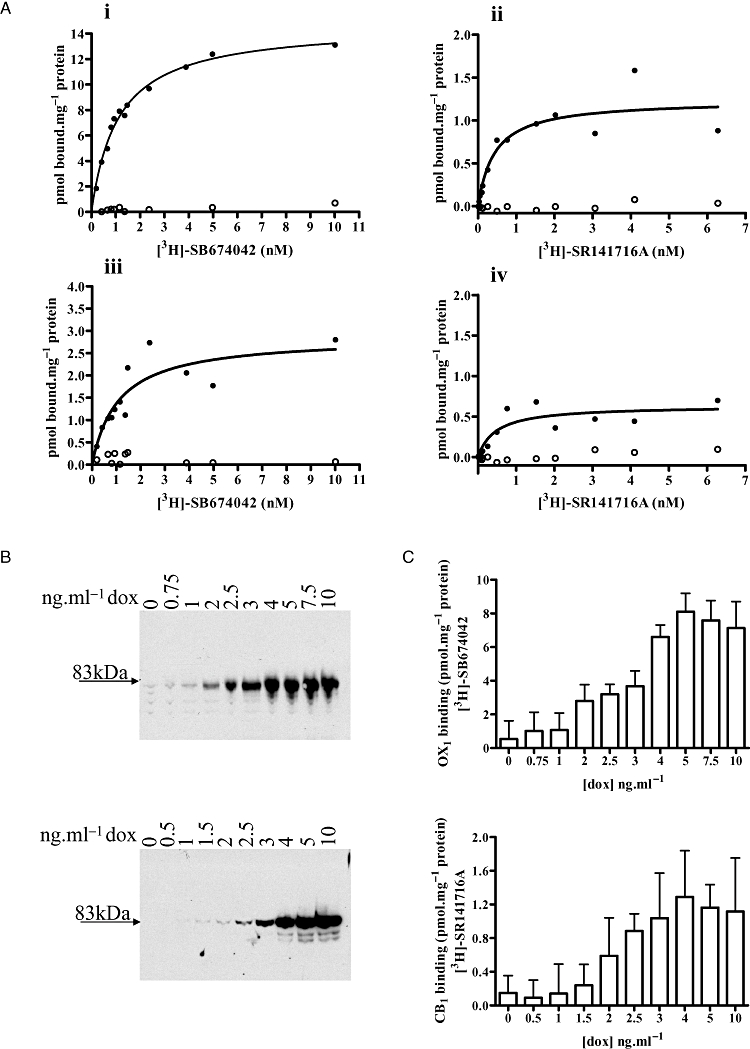
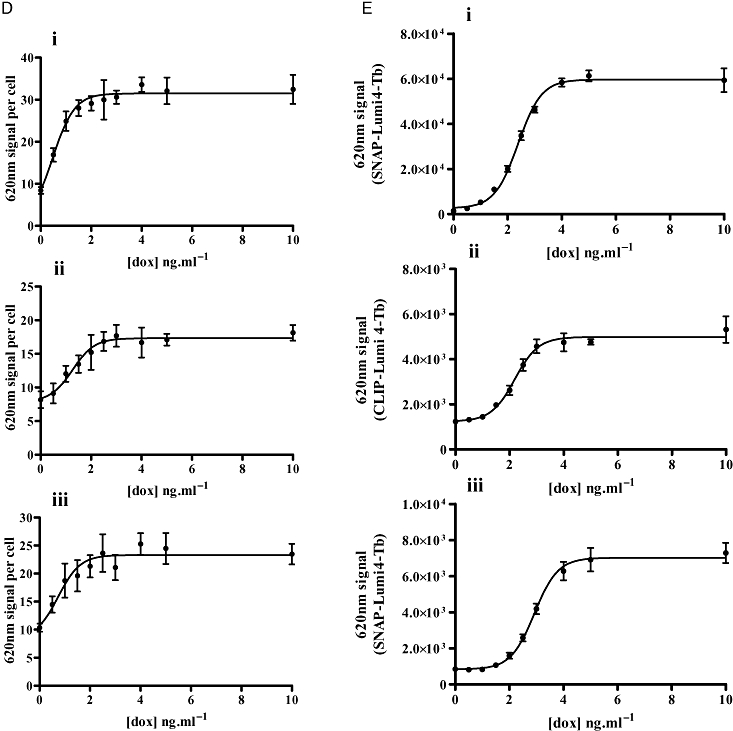
Control of the level of expression of VSV-G-SNAP-OX1 VSV-G-SNAP-CB1 or HA-CLIP-CB1 constructs in Flp-In™ T-REx™ 293 cells. (A) The expression of VSV-G-SNAP-OX1 (i), HA-CLIP-CB1 (ii), VSV-G-OX1-eYFP (iii) and CB1-CFP (iv) were measured in uninduced cells (open symbols) or those induced with a maximally effective concentration of doxycycline (filled symbols) via the specific binding of a range of concentrations of [3H]-SB674042 (i and iii) or [3H]-SR141716A (ii and iv). Data from representative experiments are shown (n≥ 3). (B) Membranes were prepared from Flp-In™ T-REx™ 293 cells harbouring VSV-G-SNAP-OX1 (upper panel) or HA-CLIP-CB1 (lower panel) that were treated with the indicated concentrations of doxycycline. Samples were resolved by SDS-PAGE and immunoblotted with anti-SNAP/CLIP as in Figure 1B. (C) Membranes were generated from cells harbouring VSV-G-SNAP-OX1 that had been treated with the indicated concentrations of doxycycline. The specific binding of 10 nM [3H]-SB674042 (upper panel) or 8 nM [3H]-SR141716A (lower panel) was assessed. (D, E) Cells induced with varying concentrations of doxycycline were employed in enzyme-linked immunosorbent assays using anti-SNAP/CLIP (D) or labelled with SNAP- or CLIP-Lumi4Tb (E). In each case, (i) VSV-G-SNAP-OX1, (ii) HA-CLIP-CB1 and (iii) VSV-G-SNAP-CB1.
Due to the inducible nature of the Flp-In™ T-REx™ locus, expression levels of each construct could be controlled by varying the concentration of doxycycline in the cell medium (Figure 2B, C, and D). Immunoblotting (Figure 2B), specific 3H ligand binding (Figure 2C) and both anti-epitope tag (see later discussion) and anti-SNAP/CLIP enzyme-linked immunosorbent assays (Figure 2D) as well as SNAP- or CLIP-Lumi-4Tb binding (Figure 2E) indicated that half-maximal expression of the receptor constructs was produced by exposure to 1–3 ng·mL−1 of doxycycline for 24 h. Specifically (Figure 2D), for VSV-G-SNAP-OX1, HA-CLIP-CB1 and VSV-SNAP-CB1, with pEC50 values of 0.48 ± 0.13, 1.27 ± 0.12 and 0.74 ± 0.24 ng·mL−1, respectively, and (Figure 2E), pEC50 values of 2.39 ± 0.04, 2.19 ± 0.07 and 2.93 ± 0.04 ng·mL−1, respectively.
Both the VSV-G-SNAP-OX1 and HA-CLIP-CB1 constructs were functional. In cells induced to express VSV-G-SNAP-OX1 the endogenous agonist orexin-A elevated [Ca2+]i in a concentration-dependent manner with a pEC50 value of 6.91 ± 0.05 whereas in uninduced cells orexin-A was unable to generate a signal (Figure 3A). Similarly, in cells able to express HA-CLIP-CB1 the CB1 agonist CP55940 caused a marked enhancement of ERK1/2 MAP kinase phosphorylation with a pEC50 value of 7.33 ± 0.06 but was without substantial effect in the absence of receptor induction (Figure 3B). Cells expressing VSV-G-OX1-eYFP (Figure 3C) or CB1-CFP (Figure 3D) were used in similar experiments and provided pEC50 values of 6.25 ± 0.07 and 7.51 ± 0.12, respectively. Thus, for HA-CLIP-CB1 and CB1-CFP there is a good agreement and slightly less so for VSV-SNAP-OX1 and VSV-G-OX1-eYFP. This slight loss of potency may be due to the larger amino terminal tag for a receptor that responds to a peptide ligand.
Figure 3.
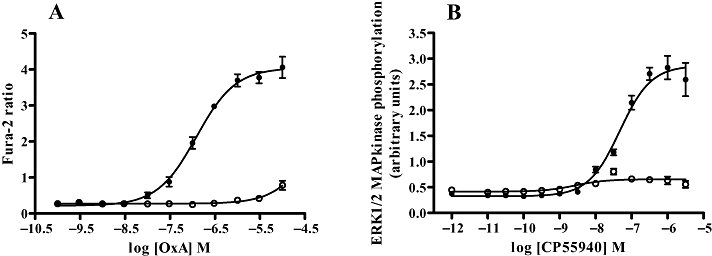
SNAP- and CLIP-tagged forms of the human OX1 and CB1 receptor generate functional responses. Flp-In™ T-REx™ 293 cells harbouring VSV-G-SNAP-OX1 (A), HA-CLIP-CB1 (B), were untreated (open symbols) or treated with doxycycline (filled symbols) to induce expression of the relevant receptor construct. Varying concentrations of orexin A (A) or CP55940 (B) were added and either alteration in [Ca2+]i (A) or phosphorylation of the ERK1/2 MAP kinases (B) assessed. Data are combined from 5 independent experiments and represent means ± SEM.
Addition of doxycycline to cells harbouring VSV-G-SNAP-OX1 resulted in a large increase in the binding of anti-VSV-G primary antibody to the cells (Figure 4A), indicative of cell surface delivery of the expressed construct. Addition of varying concentrations of orexin-A for 60 min prior to addition of the anti-VSV-G primary antibody resulted in a concentration-dependent reduction in VSV-G binding at the cell surface (Figure 4A) of these cells, consistent with orexin-A mediated internalization of VSV-G-SNAP-OX1 to a location where the anti-VSV-G antibody could no longer access the receptor construct. This was concentration-dependent with a pEC50 value of 6.67 ± 0.15 (Figure 4A). By contrast, pretreatment of the cells with varying concentrations of the OX1 receptor antagonists SB334867 or SB408124 did not result in a reduction in cell surface VSV-G-SNAP-OX1 (Figure 4A). Removal of orexin-A after 40 min and addition of fresh medium containing SB408124 resulted in a rapid, time-dependent increase in anti-VSV-G binding (Figure 4B), consistent with recycling of the receptor to the cell surface. Similar studies were performed using anti-HA on cells harbouring HA-CLIP-CB1 (Figure 4C and D). Again, addition of doxycycline to these cells resulted in a substantial, 2.5-fold increase in the binding of anti-HA primary antibody whilst addition of varying concentrations of the CB1 receptor agonist CP55940 resulted in a concentration-dependent (pEC50= 8.50 ± 0.25) decrease in anti-HA labelling (Figure 4C). Although the CB1 receptor neutral antagonist O2050 (Canals and Milligan, 2008) was without effect on cell surface anti-HA labelling, interestingly, addition of the CB1 receptor inverse agonist rimonabant increased cell surface anti-HA labelling in a concentration-dependent manner, with a pEC50 value of 6.95 ± 0.30 (Figure 4D).
Figure 4.
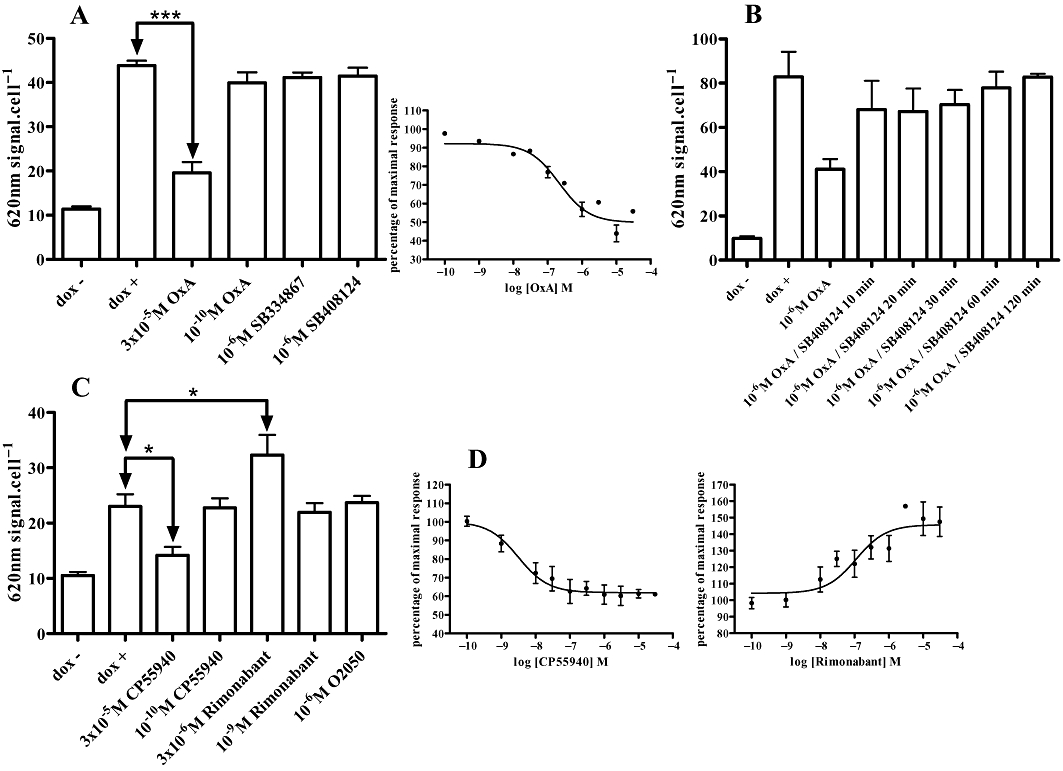
Ligand regulation of cell surface SNAP/CLIP-tagged receptors. Flp-In™ T-REx™ 293 cells harbouring VSV-G-SNAP-OX1 (A, B) or HA-CLIP-CB1 (C, D) were untreated (dox-) or induced to express the receptor constructs (dox + and all other lanes). (A) After induction cells were treated with varying concentrations of orexin A, SB334867 or SB408124 for 40 min before binding of anti-VSV-G was assessed. Such studies were quantified and the difference of signal between dox − and dox + defined as 100% of cell surface VSV-G-SNAP-OX1 (insert). (B) Cells as in (A) were treated with orexin A (1 µM, 40 min). The agonist was removed and replaced with medium containing SB408124 (1 µM) and binding of anti-VSV-G assessed at varying times. (C, D) Experiments akin to those in (A) were conducted using cells harbouring HA-CLIP-CB1 and the effects of various ligands assessed based on the binding of anti-HA. Data represent means ± SEM from at least four independent experiments.*P < 0.05; ***P < 0.001. In D, effects of varying concentrations of CP55940 or rimonabant are quantified.
SNAP- and CLIP-tags can be labelled covalently with small molecule fluorescent dyes to allow identification of the location of proteins linked to them. The cell permeant label SNAP-505 was added to cells induced to express VSV-G-SNAP-OX1. This resulted in strong green fluorescence at the plasma membrane of these cells (Figure 5A) with relatively little staining of internal structures (Figure 5A). This is consistent with a large proportion of the induced VSV-G-SNAP-OX1 being delivered successfully to the cell surface and residing there at steady state. As a control, SNAP-505 was added to cells not pretreated with doxycycline and therefore not expressing VSV-G-SNAP-OX1. No fluorescent signal was detected in these cells (Figure 5B). As a further control, cells induced to express VSV-G-SNAP-OX1 were incubated with the SNAP-blocking reagent bromothenylpteridine (BTP) followed by SNAP-505. This prevented fluorescent labelling of the cells (Figure 5C). Results from cells able to express HA-CLIP-CB1 on demand were less convincing when CLIP-505 was used as the potential substrate (not shown) because significant staining was observed in the absence of receptor induction, whilst a blocking reagent for specific CLIP labelling was unable to eliminate this staining. In contrast to SNAP-505, SNAP Surface-549 is not cell-permeant and therefore can be used to label selectively cell surface SNAP-tagged receptors and subsequently to follow movement of labelled proteins away from the cell surface. For VSV-G-SNAP-OX1 (Figure 6) addition of SNAP Surface-549 resulted in cell surface labelling when these cells were induced to express the construct (Figure 6) but no signals were generated in un-induced cells (not shown). Following labelling with SNAP Surface-549 substrate, cells were washed and then challenged with orexin A (0.5 µM) for varying times before imaging. Over time a substantial fraction of the covalently-linked fluorescent signal, reflecting the labelled SNAP-tagged receptor, was observed within the cells and this provided an alternative measure of agonist-induced internalization (Figure 6).
Figure 5.

Selective identification of VSV-G-SNAP-OX1 by covalent labelling of the SNAP tag. The cell permeant label SNAP-505 was added to cells induced (A, C) or not (B) to express VSV-G-SNAP-OX1. In (C), cells as in (A) were exposed to the SNAP-blocking reagent bromothenylpteridine (BTP) before incubation with SNAP-505. Cells were subsequently imaged.
Figure 6.
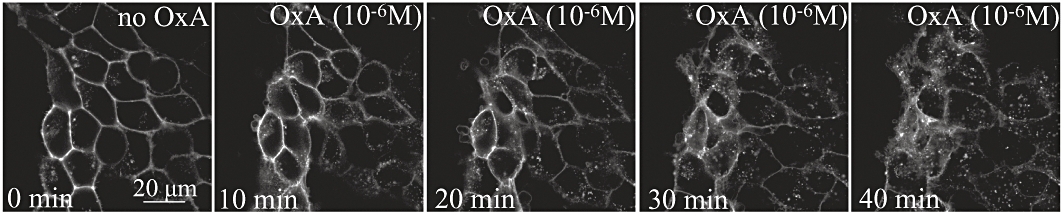
SNAP-tag labelling visualizes orexin A-mediated internalization of an OX1 receptor construct. The cell impermeant label SNAP Surface-549 was added to cells induced to express VSV-G-SNAP-OX1 Orexin A (1 µM) was added subsequently and cells were imaged at various time points. Scale bar = 20 µm.
One issue with such studies is that although providing a clear visual picture of ligand-mediated trafficking of the receptor construct, such images are a challenge to quantify without the construction of three-dimensional cell models and the use of complex algorithms that are provided by so called ‘high content’ analysis platforms (Zanella et al., 2010). An alternate means to utilize and quantify SNAP- and/or CLIP-tagged receptors takes advantage of Tag-Lite™ technology (Maurel et al., 2008; Alvarez-Curto et al., 2010). Herein, reagents such as SNAP-Lumi4Tb can be added to cells expressing a SNAP-tagged GPCR and, following exposure to 337 nm light, the time-resolved fluorescent output at 620 nm can be measured as a monitor of cell surface receptor levels (Figure 1). In Flp-In™ T-REx™ 293 cells induced maximally to express VSV-G-SNAP-OX1 addition of SNAP-Lumi4Tb resulted in a large signal (Figure 7A). Little signal was produced from un-induced Flp-In™ T-REx™ 293 cells harbouring VSV-G-SNAP-OX1, resulting in a signal to background ratio of 41.1 ± 3.6 (Figure 7A). Induction of varying amounts of VSV-G-SNAP-OX1, followed by addition of SNAP-Lumi4Tb resulted in a pattern consistent with the use of [3H]-SB674042 ligand binding and anti-SNAP/CLIP antibodies but with a greatly enhanced signal window (Figure 7A). Indeed, the signal to background ratio produced by the use of SNAP-Lumi4Tb was substantially greater than when equivalent studies used either anti-SNAP/CLIP or the specific binding of [3H]-SB674042 (Figure 7A). Similar experiments carried out with cells induced to express VSV-G-SNAP-CB1 produced a substantially lower signal to background ratio (8.53 ± 0.64), but one that was still a significant improvement over the anti-SNAP-CLIP binding. The pEC50 values for doxycycline induction of receptor expression calculated from SNAP-Lumi4Tb emission were (VSV-G-SNAP-OX1) 2.37 ± 0.03 ng·mL−1 doxycycline and (VSV-G-SNAP-CB1) 2.96 ± 0.03 ng·mL−1 doxycycline. These may be compared with the values of 1.32 ± 0.2 and 0.11 ± 1.76 ng·mL−1 doxycycline, respectively, obtained from the anti-SNAP/CLIP binding and 2.93 ± 0.20 ng·mL−1 doxycycline for [3H]-SB674042 binding to VSV-SNAP-OX1. Addition of varying concentrations of orexin-A for 40 min prior to addition of SNAP-Lumi4Tb again demonstrated concentration-dependent orexin A-mediated receptor internalization as well as a lack of effect of either SB334867 or SB408124 (Figure 7B). Quantification of the orexin-A mediated internalization generated a pEC50 value of 7.12 ± 0.14, in line with those obtained via anti-VSV-G binding in Figure 4A.
Figure 7.
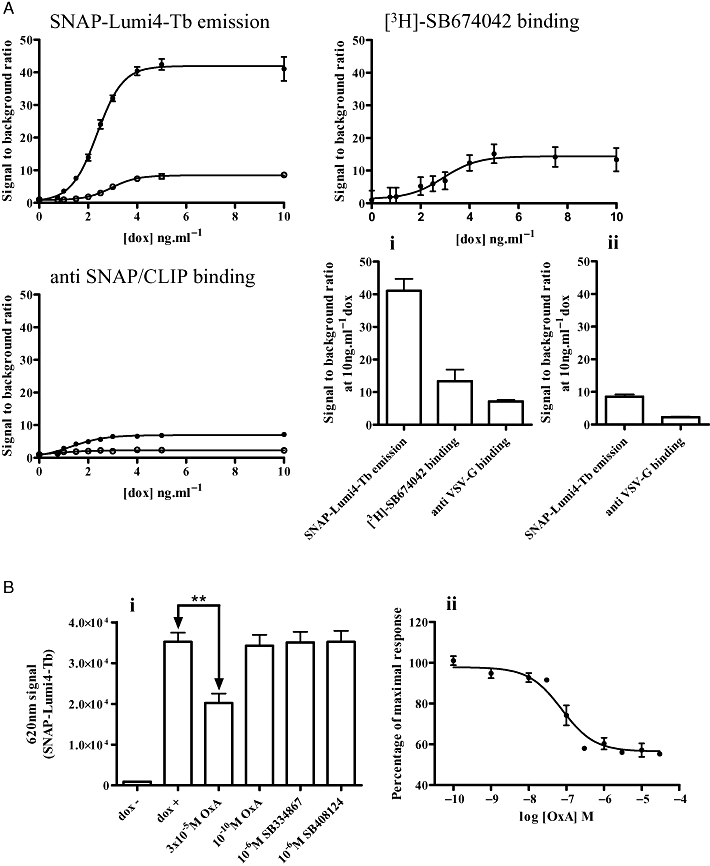
Using time-resolved fluorescence reagents to detect and to analyse OX1 receptor expression and regulation. (A) Cells harbouring VSV-G-SNAP-OX1 (closed symbols) or VSV-G-SNAP-CB1 (open symbols) were induced with varying concentrations of doxycycline. Receptor was measured using SNAP-Lumi4Tb emission, [3H]-SB674042 binding (VSV-G-SNAP-OX1 only) and anti-SNAP/CLIP binding. Data are shown as signal to background ratios. Comparisons of the signal to background ratio for each approach following treatment with 10 ng·mL−1 doxycycline are shown in panels (i) VSV-G-SNAP OX1 or (ii) VSV-G-SNAP-CB1. (B i, ii) Cells induced to express VSV-G-SNAP-OX1 were treated with varying concentrations of orexin A, SB408124 or SB334867 for 40 min then SNAP-Lumi4Tb was added. Emission at 620 nm was subsequently measured. Data represent means ± SEM from at least four independent experiments, **P < 0.01.
Equivalent studies were performed on cells harbouring HA-CLIP-CB1 (Figure 8A). However, when compared with the signal to background ratio of either the specific binding of [3H]-SR141716A or direct anti-SNAP/CLIP binding (Figure 8A), CLIP-Lumi4Tb offered no quantitative advantage, in each case being in the region of 5-fold. This may reflect the relatively high binding of CLIP-Lumi4Tb to these cells in the absence of induction of HA-CLIP-CB1 (Figure 8B). Despite, this, addition of CP55940 resulted in a concentration-dependent decrease in CLIP-Lumi4Tb binding, consistent with agonist-induced internalization of the receptor construct (Figure 8B), with a pEC50 value of 8.55 ± 0.16, almost identical to that noted in Figure 4. In this case neither O2050 nor rimonabant produced a statistically significant alteration in apparent cell surface levels of HA-CLIP-CB1 (Figure 8B), although it was possible to plot the increase produced by rimonabant to generate a pEC50 value of 6.91 ± 0.29, again very similar to the value produced in the studies depicted in Figure 4.
Figure 8.
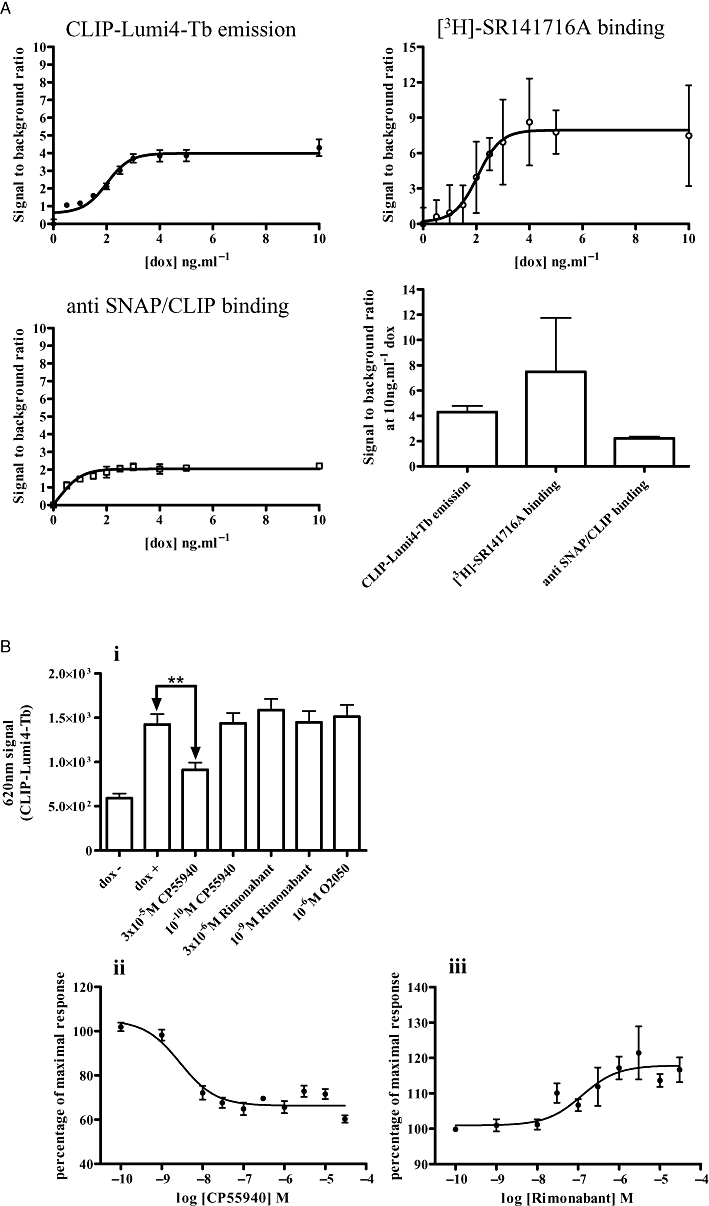
CLIP-Lumi4Tb does not provide an improved means to detect expression and regulation of HA-CLIP-CB1. (A) Cells harbouring HA-CLIP-CB1 were induced with varying concentrations of doxycycline. Receptor was measured using CLIP-Lumi4-Tb emission, [3H]-SR141716A binding and anti-SNAP/CLIP binding. Data are shown as signal to background ratios. Comparisons of the signal to background ratios for each approach following treatment with 10 ng·mL−1 doxycycline are shown in the bottom right panel. (B) Cells induced to express HA-CLIP-CB1 were treated with varying concentrations of CP55940, rimonabant or O2050 for 60 min. CLIP-Lumi4-Tb was added and emission at 620 nm measured. Data represent means ± SEM from at least four independent experiments, **P < 0.01.
Discussion and conclusions
The OX1 and OX2 orexin receptors have attracted considerable attention because of their roles in the regulation of wakefulness (Sullivan and Guilleminault, 2009; Coleman and Renger, 2010) and because their endogenous peptide ligands orexin A and orexin B are produced from a limited number of hypothalamic neurons that project throughout the central nervous system. Although there is expression of both the orexin peptides and the orexin receptors in the periphery, their roles outwith the central nervous system currently remain much less well defined (Voisin et al., 2003; Baccari, 2010). A number of studies have explored the signal transduction mechanisms that can be initiated by the orexin receptors (Smart et al., 1999; 2001; Ammoun et al., 2006) but, although selective OX2 receptor antagonists have recently become available (Malherbe et al. 2009a,b; Coleman and Renger, 2010), the majority of published studies have concentrated on the OX1 orexin receptor (Smart et al., 1999; 2001;). These have shown that this receptor links predominantly to elevation of intracellular Ca2+ levels via members of the Gq/G11 family of G proteins upon binding of orexin A (Ammoun et al., 2006), interacts strongly with β-arrestins (Evans et al., 2005; Milasta et al., 2005) and subsequently undergoes substantial internalization from the surface of cells upon binding orexin A (Milasta et al., 2005). The CB1 and CB2 cannabinoid receptors have also been studied widely, not least because of the psychoactive properties of cannabinoid CB1 agonists (Pertwee and Ross, 2002; Pertwee, 2005) and the development of the CB1 receptor antagonist/inverse agonist rimonabant as a therapy to limit appetite and promote weight loss (Burch et al., 2009; Lee et al., 2009).
Agonist-induced internalization is a common property of many GPCRs and hence has been employed to both identify novel activators of GPCRs and to more fully explore the regulation of such receptors (McLean et al., 1999; Milligan, 1999; Milasta et al., 2005; Haasen et al., 2006; Heilker, 2006). A number of approaches have been used to examine receptor internalization, and these include monitoring the loss of cell surface detection by antibodies that recognize extracellular located epitopes in the target GPCR. Although these can be authentic, native epitopes the limited availability of high affinity immunological reagents that bind to such elements of GPCRs means that, in reality, such studies largely take advantage of introduced N-terminal epitope tags. In recent times the introduction of SNAP- and CLIP-tagging of proteins (Tirat et al., 2006; Gautier et al., 2008) has provided a novel means to explore protein-protein interactions via FRET as well as many aspects of ligand regulation of GPCR internalization. This reflects the enzymatic activity of the SNAP and CLIP tags that allows covalent attachment of small molecules of choice, including a range of fluorophores. To assess the general utility of SNAP/CLIP tagging to assess regulation of GPCRs we generated both forms of the human OX1 and CB1 receptors that contained not only the SNAP tag sequence but also the VSV-G epitope tag in the N-terminal extracellular region and an equivalent form of the CB1 receptor that contained the HA epitope tag and the CLIP protein sequence. These constructs were cloned into the inducible Flp-In™ locus of Flp-In™ T-REx™ 293 cells because this system allows highly regulated induction of the protein encoded by DNA at this locus from an extremely low, generally undetectable, basal level (Ellis et al., 2006; Canals and Milligan, 2008; Lopez-Gimenez et al., 2008). Following induction of VSV-G-SNAP-OX1 addition of orexin A caused internalization of this construct that could be assessed either by imaging of a cell impermeant fluorophore bound covalently to the SNAP tag or enzyme-linked immunosorbent assays employing both anti-VSV-G and anti-SNAP antibodies. However, although both of these antibodies provided reasonable signal to background ratios when comparing uninduced cells with those induced to express the VSV-G-SNAP-OX1 construct, this was improved vastly by use of the long-lived fluorescence characteristics of SNAP-Lumi4Tb in time-resolved fluorescence format. Indeed, signal to background ratio was some six- to 10-fold higher than when using either antibody. This allows therefore receptor internalization studies to be performed and quantified at much lower levels of receptor expression. Although the HA-CLIP-CB1 construct could also be detected and quantified in similar ways, labelling of cells harbouring but not induced to express this construct with either CLIP-Lumi4Tb or a CLIP-selective fluorophore limited the signal to background ratios obtained with this system. VSV-G-SNAP-CB1 however, while having a poor signal to noise ratio with SNAP-Lumi4Tb binding compared with VSV-G-SNAP-OX1 at this endpoint, still produced a significantly improved signal compared with anti-SNAP binding. Overall, the signal to noise ratio for the CB1 receptor constructs was better for SNAP labelling than for CLIP.
It was noted that the CB1 inverse agonist rimonabant was able to increase the cell surface expression of CB1, presumably due to an effect upon the constitutive recycling of this receptor. This effect was more pronounced when measured with antibody binding in an enzyme linked immuno-absorbant assay then for CLIP-Lumi4Tb binding (or SNAP-Lumi4Tb binding in the VSV-G-SNAP-CB1, unpublished results). The effect was only seen for this inverse agonist and not other ligands widely regarded as inverse agonists such as AM281, AM251 and LY320136 (unpublished results). The compound O2050, widely regarded as a CB1 neutral antagonist (Canals and Milligan, 2008) also lacked effect in these assays. It is noteworthy, however, that a CXCR2 inverse agonist has also recently been shown to produce arrestin redistribution (Kredel et al., 2009). It is presently unclear as to why an increase in receptor density at the cell surface should be more easily detectable by antibody binding then by SNAP or CLIP-Lumi4Tb binding and this clearly requires further study.
The Tag-Lite™ (SNAP/CLIP-Lumi4Tb) technology is also highly suitable for monitoring protein-protein interactions at the surface of living cells. The concept that many, if not all, GPCRs display a propensity to form dimers or larger oligomeric complexes has gained widespread support in recent years (Milligan, 2004; 2007; 2008;). Recent use of this for both rhodopsin- and metabotropic glutamate-family receptors (Maurel et al., 2008; Alvarez Curto et al., 2010) has shown the utility of this system.
These studies demonstrate the wide-ranging suitability of SNAP- and CLIP-tagging to detect expression and explore ligand regulation of the cellular location of both the orexin OX1 and cannabinoid CB1 receptors. These are applicable to address both basic research questions and to be incorporated into ligand screening methodologies for early stage drug-discovery. We anticipate that such approaches will become widely used for both GPCRs and other membrane proteins but, based on these results and the currently available reagents, the SNAP tag may offer greater utility and flexibility than the CLIP tag.
Acknowledgments
These studies were supported by the Medical Research Council (grant G0900050).
Glossary
Abbreviations
- Almorexant
(2R)-2-[(1S)-6,7-dimethoxy-1-{2-[4-(trifluoromethyl)phenyl]ethyl}- 3,4-dihydroisoquinolin-2(1H)-yl]-N-methyl-2-phenylacetamide
- AM251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophen yl)-4-methyl-1H-pyrazole-3-carboxamide
- CP55940
(-)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- O2050
(6aR,10aR)-3-(1-methanesulphonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran
- GPCR
G protein-coupled receptor
- htrf
homogeneous time-resolved fluorescence
- SB334867
N-(2-methyl-6-benzoxazolyl)-N'-1,5-naphthyridin-4-yl urea
- SB408124
N-(6,8-difluoro-2-methyl-4-quinolinyl)-N'-[4-(dimethylamino)phenyl]urea
- SB674042
1-(5-(2-fluoro-phenyl)-2-methyl-thiazol-4-yl)-1-{[S]-2-[5-phenyl-(1,3,4)oxadiazol-2-ylmethyl]-pyrrolidin-1-yl}-methanone
- SR141716A
(rimonabant), 5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide
Conflict of interests
None.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Adie EJ, Kalinka S, Smith L, Francis MJ, Marenghi A, Cooper ME, et al. A pH-sensitive fluor, CypHer 5, used to monitor agonist-induced G protein-coupled receptor internalization in live cells. Biotechniques. 2002;33:1152–1154. doi: 10.2144/02335dd10. [DOI] [PubMed] [Google Scholar]
- Adie EJ, Francis MJ, Davies J, Smith L, Marenghi A, Hather C, et al. CypHer 5: a generic approach for measuring the activation and trafficking of G protein-coupled receptors in live cells. Assay Drug Dev Technol. 2003;1:251–259. doi: 10.1089/15406580360545062. [DOI] [PubMed] [Google Scholar]
- Alvarez-Curto E, Ward RJ, Pediani JD, Milligan G. Ligand regulation of the quaternary organization of cell surface M3 muscarinic acetylcholine receptors analyzed by fluorescence resonance energy transfer (FRET) imaging and homogenous time-resolved FRET. J Biol Chem. 2010;285:23318–23330. doi: 10.1074/jbc.M110.122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammoun S, Johansson L, Ekholm ME, Holmqvist T, Danis AS, Korhonen L, et al. OX1 orexin receptors activate extracellular signal-regulated kinase in Chinese hamster ovary cells via multiple mechanisms: the role of Ca2+ influx in OX1 receptor signaling. Mol Endocrinol. 2006;20:80–99. doi: 10.1210/me.2004-0389. [DOI] [PubMed] [Google Scholar]
- Baccari MC. Orexins and gastrointestinal functions. Curr Protein Pept Sci. 2010 doi: 10.2174/138920310790848377. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Bingham MJ, Cai J, Deehan MR. Eating, sleeping and rewarding: orexin receptors and their antagonists. Curr Opin Drug Discov Devel. 2006;9:551–559. [PubMed] [Google Scholar]
- Burch J, McKenna C, Palmer S, Norman G, Glanville J, Sculpher M, et al. Rimonabant for the treatment of overweight and obese people. Health Technol Assess. 2009;13:13–22. doi: 10.3310/hta13suppl3/03. [DOI] [PubMed] [Google Scholar]
- Canals M, Milligan G. Constitutive activity of the cannabinoid CB1 receptor regulates the function of co-expressed Mu opioid receptors. J Biol Chem. 2008;283:11424–11434. doi: 10.1074/jbc.M710300200. [DOI] [PubMed] [Google Scholar]
- Coleman PJ, Renger JJ. Orexin receptor antagonists: a review of promising compounds patented since 2006. Expert Opin Ther Pat. 2010;20:307–324. doi: 10.1517/13543770903567085. [DOI] [PubMed] [Google Scholar]
- Coman OA, Paunescu H, Coman L, Badarau A, Fulga I. Recent data on cannabinoids and their pharmacological implications in neuropathic pain. J Med Life. 2008;1:365–375. [PMC free article] [PubMed] [Google Scholar]
- Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, et al. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Bass CE, Van Horn CG, Howlett AC. Signal transduction via cannabinoid receptors. CNS Neurol Disord Drug Targets. 2009;6:422–431. doi: 10.2174/187152709789824615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J Biol Chem. 2006;281:38812–38824. doi: 10.1074/jbc.M602494200. [DOI] [PubMed] [Google Scholar]
- Evans NA, Groarke DA, Warrack J, Greenwood CJ, Dodgson K, Milligan G, et al. Visualizing differences in ligand-induced beta-arrestin-GFP interactions and trafficking between three recently characterized G protein-coupled receptors. J Neurochem. 2005;77:476–485. doi: 10.1046/j.1471-4159.2001.00269.x. [DOI] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Corrêa IR, Jr, Kindermann M, Beaufils F, et al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Haasen D, Schnapp A, Valler MJ, Heilker R. G protein-coupled receptor internalization assays in the high-content screening format. Methods Enzymol. 2006;414:121–139. doi: 10.1016/S0076-6879(06)14008-2. [DOI] [PubMed] [Google Scholar]
- Heilker R. High content screening to monitor G protein-coupled receptor internalisation. Ernst Schering Found Symp Proc. 2006;2:229–247. doi: 10.1007/2789_2006_011. [DOI] [PubMed] [Google Scholar]
- Johansson L, Ekholm ME, Kukkonen JP. Regulation of OX1 orexin/hypocretin receptor-coupling to phospholipase C by Ca2+ influx. Br J Pharmacol. 2007;150:97–104. doi: 10.1038/sj.bjp.0706959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallal L, Benovic JL. Fluorescence microscopy techniques for the study of G protein-coupled receptor trafficking. Methods Enzymol. 2002;343:492–506. doi: 10.1016/s0076-6879(02)43154-0. [DOI] [PubMed] [Google Scholar]
- Kirkham TC. Cannabinoids and appetite: food craving and food pleasure. Int Rev Psychiatry. 2009;21:163–171. doi: 10.1080/09540260902782810. [DOI] [PubMed] [Google Scholar]
- Kredel S, Wolff M, Wiedenmann J, Moepps B, Nienhaus GU, Gierschik P, et al. CXCR2 inverse agonism detected by arrestin redistribution. J Biomol Screen. 2009;14:1076–1091. doi: 10.1177/1087057109344616. [DOI] [PubMed] [Google Scholar]
- Kroeger D, de Lecea L. The hypocretins and their role in narcolepsy. CNS Neurol Disord Drug Targets. 2009;8:271–280. doi: 10.2174/187152709788921645. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Jerman JC, Brough SJ, Scott C, Porter RA, Herdon HJ. Characterisation of the binding of [3H]-SB-674042, a novel nonpeptide antagonist, to the human orexin-1 receptor. Br J Pharmacol. 2004;141:340–346. doi: 10.1038/sj.bjp.0705610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Choi EB, Pak CS. The current status and future perspectives of studies of cannabinoid receptor 1 antagonists as anti-obesity agents. Curr Top Med Chem. 2009;9:482–503. doi: 10.2174/156802609788897844. [DOI] [PubMed] [Google Scholar]
- Lopez-Gimenez JF, Vilaró MT, Milligan G. Morphine desensitization, internalization, and down-regulation of the mu opioid receptor is facilitated by serotonin 5-hydroxytryptamine2A receptor co-activation. Mol Pharmacol. 2008;74:1278–1291. doi: 10.1124/mol.108.048272. [DOI] [PubMed] [Google Scholar]
- McLean AJ, Bevan N, Rees S, Milligan G. Visualizing differences in ligand regulation of wild-type and constitutively active mutant beta(2)-adrenoceptor-green fluorescent protein fusion proteins. Mol Pharmacol. 1999;56:1182–1191. doi: 10.1124/mol.56.6.1182. [DOI] [PubMed] [Google Scholar]
- Malherbe P, Borroni E, Gobbi L, Knust H, Nettekoven M, Pinard E, et al. Biochemical and behavioural characterization of EMPA, a novel high-affinity, selective antagonist for the OX(2) receptor. Br J Pharmacol. 2009a;156:1326–1341. doi: 10.1111/j.1476-5381.2009.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe P, Borroni E, Pinard E, Wettstein JG, Knoflach F. Biochemical and electrophysiological characterization of almorexant, a dual orexin 1 receptor (OX1)/orexin 2 receptor (OX2) antagonist: comparison with selective OX1 and OX2 antagonists. Mol Pharmacol. 2009b;76:618–631. doi: 10.1124/mol.109.055152. [DOI] [PubMed] [Google Scholar]
- Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, et al. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods. 2008;5:561–567. doi: 10.1038/nmeth.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milasta S, Evans NA, Ormiston L, Wilson S, Lefkowitz RJ, Milligan G. The sustainability of interactions between the orexin-1 receptor and beta-arrestin-2 is defined by a single C-terminal cluster of hydroxy amino acids and modulates the kinetics of ERK MAPK regulation. Biochem J. 2005;387:573–584. doi: 10.1042/BJ20041745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. Exploring the dynamics of regulation of G protein-coupled receptors using green fluorescent protein. Br J Pharmacol. 1999;128:501–510. doi: 10.1038/sj.bjp.0702824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol. 2004;66:1–7. doi: 10.1124/mol.104.000497.. [DOI] [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor dimerisation: molecular basis and relevance to function. Biochim Biophys Acta. 2007;1768:825–835. doi: 10.1016/j.bbamem.2006.09.021. [DOI] [PubMed] [Google Scholar]
- Milligan G. A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol. 2008;153(Suppl 1):S216–S229. doi: 10.1038/sj.bjp.0707490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer DN. Almorexant, a dual orexin receptor antagonist for the treatment of insomnia. Curr Opin Investig Drugs. 2010;11:101–110. [PubMed] [Google Scholar]
- Pertwee RG. Pharmacological actions of cannabinoids. Handb Exp Pharmacol. 2005;168:1–51. doi: 10.1007/3-540-26573-2_1. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids. 2002;66:101–121. doi: 10.1054/plef.2001.0341. [DOI] [PubMed] [Google Scholar]
- Sakurai T. Reverse pharmacology of orexin: from an orphan GPCR to integrative physiology. Regul Pept. 2005;126:3–10. doi: 10.1016/j.regpep.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Smart D, Jerman JC, Brough SJ, Rushton SL, Murdock PR, Jewitt F, et al. Characterization of recombinant human orexin receptor pharmacology in a Chinese hamster ovary cell-line using FLIPR. Br J Pharmacol. 1999;128:1–3. doi: 10.1038/sj.bjp.0702780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA, et al. SB-334867-A: the first selective orexin-1 receptor antagonist. Br J Pharmacol. 2001;132:1179–1182. doi: 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Stoddart LA, Devine NM, Jenkins L, Milligan G. The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J Biol Chem. 2009;284:17527–17539. doi: 10.1074/jbc.M109.012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SS, Guilleminault C. Emerging drugs for insomnia: new frontiers for old and novel targets. Expert Opin Emerg Drugs. 2009;14:411–422. doi: 10.1517/14728210903171948. [DOI] [PubMed] [Google Scholar]
- Tirat A, Freuler F, Stettler T, Mayr LM, Leder L. Evaluation of two novel tag-based labelling technologies for site-specific modification of proteins. Int J Biol Macromol. 2006;39:66–76. doi: 10.1016/j.ijbiomac.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Voisin T, Rouet-Benzineb P, Reuter N, Laburthe M. Orexins and their receptors: structural aspects and role in peripheral tissues. Cell Mol Life Sci. 2003;60:72–87. doi: 10.1007/s000180300005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Hohmann AG. Cannabinoid mechanisms of pain suppression. Handb Exp Pharmacol. 2005;168:509–554. doi: 10.1007/3-540-26573-2_17. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Alvarez-Curto E, Milligan G. Using the Flp-In™ T-Rex™ system to regulate GPCR expression. In: Willars G, Challiss JA, editors. Totowa, NJ: Humana Press; 2011. Receptor Signal Transduction Protocols, 3rd Edition. Methods in Molecular Biology series. in press. [DOI] [PubMed] [Google Scholar]
- Wu DF, Yang LQ, Goschke A, Stumm R, Brandenburg LO, Liang YJ, et al. Role of receptor internalization in the agonist-induced desensitization of cannabinoid type 1 receptors. J Neurochem. 2008;104:1132–1143. doi: 10.1111/j.1471-4159.2007.05063.x. [DOI] [PubMed] [Google Scholar]
- Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends Biotechnol. 2010;5:237–245. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.