

Abstract
Oxidized low-density lipoprotein (OxLDL) causes impairment of endothelium-dependent, nitric oxide (NO)-mediated vasodilation involving L-arginine deficiency. However, the underlying mechanism remains elusive. Since arginase and endothelial NO synthase (eNOS) share the substrate L-arginine, we hypothesized that OxLDL may reduce L-arginine availability to eNOS for NO production, and thus vasodilation, by up-regulating arginase. To test this hypothesis, porcine subepicardial arterioles (70-130 μm) were isolated for vasomotor study and for immunohistochemical detection of arginase and eNOS expressions. The coronary arterioles dilated dose-dependently to the endothelium-dependent NO-mediated vasodilator serotonin. This vasodilation was inhibited in the same manner by NOS inhibitor L-NAME and by lumenal OxLDL (0.5 mg protein/ml). The inhibitory effect of OxLDL was reversed after treating the vessels with either L-arginine (3 mM) or arginase inhibitor difluoromethylornithine (DFMO; 0.4 mM). Consistent with vasomotor alterations, OxLDL inhibited serotonin-induced NO release from coronary arterioles and this inhibition was reversed by DFMO. Vascular arginase activity was significantly elevated by OxLDL. Immunohistochemical analysis indicated that Ox-LDL increased arginase I expression in the vascular wall without altering eNOS expression. Taken together, these results suggest that OxLDL up-regulates arginase I, which contributes to endothelial dysfunction by reducing L-arginine availability to eNOS for NO production and thus vasodilation.
Keywords: arteriole, endothelium, low-density lipoprotein, nitric oxide, dilation
Introduction
The impairment of endothelium-dependent relaxation of coronary conduit arteries by atherosclerotic insult [5,52] or by a high level of atherogenic molecule, oxidized low-density lipoprotein (OxLDL) [20,43,48], is well documented. The functional consequences of this pathology have been shown to extend into coronary arterioles (<150 μm in diameter) [12,35], the primary site involved in the coronary flow regulation [13]. A number of studies have shown that the endothelial NO bioavailability and NO synthase (eNOS) pathway are adversely affected by OxLDL [20,25,33,43,48]. We have previously reported that OxLDL selectively impairs endothelium-dependent, NO-mediated dilation of porcine coronary arterioles in response to pharmacological agonists [25] and to increased shear stress [27]. Interestingly, administration of NO precursor L-arginine improves OxLDL-impaired vasodilatory function [25,27]. These studies indicate that reduced L-arginine availability contributes to the vascular dysfunction elicited by OxLDL. Although the mechanism responsible for the L-arginine deficiency remains elusive, it is possible that the increase in L-arginine consumption by other enzymes might lead to the reduction of L-arginine availability for eNOS during OxLDL insult.
Arginase enzymes (both type I and type II isoforms) consume L-arginine to form L-ornithine and urea and have been shown to be constitutively expressed in the endothelial and smooth muscle cells [3,6,55]. These enzymes, at the endothelial level, can play a counteracting role in modulating NO production [8,11,58] and in NO-mediated vasodilation by competing with eNOS for their common substrate L-arginine [6,17,58]. Interestingly, accumulating evidence has shown that the expression of arginase is elevated in a variety of cells and tissues with inflammation and oxidative stress [3,10,46,54], the conditions that are known to be associated with atherogenesis. Moreover, L-arginine deficiency was recently found to be involved in the vascular dysfunction in hypercholesterolemia and atherosclerosis [51]. In view that enhanced arginase activity and/or expression can contribute to endothelial dysfunction related to NO deficiency in various cardiovascular diseases [6,29,56], it is likely that arginase may influence arteriolar vasomotor function during the development of atherosclerosis. However, it remains unclear whether the OxLDL, one of the major atherogenic molecules, plays a role in modulating vasodilatory function by manipulating NO bioavailability via the arginase pathway. In this context, we examined the effect of OxLDL on NO production and arginase activity/expression in relation to the vasomotor function of isolated coronary arterioles. We speculated that OxLDL might exert its adverse effect on the endothelium-mediated vasodilatory function by reducing L-arginine availability for NO synthesis via arginase activation.
Materials and Methods
Preparation of Coronary Arterioles and Functional Analyses
The identification and isolation of pig coronary arterioles were described in detail previously [36]. Briefly, pigs (10 to 15 kg, 22 males and 18 females) were anesthetized with pentobarbital sodium (20 mg/kg). After a left thoracotomy was performed, the heart was electrically fibrillated, quickly removed, and immediately placed in cold (5°C) physiological salt solution (PSS). The procedures followed were in accordance with guidelines set by the Laboratory Animal Care Committee at Texas A&M Health Science Center. Arterioles (60-80 μm in internal diameter in situ) were isolated from the small branches of left anterior descending artery of left ventricle. One to three vessels were isolated from each heart for functional studies described below.
The isolated coronary arterioles were cannulated with PSS-filled micropipettes in a Lucite vessel chamber. After cannulation of an arteriole, the chamber was transferred to the stage of an inverted microscope (model IM35, Zeiss, Thornwood, NY). The vessel was pressurized without flow at 60 cmH2O by an adjustable reservoir system. Internal diameter of the vessel was measured throughout the experiment using video microscopic techniques as described previously [33]. The cannulated arterioles were equilibrated in a vessel bath at 36-37°C to allow development of basal tone. Coronary arteriolar responses to the endothelium-dependent NO-mediated vasodilator serotonin (10-10 M to 10-6 M) [28,29] and to the endothelium-independent NO donor sodium nitroprusside (10-9 M to 10-5 M) [33] were examined before and after treating the vessels with NO synthase inhibitor NG-nitro-L-arginine methyl ester (L-NAME) (10 μM, 30-minute extraluminal incubation) [27] or replacing the intraluminal solution (PSS) with OxLDL (0.5 mg protein/ml; 90-minute incubation) [27]. The combination of L-NAME and OxLDL treatment was performed in another set of vessels to assess whether OxLDL and L-NAME have an additive inhibitory effect on serotonin-induced dilation. To rule out time-dependent and non-specific effects of the intraluminal incubation procedure, the vasodilatory responses were also examined in another series of experiments after treating the vessels with OxLDL vehicle solution (i.e., PSS, 90-minute intraluminal incubation). To determine whether L-arginine deficiency contributes to the effect of OxLDL, the vasodilation to serotonin was re-examined after incubating the vessels with L-arginine (3 mM, extraluminal treatment) for 30 minutes. The specificity of L-arginine was examined by extraluminally incubating the vessels with D-arginine (3 mM). In some vessels, the role of arginase in altered vasodilation to serotonin was assessed by intraluminal co-administration of OxLDL (0.5 mg protein/ml) and arginase inhibitors α-difluoromethylornithine (DFMO, 0.4 mM; a gift from Ilex Oncology, San Antonio, TX) [47] or Nw-hydroxy-nor-L-arginine (nor-NOHA; Alexis, San Diego, CA) (0.1 mM) [49] for 90 minutes. All chemicals and drugs were purchased from Sigma-Aldrich (St. Louis, MO), except as specifically stated. All the drugs used for the functional study were dissolved in PSS.
At the end of each experiment, the vessels were relaxed with sodium nitroprusside (10-4 M) to obtain their maximal diameter at 60 cmH2O intraluminal pressure. We have previously shown that this concentration of sodium nitroprusside produced maximal relaxation of isolated vessels because their diameters were not further increased by a calcium free solution containing EDTA (1 mM) [25]. All diameter changes in response to agonists were normalized to the vasodilation in response to 10-4 M sodium nitroprusside and expressed as a percentage of maximal dilation. All data are presented as means±SEM and one or two vessels were used from each heart. Statistical comparisons of vasomotor responses under different treatments were performed with two-way ANOVA and tested with Fisher's protected least significant difference multiple range test. Differences in resting diameter before and after pharmacological interventions were compared by paired Student's t-tests. Significance was accepted at P<0.05.
Oxidation of Low Density Lipoprotein
OxLDL was prepared as reported previously [25,27]. Briefly, human native LDL (5 mg protein/ml) was oxidized by exposure to 10 μM CuCl2 for 24 hours at room temperature. The degree of LDL oxidation was spectrophotometrically measured [18] by the thiobarbituric acid-reactive substances (TBARS) assay [9]. TBARS data were expressed as nanomoles malondialdehyde per mg of LDL protein. OxLDL was dialyzed against Dulbecco's phosphate-buffered saline for 24 hours. The OxLDL was stored at 4°C and used within 2 weeks. Before each experiment, OxLDL was filtered with a 0.2 μm pore filter (Corning, Corning, NY) and tested for endotoxin (<0.06 endotoxin unit/ml, Limulus assay, Cambrex) at levels insufficient to affect vasomotor function [34]. OxLDL was then diluted to the final concentration, 0.5 mg protein/ml, in PSS. The protein concentration of LDL was determined by using the modified Lowry assay [38]. OxLDL used in this study exhibited extensive oxidation (12.1±1.8 nmol malondialdehyde/mg LDL protein; n=6).
NO Assay
NO production from coronary arterioles was evaluated by measuring nitrite with a chemiluminescence NO analyzer (Sievers Instruments, Boulder, CO) as described previously [26]. Vessels (∼70 μm in diameter with 1-2 mm in length) were isolated and placed in a microcentrifuge tube (5 vessels/tube) containing 150 μl PSS. After a 90-minute initial incubation at 37°C, serotonin (0.1 μM final concentration) was added to the vessel bath for 30 minutes. The bathing solution was then collected for nitrite measurement. In parallel to the control experiments, another series of studies were performed by pre-incubating the vessels with OxLDL (final concentration 0.5 mg protein/ml in PSS at 37°C) with or without arginase inhibitor (DFMO 0.4 mM or nor-NOHA 0.1 mM) for 90 minutes and the serotonin (0.1 μM final concentration) was subsequently added to the vessel bath. Following a 30-minute incubation with serotonin, nitrite was assayed. In addition, three separate experiments were ran in parallel to the above protocols as negative control by adding corresponding vehicle solution (PSS) and agent in each tube without the vessels. The protein levels in each tube were quantified by bicinchoninic acid protein assay (Pierce, Rockford, IL) and were used as a basis to normalize the NO production. Differences in nitrite concentration were compared by using Student's t-test. Significance was accepted at P < 0.05.
Arginase Activity Assay
Coronary arterioles (5 to 7 vessels/sample, ∼70 μm in diameter, 1 to 2 mm in length) were isolated and incubated with or without OxLDL (0.5 mg protein/ml) for 90 minutes at 37°C. In another set of experiments, isolated vessels were treated with OxLDL in combination with DFMO under conditions described above. The vessels were than transferred to the lysis buffer for an arginase activity assay as described previously [58]. Briefly, vessel lysate (50 μl) was added into 75 μl of Tris·HCl (50 mM, pH 7.5) containing 10 mM MnCl2. Heating the lysate at 55°C to 60°C for 10 minutes activated arginase. The hydrolysis reaction of L-arginine by arginase was performed by incubating the mixture containing activated arginase with 50 μL of L-arginine (0.5 M, pH 9.7) at 37°C for 1 hour and was stopped by adding 400 μl of the acid solution mixture (H2SO4:H3PO4:H2O=1:3:7). For calorimetric determination of urea, α-isonitrosopropiophenone (25 μl, 9% in absolute ethanol) was then added and the mixture was heated at 100°C for 45 minutes. After placing the sample in the dark for 10 minutes at room temperature, the urea concentration was determined spectrophotometrically by the absorbance at 550 nm measured with a microplate reader (Bio-Tek Instruments, Winooski, VT). The amount of urea produced, after normalization with protein, was used as an index for arginase activity. Differences in arginase activity were compared by using Student's t-test. Significance was accepted at P < 0.05.
Immunohistochemical Analysis
Coronary arterioles (∼100 μm inner diameter) were prepared for immunohistochemical analysis as described previously [29,58]. The vessels treated with PSS (vehicle) and OxLDL were prepared for cryosection (12 μm thick) and then immunolabeled with mouse anti-arginase I (1:40 dilution; BD Biosciences Transduction Laboratories, Lexington, KY) or anti-eNOS monoclonal antibodies (1:60 dilution; BD Biosciences Transduction Laboratories, Lexington, KY). Staining control tissues were exposed to corresponding non-immune mouse immunoglobulin (Jackson ImmunoResearch Laboratories, West Grove, PA) in place of the primary antibody. The slides were then incubated with the secondary antibody, goat anti-mouse monoclonal antibody conjugated with biotin (1:60 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA) and then fluorescein avidin D (1:35 dilution; Vector Laborotories, Burlingame, CA). The control and experimental tissues were placed on the same slide and processed under the same conditions. The slides were observed and analyzed using the Ultima-Z 312 Confocal Microscope (Meridian Instruments, Okemos, MI). The scanning parameters for image acquisition were identical for both control and experimental tissues. The resulting images (both arginase and eNOS) were quantitatively analyzed for arginase or eNOS fluorescence intensity using ImageJ software (National Institutes of Health) with the following procedures. The images were thresholded to determine the background control vessel signals. The average fluorescent intensity of the staining-control vessels for each slide group was used to subtract out the signals corresponding to the non-specific staining for each slide group. Then the background-subtracted signal intensity for each experimental data-point in each slide group was normalized to the average background-subtracted fluorescent intensity of the vehicle-treated tissues from that slide group. Student's t-tests were then conducted on the resulting normalized and background-subtracted data for arginase and eNOS independently. Significance was accepted at P < 0.05.
Results
Effect of OxLDL on Coronary Arteriolar Dilation to Serotonin
All vessels developed a similar level of basal tone (∼66±2% of their maximal diameter) within 40 minutes at 36-37°C bath temperature with 60 cmH2O intraluminal pressure. The average resting and maximal diameters of vessels for vasomotor studied were 64±3 μm and 97±4 μm, respectively. Under control conditions, serotonin dilated coronary arterioles in a dose-dependent manner and produced 87±5% of maximal dilation at the highest concentration studied (Figure 1). In the presence of L-NAME, there was a tendency to increase resting tone of the vessel but did not reach statistical significance (before L-NAME: 63±2% of maximal diameter; after L-NAME: 61±3% of maximal diameter). However, the vasodilation to serotonin was significantly attenuated by L-NAME (Figure 1). Similarly, OxLDL (0.5 mg protein/ml) did not significantly change basal tone (before OxLDL: 64±1% of maximal diameter; after OxLDL: 62±2% of maximal diameter) at the end of 90-minute incubation, but the dilation to serotonin was significantly inhibited in a fashion comparable to that produced by L-NAME. Combined OxLDL and L-NAME treatment did not further inhibit serotonin-induced dilation compared to the treatment of OxLDL or L-NAME alone (Figure 1). In a separate series of studies, intraluminal incubation of vessels with OxLDL vehicle solution (n=3) did not alter serotonin-induced vasodilation (data not shown).
Figure 1.
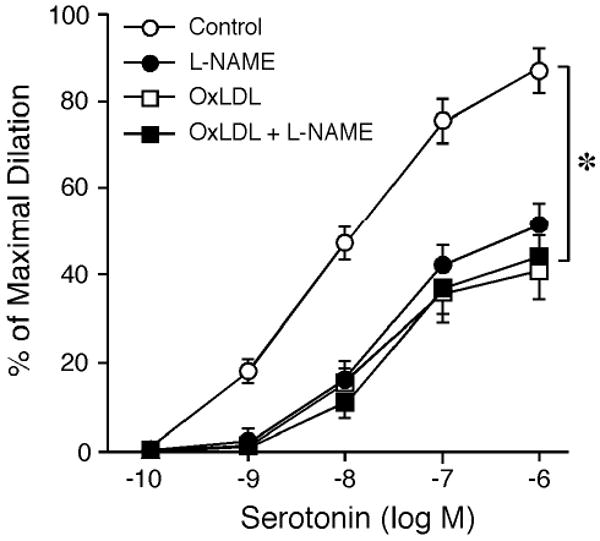
Effect of OxLDL on coronary arteriolar dilation to serotonin. The coronary arterioles dilated in a concentration-dependent manner to serotonin (Control; n=15). The serotonin-induced vasodilation was significantly inhibited by either L-NAME (10 μM; n=5) or OxLDL (0.5 mg protein/ml; n=6). The inhibitory effect of L-NAME was comparable to that of OxLDL. The combination of OxLDL and L-NAME treatment did not further inhibit vasodilation to serotonin (n=4). *P<0.05 vs. Control.
The inhibitory effect of OxLDL on serotonin-induced dilation was restored by a subsequent incubation of OxLDL-treated vessels with L-arginine (Figure 2), but not with D-arginine (n=4, data now shown). The L-arginine or D-arginine treatment did not affect resting vascular tone. Treating the vessels with DFMO did not alter resting tone, but the inhibitory effect of OxLDL on serotonin-induced dilation was not observed (Figure 2). Another arginase inhibitor, nor-NOHA (0.1 mM), produced similar effect as DFMO (n=4, data not shown). Co-incubation of the vessels with OxLDL and arginase inhibitors (DFMO or nor-NOHA) had a tendency to reduce vascular resting tone, but did not reach statistical significance (before DFMO: 61±2% of maximum diameter vs. after DFMO: 64±3% of maximum diameter). The dose-dependent vasodilations in response to serotonin were not altered by L-arginine or DFMO (Figure 3A). In addition, the vasodilations induced by sodium nitroprusside were not altered by OxLDL, L-arginine, or DFMO (Figure 3B).
Figure 2.
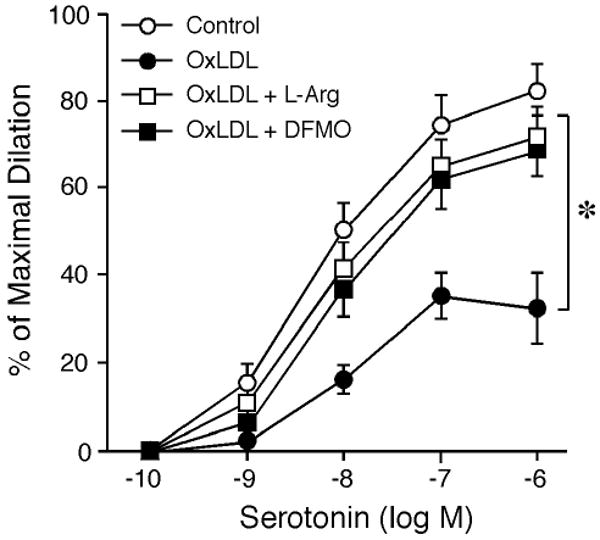
Effect of L-arginine administration and arginase inhibition on vasomotor function. Coronary arteriolar dilation to serotonin (Control; n=14) was inhibited by OxLDL (0.5 mg protein/ml; n=5). Administration of L-arginine (L-Arg, 3 mM; n=4) or arginase inhibitor DFMO (0.4 mM; n=5) restored vasodilation to serotonin. *P<0.05 vs. Control.
Figure 3.
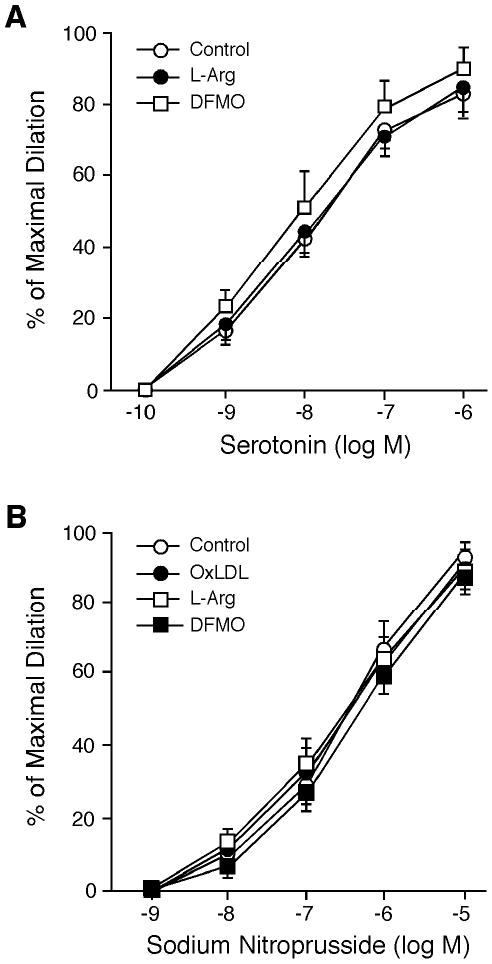
Effect of L-arginine and DFMO on vasomotor function. A: Coronary arteriolar dilation to serotonin (Control; n=8) was not altered in the presence of L-arginine (3 mM; n=4) or DFMO (0.4 mM; n=4). B: Coronary arteriolar dilation in response to sodium nitroprusside (Control; n=14) was not altered by OxLDL (0.5 mg protein/ml; n=5), L-arginine (L-Arg, 3 mM; n=4), or DFMO (0.4 mM; n=5).
Effect of OxLDL on NO Production and Arginase Activity
Incubation of the vessels with OxLDL did not alter their NO production under resting conditions. However, the NO production evoked by serotonin was significantly reduced in the vessels treated with OxLDL (Figure 4). In the presence of DFMO, there was a tendency to increase serotonin-stimulated NO production, but the level did not reach statistical significance. The inhibition of NO production by OxLDL was not observed in the vessels treated with DFMO (Figure 4). In addition, arginase activity was elevated by 3-fold in the coronary arterioles treated with OxLDL, and this increase was abolished in the presence of DFMO (Figure 5).
Figure 4.
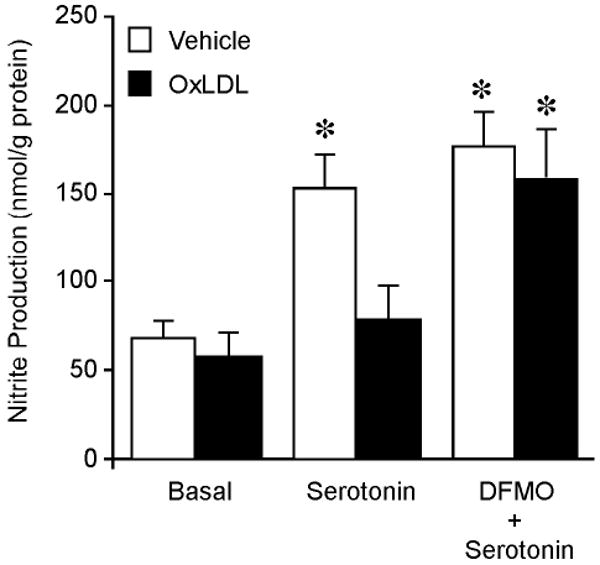
NO production by coronary arterioles in the presence and absence of OxLDL. NO production was measured by chemiluminescence detection of nitrite. The resting NO production (Basal; n=6) was not altered by OxLDL (0.5 mg protein/ml; n=6). In the presence of OxLDL (0.5 mg protein/ml; n=4), the NO production stimulated by serotonin (0.1 μM; n=4) was significantly inhibited. In the presence of arginase inhibitor DFMO (0.4 mM), the serotonin-induced NO production (0.1 μM; n=5) was comparable to that without DFMO and the OxLDL (0.5 mg protein/ml; n=5) did not affect this NO production. *P<0.05 vs. Basal.
Figure 5.
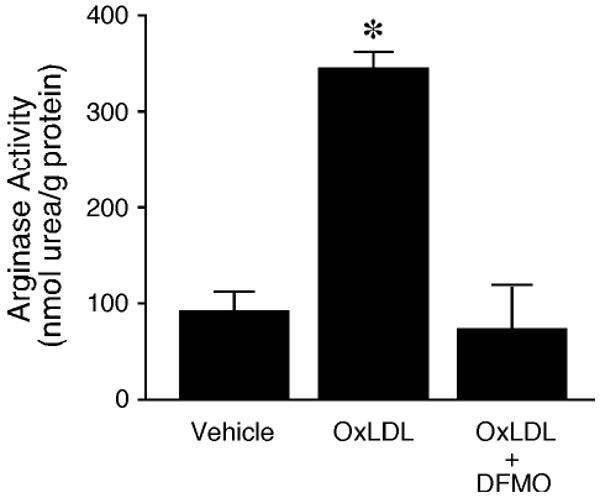
Effect of OxLDL on arginase activity of porcine coronary arterioles. Arginase activity (i.e., urea production) was significantly elevated in the OxLDL-treated vessels (0.5 mg protein/ml; n=5) compared with vessels without OxLDL treatment (Control; n=5). The OxLDL-increased arginase activity was blocked in the presence of DFMO (OxLDL + DFMO; n=5). *P<0.05 vs. Control.
Effect of OxLDL on Arginase I and eNOS Expression
To detect the effects of OxLDL on arginase I and eNOS expressions, the coronary arterioles incubated with or without OxLDL (0.5 mg protein/ml, 90 minutes) were subjected to immunohistochemical study. Arginase I was found to localize mainly in the endothelial cells with a low level of expression in smooth muscle cells of coronary arterioles (Figure 6Ab), whereas the eNOS was observed in the endothelium only (Figure 6Ae). In the vessels treated with OxLDL, the level of positive immunostaining for arginase I was significantly increased in both endothelial and smooth muscle cells (Figure 6Ac). The fluorescence signals for arginase I were increased by 2.8-fold in vessels treated with OxLDL (Figure 6B). There was no significant change in the eNOS staining intensity after OxLDL treatment (Figure 6Af and Figure 6B).
Figure 6.
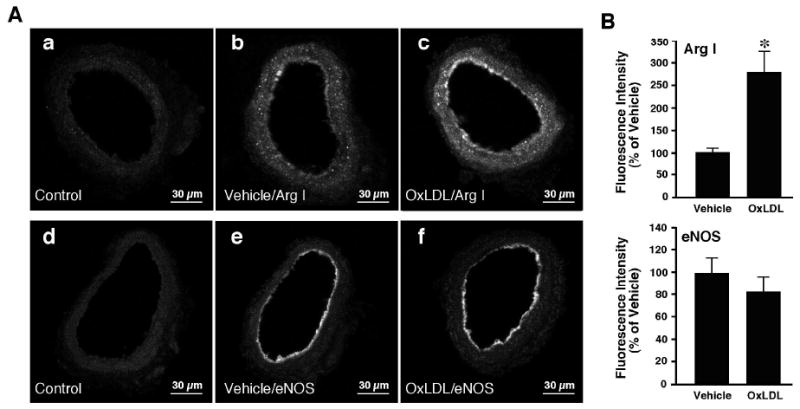
Immunohistochemical detection of arginase I and eNOS in coronary arterioles. A: Cross-sectional view of fluorescein-labeled vessels after treatment with non-immune immunoglobulin, anti-arginase I or anti-eNOS primary antibodies. Panels a and d: Images obtained from vessels treated with non-immune immunoglobulin (Control; n=6). Panels b and e: images obtained from vessels treated with PSS (Vehicle) and immunolabeled with arginase I (Arg I) (panel b; n=6) or eNOS (panel e; n=6) antibody. Panels c and f: Images were obtained from vessels treated with OxLDL (0.5 mg protein/ml) and immunolabeled with arginase I (Arg I) (panel c; n=6) or eNOS (panel f; n=6) antibody. Fluorescence signals for arginase I were detected in both the endothelium and smooth muscle of the vessels (panels b and c). eNOS fluorescence signals were detected in the endothelium only (panels e and f). B: Quantitative analyses of arginase I and eNOS fluorescence signals in vessels treated with vehicle or OxLDL. The arginase I signal intensity was significantly increased in both the endothelial and smooth muscle cells of the vessel treated with OxLDL. OxLDL did not alter eNOS signal intensity.
Discussion
In the present study, we have demonstrated that endothelium-dependent, NO-mediated vasodilation of coronary arterioles to serotonin is compromised by OxLDL. The reduction of serotonin-stimulated NO release from coronary arterioles is related to the increase of arginase activity in OxLDL-treated vessels. Inhibition of arginase activity restores NO production of coronary arterioles treated with Ox-LDL. Administration of NO precursor L-arginine or inhibition of arginase activity restores vasodilation to serotonin. Moreover, OxLDL up-regulates arginase I expression without altering eNOS expression. These results suggest that OxLDL impairs endothelium-dependent, NO-mediated dilation by reducing L-arginine availability, and thus NO production, through upregulated arginase I.
Although an altered vasomotor reactivity is generally reported in the clinical and experimental models of atherosclerosis with enhanced vasoconstrictor action and reduced endothelium-dependent vasodilation [5,19,30,37,52], a number of investigators have demonstrated that atherosclerosis is not necessary to impair endothelium-dependent vasodilations; rather, hypercholesterolemia is sufficient to exert the adverse effect [15,41]. In fact, endothelial dysfunction, especially the reduced NO bioavailability, has been regarded as a risk factor for atherogenesis [21,53]. In line with the present finding that the atherogenic molecule OxLDL exerts an adverse effect on endothelium-dependent NO-mediated vasodilation, our previous studies have shown the impairment of endothelial function in response to pharmacological stimuli or to an elevated shear stress in coronary arterioles from atherosclerotic animals [35] or by exposing the vessels to a high level of OxLDL [25,27]. It is worth noting that OxLDL selectively impairs NOS-related endothelial function without altering vasodilations mediated by cyclooxygenase or cytochrome P-450 monooxygenase [27]. Interestingly, administering a high level of NO precursor, L-arginine, improves endothelium-dependent vasomotor function [25,27,35] as shown in the present in-vitro study (Figure 2) and in a number of other studies using in-vivo hypercholesterolemia models [14,16,22]. However, the underlying mechanism for vascular endothelial dysfunction in relation to the beneficial effect of L-arginine on hypercholesterolemia and OxLDL insults remains incompletely understood.
Since L-arginine is an important limiting factor for NO production in addition to the regulation of cofactors, gene expression and enzymatic activity for eNOS [23], the reduction of L-arginine availability during OxLDL insult might affect endothelial production of NO. Interestingly, it was recently reported that the activity of the L-arginine consuming enzyme arginase is up-regulated in the atherosclerotic aorta of apoE knockout mice [39]. We speculated that OxLDL might exert its adverse effect on vasodilation to serotonin by up-regulating endothelial arginase. Indeed, our study showed that exposure of coronary arterioles to OxLDL increased arginase activity and also reduced serotonin-stimulated NO production (Figures 4 and 5). This is consistent with the idea that arginase in endothelial cells reciprocally regulates NO-mediated vasomotor function by competing with eNOS for their common substrate L-arginine [58]. In the present study, OxLDL reduced NO production (Figure 4) without affecting vasodilatory function in response to NO donor sodium nitroprusside (Figure 3B), suggesting that OxLDL exerts its adverse effect at the endothelial level. The effect of OxLDL appears to be specifically related to NO deficiency because inhibition of NOS activity by L-NAME did not further aggravate the impact of OxLDL (Figure 1). These results indicate that OxLDL affects the same pathway as L-NAME does in the present preparation. Administering a high level of L-arginine (but not D-arginine) or treating the arterioles with arginase inhibitors (DFMO or nor-NOHA) effectively improved vasodilation (Figure 2) and NO release (Figure 4) in response to serotonin in OxLDL-treated vessels. The little effect of extraluminal L-arginine on serotonin-induced vasodilation in untreated control vessels is consistent with our previous reports that the endothelium-dependent vasodilations to increased flow [27,35] and to serotonin and ATP [25] are not affected by L-arginine. In the presence of arginase inhibitor DFMO, there is a tendency, but not statistical significance, to increase serotonin-induced dilation of control vessels (Figure 3A). This result is different from our pervious findings that DFMO significantly enhances serotonin-induced dilation in normal coronary arterioles [29,59]. This discrepancy may be explained by 2- to 3-fold higher vascular arginase activity in previous studies. Therefore, inhibition of arginase in the present study has little effect on vasodilation to serotonin. The reason for the observed higher arginase activity in the previously studies is unclear. The younger age of animal and smaller size of vessel used in the present study may contribute to the inconsistent results. Nevertheless, the absence of vascular effect of L-arginine and DFMO in control vessels reflects the specific action of these two molecules in improving endothelium-dependent vasodilatory function in vessels challenged with OxLDL. In addition, L-arginine, DFMO and OxLDL did not alter vasodilatory function in response to smooth muscle vasodilator sodium nitroprusside (Figure 3B). These corroborative results suggest the contribution of L-arginine deficiency to the reduced NO bioavailability and endothelial dysfunction and the improvement of vasomotor function via L-arginine/NOS pathway as a result of arginase inhibition.
Up-regulation of arginase has been shown to be involved in various cardiovascular diseases, including atherosclerosis [56]. Immunohistochemical studies have shown that vascular endothelial and smooth muscle cells express both arginase I and II isoforms, but the relative level of expression appears to be species dependent [6,24,39,55]. We have previously demonstrated that porcine coronary arterioles express mainly arginase I in both endothelial and smooth muscle cells [58] and its expression level is significantly elevated by ischemia-reperfusion injury [29], oxidative stress [50] and pressure overload [59]. In the present study, arginase I expression in both endothelial and smooth muscle cells was up-regulated in coronary arterioles challenged with OxLDL (Figure 6). This may explain the observed increase in arginase activity in these vessels (Figure 5). Interestingly, recent studies found that vascular arginase expression is increased in animals with hypercholesterolemia and atherosclerosis in association with impaired NO-mediated vascular function [24,39]. Since OxLDL is an active atherogenic factor and contributes significantly to the development of endothelial dysfunction, it is speculated that OxLDL is responsible for the vascular arginase up-regulation in vivo leading to vasomotor dysfunction as seen in the present study.
Both animal and clinical studies have shown a strong correlation between extent of atherosclerosis and titers of autoantibodies to epitopes of OxLDL [42,45]. The eNOS expression was found to be reduced in the atheroma of human [40] and animal [1] vessels correlated with OxLDL level. However, the direct effect of OxLDL on eNOS expression is controversial. Exposure of cultured endothelial cells to a low concentration of OxLDL (10 μg protein/ml) for 24 hours appears to up-regulate eNOS protein [31], but higher concentrations of OxLDL (40 to 100 μg/ml, 24 hours) down-regulate eNOS expression, depending upon the incubation time and extent of LDL oxidation [4,57]. Therefore, the observed reduction of NO production by OxLDL can be attributable to the down-regulation of eNOS protein. Although the concentration of OxLDL contributing to pathogenesis of vascular disease in vivo is not known, it has been predicted to be 0.5 to 2 mg protein/ml in human atherosclerotic lesions [32]. In the present study, OxLDL (0.5 mg protein/ml, 90 minutes), at the level within the pathophysiological range as reported previously in both in-vitro [25,27,32,43] and in-vivo [44] models, did not alter eNOS expression in coronary arterioles (Figure 6B). Although it is unclear whether a prolonged exposure (>90 minutes) of OxLDL will down-regulate eNOS, the endothelial dysfunction observed in the present study appears to be independent of eNOS expression. Interestingly, a biochemical study of eNOS activity indicates that OxLDL can serve as a cholesterol acceptor to displace eNOS and caveolin from plasmalemmal caveolae to reduce eNOS activity [7]. This inhibitory effect is reversible if OxLDL is removed from the system [7]. However, our results seem to exclude the involvement of this inhibitory pathway in the intact vessel settings since L-arginine supplementation and arginase inhibition effectively restored NO production and NO-mediated dilation in the presence of OxLDL. It appears that the eNOS remains intact in the presence of OxLDL and is readily available to function if arginase activity is suppressed or a sufficient amount of L-arginine is supplied.
There are several limitations in the present study. First, the contribution of other NOS isoforms, other than eNOS, to serotonin-induced dilation of coronary arterioles were not studied in view that L-NAME is a non-specific inhibitor of NOS, thus, the inhibition by L-NAME is not specific to eNOS and the endothelium. Therefore, the role of arginase in counteracting eNOS may be overestimated with the consideration that serotonin-induced dilation may occur through stimulation of nNOS in the vascular smooth muscle. Although nNOS expression in porcine coronary arterioles is not detectable (unpublished observations), the possible existence of nNOS in our vessels cannot be excluded. Nevertheless, we have previously reported that serotonin causes vasoconstriction in the denuded small coronary arterioles with the vessel size similar to that used for the present study [35]. This finding suggests that activation of serotonin receptors in the smooth muscle of small porcine coronary arterioles elicits vasoconstriction rather then vasodilation. It appears that activation of nNOS by serotonin might not be significant, if this isoform exists in the coronary arterioles. Another limitation of the present study is the inability to determine intracellular level of L-arginine due to technical difficulty in collecting sufficient amount of viable microvessels for amino acid analysis. Nevertheless, the utilization of pharmacological tools and with L-arginine vs. D-arginine administration along with immunohistochemical results support the conclusions summarized below.
Collectively, in the present study we have demonstrated for the first time that OxLDL inhibits endothelium-dependent, NO-mediated vasodilatory function in small coronary arterioles without altering eNOS expression. The up-regulated arginase appears to reduce L-arginine availability for eNOS and thus compromises NO synthesis for vasodilation. Since platelet aggregation and the release of serotonin and other endothelium-dependent vasoactive substances from activated platelets are enhanced by atherogenic lipoproteins [2], it is speculated that the up-regulation of arginase by OxLDL linking to NO deficiency and vasomotor dysfunction may contribute to the development of coronary ischemic events in association with hyperlipidemia/atherosclerosis and oxidative stress.
Acknowledgments
This study was supported by National Heart, Lung and Blood Institute grant HL-491351 to JCL and LK.
References
- 1.Aikawa M, Sugiyama S, Hill CC, Voglic SJ, Rabkin E, Fukumoto Y, Schoen FJ, Witztum JL, Libby P. Lipid lowering reduces oxidative stress and endothelial cell activation in rabbit atheroma. Circulation. 2002;106:1390–1396. doi: 10.1161/01.cir.0000028465.52694.9b. [DOI] [PubMed] [Google Scholar]
- 2.Aviram M, Brook JG. Platelet activation by plasma lipoproteins. Prog Cardiovasc Dis. 1987;30:61–72. doi: 10.1016/0033-0620(87)90011-9. [DOI] [PubMed] [Google Scholar]
- 3.Bachetti T, Comini L, Francolini G, Bastianon D, Valetti B, Cadei M, Grigolato P, Suzuki H, Finazzi D, Albertini A, Curello S, Ferrari R. Arginase pathway in human endothelial cells in pathophysiological conditions. J Mol Cell Cardiol. 2004;37:515–523. doi: 10.1016/j.yjmcc.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Bao M, Lou Y. Isorhamnetin prevent endothelial cell injuries from oxidized LDL via activation of p38MAPK. Eur J Pharmacol. 2006;547:22–30. doi: 10.1016/j.ejphar.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Barbato E, Piscione F, Bartunek J, Galasso G, Cirillo P, De Luca G, Iaccarino G, De Bruyne B, Chiariello M, Wijns W. Role of β2 adrenergic receptors in human atherosclerotic coronary arteries. Circulation. 2005;111:288–294. doi: 10.1161/01.CIR.0000153270.25541.72. [DOI] [PubMed] [Google Scholar]
- 6.Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- 7.Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem. 1999;274:32512–32519. doi: 10.1074/jbc.274.45.32512. [DOI] [PubMed] [Google Scholar]
- 8.Buga GM, Singh R, Pervin S, Rogers NE, Schmitz DA, Jenkinson CP, Cederbaum SD, Ignarro LJ. Arginase activity in endothelial cells: inhibition by NG-hydroxy-L-arginine during high-output NO production. Am J Physiol. 1996;271:H1988–H1998. doi: 10.1152/ajpheart.1996.271.5.H1988. [DOI] [PubMed] [Google Scholar]
- 9.Chait Methods for assessing lipid and lipoprotein oxidation. Curr Opin Lipidol. 1992;3:389–394. [Google Scholar]
- 10.Chang CI, Liao JC, Kuo L. Arginase modulates nitric oxide production in activated macrophages. Am J Physiol. 1998;274:H342–H348. doi: 10.1152/ajpheart.1998.274.1.H342. [DOI] [PubMed] [Google Scholar]
- 11.Chicoine LG, Paffett ML, Young TL, Nelin LD. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L60–L68. doi: 10.1152/ajplung.00194.2003. [DOI] [PubMed] [Google Scholar]
- 12.Chilian WM, Dellsperger KC, Layne SM, Eastham CL, Armstrong MA, Marcus ML, Heistad DD. Effects of atherosclerosis on the coronary microcirculation. Am J Physiol. 1990;258:H529–H539. doi: 10.1152/ajpheart.1990.258.2.H529. [DOI] [PubMed] [Google Scholar]
- 13.Chilian WM, Eastham CL, Marcus ML. Microvascular distribution of coronary vascular resistance in beating left ventricle. Am J Physiol. 1986;251:H779–H788. doi: 10.1152/ajpheart.1986.251.4.H779. [DOI] [PubMed] [Google Scholar]
- 14.Clarkson P, Adams MR, Powe AJ, Donald AE, McCredie R, Robinson J, McCarthy SN, Keech A, Celermajer DS, Deanfield JE. Oral L-arginine improves endothelium-dependent dilation in hypercholesterolemic young adults. J Clin Invest. 1996;97:1989–1994. doi: 10.1172/JCI118632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen RA, Zitnay KM, Haudenschild CC, Cunningham LD. Loss of selective endothelial cell vasoactive functions caused by hypercholesterolemia in pig coronary arteries. Circ Res. 1988;63:903–910. doi: 10.1161/01.res.63.5.903. [DOI] [PubMed] [Google Scholar]
- 16.Cooke JP, Andon NA, Girerd XJ, Hirsch AT, Creager MA. Arginine restores cholinergic relaxation of hypercholesterolemic rabbit thoracic aorta. Circulation. 1991;83:1057–1062. doi: 10.1161/01.cir.83.3.1057. [DOI] [PubMed] [Google Scholar]
- 17.Ensunsa JL, Symons JD, Lanoue L, Schrader HR, Keen CL. Reducing arginase activity via dietary manganese deficiency enhances endothelium-dependent vasorelaxation of rat aorta. Exp Biol Med. 2004;229:1143–1153. doi: 10.1177/153537020422901109. [DOI] [PubMed] [Google Scholar]
- 18.Esterbauer H, Striegl G, Puhl H, Rotheneder M. Continuous monitoring of in vitro oxidation of human low density lipoprotein. Free Radic Res Commun. 1989;6:67–75. doi: 10.3109/10715768909073429. [DOI] [PubMed] [Google Scholar]
- 19.Freiman PC, Mitchell GG, Heistad DD, Armstrong ML, Harrison DG. Atherosclerosis impairs endothelium-dependent vascular relaxation to acetylcholine and thrombin in primates. Circ Res. 1986;58:783–789. doi: 10.1161/01.res.58.6.783. [DOI] [PubMed] [Google Scholar]
- 20.Galle J, Bassenge E. Effects of native and oxidized low-density lipoproteins on endothelium-dependent and endothelium-independent vasomotion. Basic Res Cardiol. 1991;86 2:127–142. doi: 10.1007/978-3-642-72461-9_14. [DOI] [PubMed] [Google Scholar]
- 21.Giannotti G, Landmesser U. Endothelial dysfunction as an early sign of atherosclerosis. Herz. 2007;32:568–572. doi: 10.1007/s00059-007-3073-1. [DOI] [PubMed] [Google Scholar]
- 22.Girerd XJ, Hirsch AT, Cooke JP, Dzau VJ, Creager MA. L-arginine augments endothelium-dependent vasodilation in cholesterol-fed rabbits. Circ Res. 1990;67:1301–1308. doi: 10.1161/01.res.67.6.1301. [DOI] [PubMed] [Google Scholar]
- 23.Hallemeesch MM, Lamers WH, Deutz NE. Reduced arginine availability and nitric oxide production. Clin Nutr. 2002;21:273–279. doi: 10.1054/clnu.2002.0571. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi T, Esaki T, Sumi D, Mukherjee T, Iguchi A, Chaudhuri G. Modulating role of estradiol on arginase II expression in hyperlipidemic rabbits as an atheroprotective mechanism. Proc Natl Acad Sci U S A. 2006;103:10485–10490. doi: 10.1073/pnas.0603918103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hein TW, Kuo L. LDLs impair vasomotor function of the coronary microcirculation: role of superoxide anions. Circ Res. 1998;83:404–414. doi: 10.1161/01.res.83.4.404. [DOI] [PubMed] [Google Scholar]
- 26.Hein TW, Kuo L. cAMP-independent dilation of coronary arterioles to adenosine: Role of nitric oxide, G proteins, and KATP channels. Circ Res. 1999;85:634–642. doi: 10.1161/01.res.85.7.634. [DOI] [PubMed] [Google Scholar]
- 27.Hein TW, Liao JC, Kuo L. oxLDL specifically impairs endothelium-dependent, NO-mediated dilation of coronary arterioles. Am J Physiol Heart Circ Physiol. 2000;278:H175–H183. doi: 10.1152/ajpheart.2000.278.1.H175. [DOI] [PubMed] [Google Scholar]
- 28.Hein TW, Qamirani E, Ren Y, Kuo L. C-reactive protein impairs coronary arteriolar dilation to prostacyclin synthase activation: role of peroxynitrite. J Mol Cell Cardiol. 2009;47:196–202. doi: 10.1016/j.yjmcc.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hein TW, Zhang C, Wang W, Chang CI, Thengchaisri N, Kuo L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- 30.Heistad DD, Armstrong ML, Marcus ML, Piegors DJ, Mark AL. Augmented responses to vasoconstrictor stimuli in hypercholesterolemic and atherosclerotic monkeys. Circ Res. 1984;54:711–718. doi: 10.1161/01.res.54.6.711. [DOI] [PubMed] [Google Scholar]
- 31.Hirata K, Miki N, Kuroda Y, Sakoda T, Kawashima S, Yokoyama M. Low concentration of oxidized low-density lipoprotein and lysophosphatidylcholine upregulate constitutive nitric oxide synthase mRNA expression in bovine aortic endothelial cells. Circ Res. 1995;76:958–962. doi: 10.1161/01.res.76.6.958. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs M, Plane F, Bruckdorfer KR. Native and oxidized low-density lipoproteins have different inhibitory effects on endothelium-derived relaxing factor in the rabbit aorta. Br J Pharmacol. 1990;100:21–26. doi: 10.1111/j.1476-5381.1990.tb12045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol. 1991;261:H1706–H1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- 34.Kuo L, Chilian WM, Davis MJ, Laughlin MH. Endotoxin impairs flow-induced vasodilation of porcine coronary arterioles. Am J Physiol. 1992;262:H1838–H1845. doi: 10.1152/ajpheart.1992.262.6.H1838. [DOI] [PubMed] [Google Scholar]
- 35.Kuo L, Davis MJ, Cannon MS, Chilian WM. Pathophysiological consequences of atherosclerosis extend into the coronary microcirculation: Restoration of endothelium-dependent responses by L-arginine. Circ Res. 1992;70:465–476. doi: 10.1161/01.res.70.3.465. [DOI] [PubMed] [Google Scholar]
- 36.Kuo L, Davis MJ, Chilian WM. Myogenic activity in isolated subepicardial and subendocardial coronary arterioles. Am J Physiol. 1988;255:H1558–H1562. doi: 10.1152/ajpheart.1988.255.6.H1558. [DOI] [PubMed] [Google Scholar]
- 37.Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, Ganz P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–1051. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- 38.Markwell MA, Haas SM, Tolbert NE, Bieber LL. Protein determination in membrane and lipoprotein samples: manual and automated procedures. Methods Enzymol. 1981;72:296–303. doi: 10.1016/s0076-6879(81)72018-4. [DOI] [PubMed] [Google Scholar]
- 39.Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, Hayoz D, Ruffieux J, Rusconi S, Montani JP, Yang Z. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- 40.Oemar BS, Tschudi MR, Godoy N, Brovkovich V, Malinski T, Luscher TF. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation. 1998;97:2494–2498. doi: 10.1161/01.cir.97.25.2494. [DOI] [PubMed] [Google Scholar]
- 41.Osborne JA, Siegman MJ, Sedar AW, Mooers SU, Lefer AM. Lack of endothelium-dependent relaxation in coronary resistance arteries of cholesterol-fed rabbits. Am J Physiol. 1989;256:C591–C597. doi: 10.1152/ajpcell.1989.256.3.C591. [DOI] [PubMed] [Google Scholar]
- 42.Palinski W, Tangirala RK, Miller E, Young SG, Witztum JL. Increased autoantibody titers against epitopes of oxidized LDL in LDL receptor-deficient mice with increased atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:1569–1576. doi: 10.1161/01.atv.15.10.1569. [DOI] [PubMed] [Google Scholar]
- 43.Plane F, Bruckdorfer KR, Kerr P, Steuer A, Jacobs M. Oxidative modification of low-density lipoproteins and the inhibition of relaxations mediated by endothelium-derived nitric oxide in rabbit aorta. Br J Pharmacol. 1992;105:216–222. doi: 10.1111/j.1476-5381.1992.tb14237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rangaswamy S, Penn MS, Saidel GM, Chisolm GM. Exogenous oxidized low-density lipoprotein injures and alters the barrier function of endothelium in rats in vivo. Circ Res. 1997;80:37–44. doi: 10.1161/01.res.80.1.37. [DOI] [PubMed] [Google Scholar]
- 45.Salonen JT, Yla-Herttuala S, Yamamoto R, Butler S, Korpela H, Salonen R, Nyyssonen K, Palinski W, Witztum JL. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet. 1992;339:883–887. doi: 10.1016/0140-6736(92)90926-t. [DOI] [PubMed] [Google Scholar]
- 46.Sankaralingam S, Xu H, Davidge ST. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc Res. 2010;85:194–203. doi: 10.1093/cvr/cvp277. [DOI] [PubMed] [Google Scholar]
- 47.Selamnia M, Mayeur C, Robert V, Blachier F. Alpha-difluoromethylornithine (DFMO) as a potent arginase activity inhibitor in human colon carcinoma cells. Biochem Pharmacol. 1998;55:1241–1245. doi: 10.1016/s0006-2952(97)00572-8. [DOI] [PubMed] [Google Scholar]
- 48.Tanner FC, Noll G, Boulanger CM, Luscher TF. Oxidized low density lipoproteins inhibit relaxations of porcine coronary arteries: Role of scavenger receptor and endothelium-derived nitric oxide. Circulation. 1991;83:2012–2020. doi: 10.1161/01.cir.83.6.2012. [DOI] [PubMed] [Google Scholar]
- 49.Tenu JP, Lepoivre M, Moali C, Brollo M, Mansuy D, Boucher JL. Effects of the new arginase inhibitor Nω-hydroxy-nor-L-arginine on NO synthase activity in murine macrophages. Nitric Oxide. 1999;3:427–438. doi: 10.1006/niox.1999.0255. [DOI] [PubMed] [Google Scholar]
- 50.Thengchaisri N, Hein TW, Wang W, Xu X, Li Z, Fossum TW, Kuo L. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2006;26:2035–2042. doi: 10.1161/01.ATV.0000233334.24805.62. [DOI] [PubMed] [Google Scholar]
- 51.Tousoulis D, Boger RH, Antoniades C, Siasos G, Stefanadi E, Stefanadis C. Mechanisms of disease: L-arginine in coronary atherosclerosis - a clinical perspective. Nat Clin Pract Cardiovasc Med. 2007;4:274–283. doi: 10.1038/ncpcardio0878. [DOI] [PubMed] [Google Scholar]
- 52.Turk JR, Henderson KK, Vanvickle GD, Watkins J, Laughlin MH. Arterial endothelial function in a porcine model of early stage atherosclerotic vascular disease. Int J Exp Pathol. 2005;86:335–345. doi: 10.1111/j.0959-9673.2005.00446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73:595–601. doi: 10.1253/circj.cj-08-1169. [DOI] [PubMed] [Google Scholar]
- 54.Waddington SN, Mosley K, Cook HT, Tam FW, Cattell V. Arginase AI is upregulated in acute immune complex-induced inflammation. Biochem Biophys Res Commun. 1998;247:84–87. doi: 10.1006/bbrc.1998.8755. [DOI] [PubMed] [Google Scholar]
- 55.Wei LH, Jacobs AT, Morris SM, Jr, Ignarro LJ. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2000;279:C248–C256. doi: 10.1152/ajpcell.2000.279.1.C248. [DOI] [PubMed] [Google Scholar]
- 56.Yang Z, Ming XF. Endothelial arginase: a new target in atherosclerosis. Curr Hypertens Rep. 2006;8:54–59. doi: 10.1007/s11906-006-0041-8. [DOI] [PubMed] [Google Scholar]
- 57.Yoshida H, Sasaki K, Namiki Y, Sato N, Tada N. Edaravone, a novel radical scavenger, inhibits oxidative modification of low-density lipoprotein (LDL) and reverses oxidized LDL-mediated reduction in the expression of endothelial nitric oxide synthase. Atherosclerosis. 2005;179:97–102. doi: 10.1016/j.atherosclerosis.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 58.Zhang C, Hein TW, Wang W, Chang CI, Kuo L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J. 2001;15:1264–1266. doi: 10.1096/fj.00-0681fje. [DOI] [PubMed] [Google Scholar]
- 59.Zhang C, Hein TW, Wang W, Miller MW, Fossum TW, McDonald MM, Humphrey JD, Kuo L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension. 2004;44:935–943. doi: 10.1161/01.HYP.0000146907.82869.f2. [DOI] [PubMed] [Google Scholar]