

Abstract
Covalent modification of the promyelocytic leukaemia protein (PML) by SUMO-1 is a prerequisite for the assembly of nuclear bodies (NBs), subnuclear structures disrupted in various human diseases and linked to transcriptional and growth control. Here we demonstrate that p53 is recruited into NBs by a specific PML isoform (PML3) or by coexpression of SUMO-1 and hUbc9. NB targeting depends on the direct association of p53, through its core domain, with a C-terminal region of PML3. The relocalization of p53 into NBs enhances p53 transactivation in a promoter-specific manner and affects cell survival. Our results indicate the existence of a cross-talk between PML- and p53-dependent growth suppression pathways, implying an important role for NBs and their resident proteins as modulators of p53 functions.
Keywords: nuclear bodies/p53/PML3/sumolation/transcription
Introduction
The tumour suppressor protein p53 is a key element in the control of human cell growth and differentiation and plays an important role in the maintenance of genome integrity (Ko and Prives, 1996; Levine, 1997). Most of its functions are exerted by transcriptional activation of genes involved in cell cycle, apoptosis and DNA repair (Ko and Prives, 1996). Under various stress conditions p53 becomes activated by post-translational modifications that affect its conformation and binding to several proteins, resulting in its stabilization and increased DNA-binding potential (Giaccia and Kastan, 1998).
Another way to modulate p53 activity involves changes in its subcellular distribution. Certain tumours constitutively accumulate wild-type p53, which is functionally inactive because it is sequestered in the cytoplasm (Moll et al., 1996; Ostermayer et al., 1996). In addition, treatment of human primary cells with Leptomycin B, a drug that specifically blocks nuclear export, induces the relocalization of p53 into punctate subnuclear structures, reminiscent of the so-called nuclear bodies (NBs) (Lain et al., 1999). A similar p53 distribution has also been observed upon coexpression with Mdm2 and ARF and this relocalization has been correlated with ARF-mediated inhibition of Mdm2–p53 nuclear export (Zhang and Xiong, 1999).
NBs are cell cycle-regulated, matrix-associated subnuclear structures that appear as punctate foci in the interphase nuclei (Seeler and Dejean, 1999). The structural integrity of these large multiprotein complexes appears to be important for normal cell growth and development, since in some human diseases, like acute promyelocytic leukaemia (APL) and spinocerebellar ataxia type I (SCA1), disruption of NBs leads to malignancy or neurodegenerative disorder, respectively (Hodges et al., 1998). Moreover, these structures are targeted and subsequently destroyed by numerous immediate early viral proteins (Maul, 1998).
Promyelocytic leukaemia protein (PML), the most prominent component of the NBs (also referred to as PML oncogenic domains, PODs), was first identified in APL patients, where, as a result of a reciprocal translocation event, it is fused to the retinoic acid receptor α (RARα) (de The et al., 1991; Kakizuka et al., 1991). The fundamental role of PML in directing the complex protein–protein interactions that mediate PODs formation is underlined by recent findings that the organization of several NB-associated components is impaired in PML–/– cells (Zhong et al., 2000a). The possible role of PML and NBs in control of cell growth is suggested by studies on PML knockout mice, revealing tumour suppressor and pro-apoptotic functions for PML (Quignon et al., 1998; Wang et al., 1998b). In APL cells, the expression of the PML–RARα fusion compromises the integrity of PODs, while treatment with therapeutic agents, such as arsenic trioxide (As2O3) or interferons, leads to the normalization of NB pattern and simultaneously induces differentiation or apoptosis of the malignant cells (Lavau et al., 1995; Muller et al., 1998b).
Other evidence implies NBs in the control of gene expression (Zhong et al., 2000b). The reported interaction with pRb (Alcalay et al., 1998) and the direct binding to the histone acetyltransferase CBP (LaMorte et al., 1998) suggest a relevant role for PML in the regulation of transcription.
PML, Sp100 and probably other NB-resident proteins are post-translationally modified by SUMO-1, a small ubiquitin-related modifier (Sternsdorf et al., 1997), and recently we and others reported that p53 is also conjugated to SUMO-1 (Gostissa et al., 1999; Rodriguez et al., 1999; Muller et al., 2000). It has been demonstrated that sumolation of PML is absolutely required for NB formation and for recruitment of other factors to these structures (Ishov et al., 1999; Zhong et al., 2000a).
Here we provide evidence that PML recruits p53 into NBs. The relocalization of p53 depends on its direct association with a specific PML splice variant, PML3. Moreover, we demonstrate that binding to PML3 and NB targeting of p53 result in increased transcriptional activation of a p53-regulated pro-apoptotic gene and affect cell survival.
Results
PML mediates the relocalization of p53 into NBs
Recently we and others reported that p53 can be covalently modified by conjugation to the small ubiquitin-like protein SUMO-1 (Gostissa et al., 1999; Rodriguez et al., 1999; Muller et al., 2000) and we noticed that upon coexpression of SUMO-1 and hUbc9, p53 was relocalized to subnuclear structures reminiscent of NBs. Since SUMO-1 conjugation has been proposed to modulate the subcellular localization of several proteins (Mahajan et al., 1997), we wanted to investigate whether it may assist the relocalization of p53 into NBs. Human p53-null SaOS-2 cells were microinjected with plasmids encoding wild-type p53 (p53 wt), green fluorescent protein (GFP)–SUMO-1 and haemagglutinin (HA)-hUbc9, and analysed by immunofluorescence and confocal laser microscopy. In a fraction of the injected cells (40%) the typical nucleoplasmic staining of p53 (Figure 1A, a) became organized in distinct GFP–SUMO-1-positive NBs (Figure 1A, b–d). To our surprise, the conjugation-deficient mutant p53 K386R, when microinjected in the same conditions, was relocalized to NBs to a similar extent to the wt protein (Figure 1A, f–h), thus demonstrating that sumolation of p53 is dispensable for its delivery to these structures. Notably, a similar independence of NB localization from SUMO-1 conjugation has also been described for Sp100 (Sternsdorf et al., 1999) and the transactivator protein IE2-p86 of human cytomegalovirus (Hofmann et al., 2000). NB targeting was p53 specific, since under the same conditions a construct encoding β-galactosidase fused to a nuclear localization signal (βgal-NLS) was excluded from these structures (Figure 1A, i–k). Moreover, the observed p53 relocalization was dependent on the simultaneous expression of both GFP–SUMO-1 and HA-hUbc9 (not shown). Of note, when expressed individually, HA-hUbc9 revealed both nuclear and cytoplasmic distribution, while GFP–SUMO-1 showed a nuclear diffuse staining with the protein concentrating in small dots (not shown). Coexpression of the two proteins, instead, resulted in a dramatic change of subcellular distribution and in the formation of large NBs (Figure 1A, c, g and j) where the two proteins colocalized (not shown), suggesting that both factors are rate-limiting in a process that led to the formation of these structures.


Fig. 1. p53 relocalizes into NBs. (A) SaOS-2 cells were microinjected with either 10 ng/µl pcDNA3p53wt (b–d), pRcCMVp53K386R (f–h) or pNLS-βgal (i–k) together with 30 ng/µl pGFPSUMO-1 and 50 ng/µl pcDNA3HAhUbc9. p53 staining was analysed using a rabbit antiserum, while the localization of βgal-NLS was detected by a monoclonal anti-β-galactosidase antibody. Primary antibodies and GFP–SUMO-1 staining were revealed by incubation with TRITC-conjugated secondary antibodies or by the intrinsic green fluorescence of GFP, respectively. Merging of the two colours results in a yellow signal, corresponding to colocalized proteins. (a and e) Staining of p53 wt and K386R, respectively, in the absence of coexpressed GFP–SUMO-1 and HA-hUbc9. (B) SaOS-2 cells (a–c) microinjected with 10 ng/µl pcDNA3p53wt and 30 ng/µl pcDNA3PML3 were analysed for p53 expression as above and for PML staining using the anti-PML monoclonal antibody PG-M3 followed by incubation with an FITC-conjugated secondary antibody. U2OS cells (d–f) were microinjected with pcDNA3PML3, and localization of endogenous p53 and overexpressed PML3 was examined as above. (C) LOVO cells were treated with UV light and As2O3 and expression of endogenous p53 was detected by a mixture of DO-1 and 1801 monoclonal antibodies followed by incubation with a TRITC-conjugated secondary antibody (a and c). Endogenous PML3 staining was revealed with a rabbit polyclonal serum specific for PML3 and FITC-conjugated secondary antibody (b and c).
Since it has been shown that PML plays a crucial role in the assembly of NBs by recruiting other components (Ishov et al., 1999) and that SUMO-1 conjugation of PML is necessary for this process (Zhong et al., 2000a), we next tested whether PML could be involved in the observed relocalization of p53. Immunofluorescence analysis of SaOS-2 cells microinjected with p53 wt together with PML3 (Fagioli et al., 1992) revealed that a fraction of p53 was segregated into PML3-positive NBs in almost all microinjected cells (Figure 1B, a–c). This PML3-dependent recruitment of ectopically expressed p53 into NBs was also observed in MG63, another p53-null cell line, demonstrating that this effect was not cell line specific (not shown). A similar relocalization was obtained for endogenous wt p53 as well, when PML3 expression vector was microinjected in U2OS cells (Figure 1B, d–f).
From these observations we can conclude that at least one isoform of PML is required for targeting p53 into NBs. Since overexpression of SUMO-1 and hUbc9 has the same final outcome, it is likely that this enhances the sumolation of endogenous PML and thus augments assembly of NBs and recruitment of p53 into these structures. The presence of PML3 mRNA, as detected by RT–PCR analysis in the employed cell lines (not shown), and the previous evidence that NB recruitment of PML depends on its sumolation (Zhong et al., 2000a), corroborate this interpretation.
As2O3 has been shown to increase specifically the modification of PML by SUMO-1 (Muller et al., 1998a), therefore we wanted to test whether, upon treatment with this drug, endogenous p53 could be recruited into PML3-containing NBs. For this aim, LOVO human colon carcinoma cells were treated with As2O3 and prior to fixation were subjected to a hypotonic pre-extraction to remove some of the diffuse nucleoplasmic p53. Although As2O3 treatment increased the number and size of PML3-containing NBs as expected when detected by using a polyclonal serum specific for PML3, endogenous wt p53 was not recruited into these structures (not shown). When cells were irradiated with UV light and treated with As2O3, we clearly detected colocalization between endogenous PML3 and p53 in NBs (Figure 1C). UV treatment alone was not sufficient to change the diffuse nucleoplasmic staining of p53 (not shown). These results therefore demonstrate that colocalization of endogenous p53 and PML3 can be induced under conditions that simultaneously enhance the sumolation of PML and trigger p53 activation.
Dissection of the p53 region required for NB localization
To identify the region of p53 required for NB targeting, several p53 deletion constructs were generated (Figure 2A) and microinjected into SaOS-2 cells together with PML3. The different p53 proteins were expressed at comparable levels, as judged by western blot analysis after transient transfection (not shown), and showed the typical homogeneous nuclear staining (Figure 2B, a, e, i and m). We found that N-terminal deletions missing the transactivation domain (amino acids 12–69) or the Pro-rich region (amino acids 63–91) of p53 displayed both a nuclear diffuse staining and accumulation in PML3-containing NBs (not shown), as observed for the full-length protein (see Figure 1B). In contrast, p53 294–393, which lacks the N-terminal and core domains did not change its homogeneous nucleoplasmic localization upon coexpression of PML3 (Figure 2B, n–p). These findings suggested that the core domain is required for the relocalization of p53 into NBs. Accordingly, p53 1–298, a protein that contains this domain as well as upstream N-terminal regions, efficiently relocalized into NBs (Figure 2B, j–l). Interestingly, unlike p53 wt and the deletions tested so far, this protein exclusively accumulated in NBs, indicating that sequences in the C-terminus of p53 may possess a negative regulatory role on NB targeting. Deletion of a C-terminal segment until amino acid 363 (Figure 2B, b–d) resulted in a protein that showed a staining pattern similar to the wt protein. On the contrary, p53 1–355, a deletion lacking the last 38 residues of p53, totally relocalized into NBs upon PML3 coexpression (Figure 2B, f–h). Similar results were obtained in MG63 and also when the forementioned p53 deletions were coexpressed with SUMO-1 and hUbc9 (not shown).

Fig. 2. Direct interaction between PML3 and the core domain of p53 mediates the relocalization of p53 into NBs. (A) Schematic representation of the various p53 deletion mutants and summary of their ability to bind PML3 and to relocalize into NBs. Transactivation (Tr), polyproline (PP), DNA-binding (DBD) and tetramerization and non-specific DNA-binding (Tm/NSDB) domains are indicated. Numbers refer to amino acids. NA, not assessed. (B) SaOS-2 cells were microinjected with 10 ng/µl expression vectors encoding various p53 deletions alone (a, e, i and m) or together with pcDNA3PML3 (30 ng/µl) and analysed for p53 and PML3 staining as described in Figure 1B. (C) SaOS-2 cells were transfected as indicated and immunoprecipitated with an anti-PML antibody (PG-M3). Immunoblotting was performed with the anti-p53 antibody DO-1 (lanes 1–6) or with the anti-HA antibody to detect the HA-tagged p53 294–393 protein (lanes 7 and 8). Lower panels show expression levels of the various overexpressed proteins.
These results indicate that the core domain (amino acids 90–298) of p53 is required for NB targeting. However, the evidence that the p53 H175 mutant is also localized to NBs (not shown) demonstrates that the wt conformation of the p53 DNA-binding region is dispensable. Furthermore, the massive PML3-induced relocalization of p53 1–355 as compared with p53 1–363 strongly suggests that eight residues between 356 and 363 exert a regulatory function on this process.
p53 binds to PML3 with the domain required for NB targeting
Next we investigated whether the PML3-dependent change in p53 subcellular distribution was mediated by direct association between the two proteins. SaOS-2 cells were transfected with plasmids encoding p53 wt or various deletions together with PML3, and cell lysates were immunoprecipitated with the anti-PML antibody PG-M3. The bound protein complexes were analysed by western blotting using the anti-p53 antibody, DO-1, for p53 wt and C-terminal deletion mutants (p53 1–355 and p53 1–298), or anti-HA antibody for the HA-tagged p53 294–393 protein. As shown in Figure 2C, only p53 proteins able to relocalize to NBs were immunoprecipitated from cells expressing PML3. These results clearly indicate that p53 binds to PML3 via its core domain and that this binding mediates p53 targeting to NBs in cells overexpressing PML3.
NB targeting of p53 is mediated by a specific PML isoform
The integrity of NB structures is disrupted in APL cells, in which expression of the PML–RARα fusion protein leads to the disorganization of PODs into numerous and aberrant microstructures (Hodges et al., 1998). To assess whether p53 recruitment into NBs was specific for PML3 and not for the oncogenic PML–RARα product, SaOS-2 cells were microinjected with plasmids encoding PML–RARα and p53 wt and analysed by immunofluorescence and confocal microscopy. The injected cells showed the typical microspeckled pattern for PML–RARα (Figure 3A, b and c), but the homogeneous nuclear diffuse staining of p53 was not affected (Figure 3A, a and c).

Fig. 3. Recruitment of p53 to NBs depends on its interaction with a specific PML isoform. (A) SaOS-2 cells microinjected with p53 wt (10 ng/µl) and 30 ng/µl PML–RARα (a–c) or PML-L (d–f) were analysed by immunofluorescence as in Figure 1B. (B) Schematic representation of the various PML proteins showing their functional domains. Upper numbers refer to PML-specific amino acids, lower numbers correspond to residues in RARα. Sumolation sites (S) are indicated. (C) Lysates from SaOS-2 cells transfected with PML3, PML-L or PML–RARα and precipitated with GST–p53 or GST, as indicated, were analysed by western blotting with an anti-PML polyclonal antibody. (D) Pull down experiment performed with in vitro translated p53 wt or different deletions, as indicated, and GST–PML3Ct or GST. Complexes were resolved by SDS–PAGE and visualized by autoradiography.
Since the PML–RARα fusion protein is lacking the PML C-terminal region (Figure 3B), we hypothesized that this domain is required to target p53 into NBs. PML-L, another PML splice variant, which differs only in its short C-terminal tail from the PML3 protein employed so far (Figure 3B), was coinjected with p53 wt into SaOS-2 cells and the immunostaining pattern was analysed as above. Although PML-L, as expected, formed NBs where other resident proteins, like Sp100 and SUMO-1, were found to localize (not shown), the distribution of p53 remained diffuse in the injected cells (Figure 3A, d–f). Parallel experiments performed with p53 1–355 gave similar results (not shown).
In vitro binding experiments with glutathione S-transferase (GST)–p53 on lysates from cells expressing PML3, PML-L or PML–RARα demonstrated that p53 wt binds efficiently only to PML3, while the interaction with PML-L and with PML–RARα was severely impaired (Figure 3C). A GST fusion containing the 61-residue-long C-terminal tail specific for PML3 (Figure 3B), efficiently bound to in vitro translated p53 wt, p53 1–355 and p53 1–298 (Figure 3D). These results indicate that a region outside the central domain of PML, where the three known sumolation sites have been mapped (Kamitani et al., 1998) and that is present in the PML3 isoform, is necessary and sufficient to mediate p53 binding.
PML3 affects cell survival in a p53-dependent way
Since NBs and PML have been linked to regulation of cell growth and differentiation (Lin et al., 1999) and p53 is a well established tumour suppressor (Sionov and Haupt, 1999), we examined whether recruitment of p53 into NBs can modulate cell survival. U2OS and MG63 cells were microinjected with plasmids encoding PML3 or human placental alkaline phosphatase (PLAP) as a negative control together with a GFP expression vector as marker. Twenty-four hours later cell survival was scored as the number of recovered cells positive for the intrinsic green fluorescence of GFP. U2OS cells express wt p53 that, upon introduction of PML3, was efficiently recruited into NBs (Figure 1B, d–f), while MG63 cells lack endogenous p53. Upon overexpression of PML3, we consistently observed a significant reduction of survival in U2OS (Figure 4A, left panel) but not in MG63 cells (Figure 4A, right panel). To correlate the observed phenotype with PML3-mediated recruitment of p53 into NBs, we analysed whether overexpression of p53 1–355 (the C-terminal deletion that totally relocalized to NBs upon PML3 expression, see Figure 2B) could restore the PML3-dependent effect on MG63 cells. Cell survival was analysed by microinjecting MG63 cells either with PML3 and p53 1–355 alone or with a combination of the two plasmids, and was scored as above. As plotted in Figure 4B, recovery of GFP-positive cells was severely impaired when both PML3 and p53 1–355 were simultaneously expressed (bar 4), while no effect was observed when the two proteins were individually expressed (bars 2 and 3). Under the same conditions, PML-L, the isoform impaired in binding and relocalizing p53 into NBs, did not affect cell survival when coexpressed with p53 1–355 (Figure 4B, bar 6).

Fig. 4. PML3 affects cell survival in a p53-dependent way. (A) U2OS (left panel) and MG63 (right panel) cells were microinjected with the indicated plasmids (30 ng/µl) together with pGFP (15 ng/µl) as marker. Twenty-four hours later cell survival was scored as the percentage of recovered cells positive for GFP. (B) Cell survival assay was performed in MG63 cells after ectopic expression of p53 1–355 (20 ng/µl), PML3 (30 ng/µl) and PML-L (30 ng/µl), either alone (bars 2, 3 and 5) or in combinations (bars 4 and 6). PLAP was used as negative control and to adjust total DNA amount in all the samples. Graphs represent the mean of at least seven independent experiments.
These results therefore suggest that recruitment of p53 into NBs by a specific PML isoform modulates the survival functions linked to these structures.
Binding of p53 to a specific PML isoform, PML3, increases its transcriptional activity
The evidence that PML associates with several transcription factors and coactivators, like p300/CBP and recruits them into NBs, suggests a relevant role for PML and the whole NB structure in transcriptional control (Zhong et al., 2000b). We therefore analysed whether the observed effect on cell survival upon ectopic expression of PML3 was linked to changes in p53 transactivation ability. Transient transfection assays with constructs containing two well established p53 responsive promoters, PIG3 (Polyak et al., 1997) and p21 (El-Deiry et al., 1993), cloned upstream of the luciferase reporter were performed in MG63 cells with combinations of p53 wt, p53 1–355, PML3 and PML-L expression vectors. Coexpression of p53 wt with PML3 strongly increased the transcriptional activity of p53 toward the PIG3 promoter (Figure 5A, bars 6 and 8), while under the same conditions PML-L overexpression showed no significant effect (Figure 5A, bars 6 and 10). Consistent with previous reports (Tarunina and Jenkins, 1993), p53 1–355 alone transactivated the p53-responsive promoters, although to a lesser extent as compared with p53 wt (Figure 5A and B, bars 5 and 6). However, upon coexpression of PML3, the transcriptional activity of p53 1–355 was increased to values comparable to the ones obtained with p53 wt under the same conditions (Figure 5B, bar 8). This was directly dependent on the ability of p53 1–355 to bind to PML3, since a significantly reduced effect was observed upon coexpression of p53 1–355 and PML-L (Figure 5B, bar 10). Interestingly, the PML3-mediated enhancement of p53 transactivation ability was much less evident when tested on the p21 promoter. As shown in Figure 5A and B, only a slight increase in p21 luciferase activity was detected when PML3 was coexpressed with either p53 wt or p53 1–355 (compare bars 5 and 7). The differential promoter transactivations were not due to differences in the level of expression of the various p53 proteins, as judged by western blot analysis (Figure 5, lower panels).
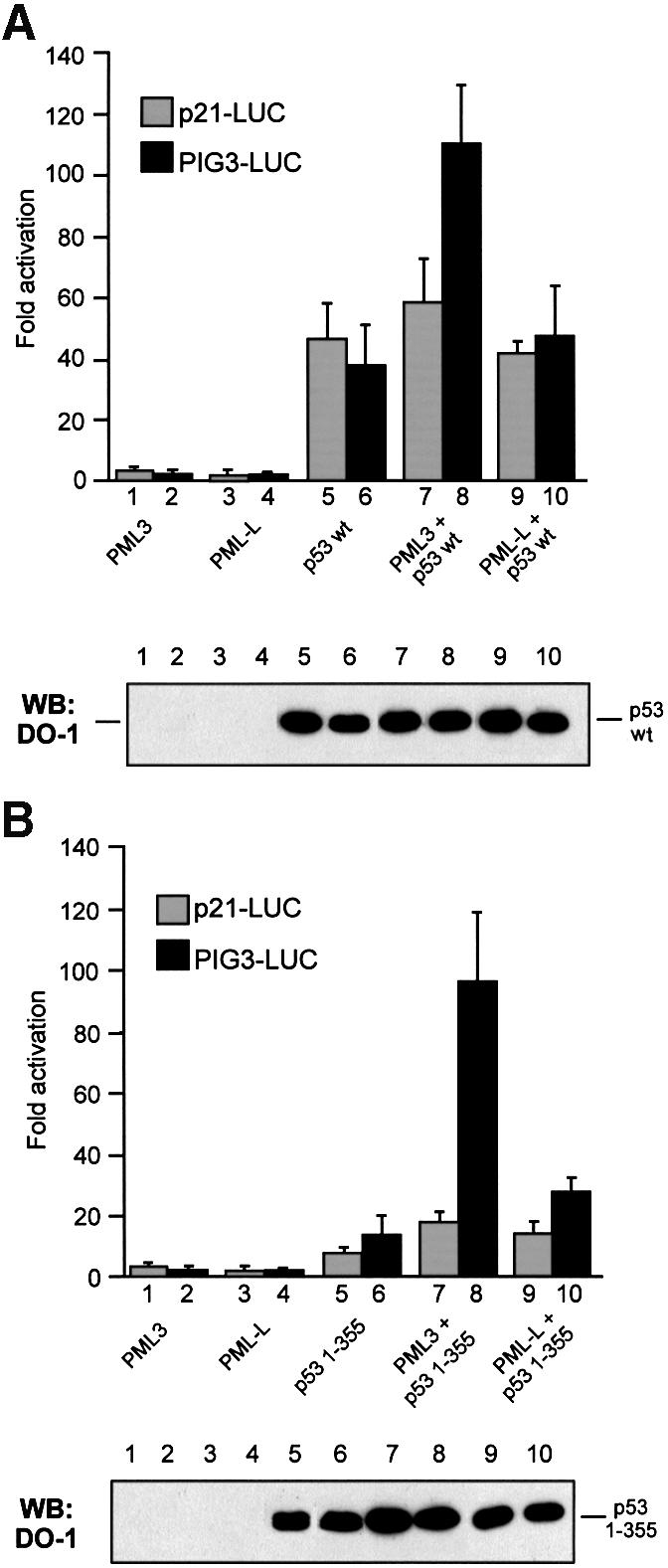
Fig. 5. PML3 enhances p53 transcriptional activity in a promoter-specific manner. Transactivation of p53 wt (A) and p53 1–355 (B) toward the p21-LUC or the PIG3-LUC reporters was assayed by transfecting MG63 cells with pcDNA3p53wt or pcDNA3p53 1–355, either alone or together with PML3 or PML-L, as indicated. Renilla luciferase reporter (pRL-CMV; 50 ng) was cotransfected in each case to normalize transfection efficiency. Graphs represent the mean of at least four independent experiments. An aliquot of each lysate was analysed by western blotting with DO-1 antibody to demonstrate comparable levels of expression of p53 in all the samples (lower panels).
These findings therefore demonstrate that PML3 is able to enhance p53-dependent transactivation in a promoter-specific manner. Of note, PIG3 belongs to a group of p53-regulated genes with the potential to induce oxidative stress and apoptosis (Polyak et al., 1997), thus providing a possible link between the observed decrease in cell survival and the specific activation of the PIG3 promoter following PML3-mediated recruitment of p53 into NBs.
Relocalization of p53 into NBs is necessary to enhance its transcriptional activity
The results shown above correlate the relocalization of p53 into NBs with its enhanced transcriptional activity. To rule out the possibility that this effect is due to interaction of p53 and PML3 within the nucleoplasm rather than in NBs, the components of which are firmly bound to the nuclear matrix, we performed nuclear fractionation analysis in MG63 cells overexpressing p53 wt and p53 1–355 either alone or together with PML3. After nuclei isolation, the nuclear soluble fraction was extracted by lysis in high salt buffer containing NP-40. Under these conditions, all nuclear PML3 was found within the insoluble matrix fraction (Figure 6, compare lanes 2 and 3), while wt p53 was mostly detected in the soluble nucleoplasmic fraction (Figure 6, lanes 5 and 6). When expressed alone, the relative amount of p53 1–355 in the insoluble fraction was higher than that of p53 wt (Figure 6, lanes 11 and 12). Upon PML3 coexpression, a significantly higher fraction of p53 wt was associated with the nuclear matrix than when expressed alone (Figure 6, lanes 8 and 9), and p53 1–355 was almost exclusively detected in the insoluble fraction (Figure 6, lanes 14 and 15). These subcellular fractionation and our immunofluorescence data (Figure 2B), as well as the absence of PML3 from the nucleoplasm, strongly argue for modulation of the p53 transactivation activity predominantly or exclusively in the NBs. Further more, the data also corroborate our indirect evidence for the regulatory function of the eight amino acids of p53 located between positions 355 and 363 in recruitment to NBs.

Fig. 6. p53 is associated with the nuclear matrix fraction in the presence of PML3. Nuclear extract from MG63 cells transfected as indicated was separated into a soluble (S) and an insoluble (I) fraction by high salt buffer lysis in the presence of NP-40. Comparable amounts of proteins for each fraction were separated by SDS–PAGE and immunoblotting was performed with anti-p53 DO-1 antibody or with anti-PML polyclonal serum. The efficiency of transfection was controlled by loading an aliquot of total lysates (T) obtained before nuclear fractionation for each sample.
To confirm further that the enhancement in p53 transcriptional activity requires both binding to PML3 and relocalization into NBs, we performed reporter assays in PML–/– MEFs with PML3S–, a mutant form of PML3 that cannot be sumolated due to mutations in all the three known sumolation sites (Kamitani et al., 1998). Lack of conjugation of PML3S– was demonstrated biochemically by an in vitro sumolation assay (not shown) and also functionally in PML–/– MEFs, where this protein did not associate with the classical NB structures upon overexpression (Figure 7A, b and e). However, PML3S– was still able to bind to p53, as shown by in vitro binding experiments (Figure 7B) and indeed it colocalized with both p53 wt and 1–355 in aberrant subnuclear structures (Figure 7A). When PML–/– MEFs were transfected with the PIG3-LUC reporter plasmid and p53 1–355 together with PML3 or PML3S, the p53 transcriptional activity was significantly increased by PML3 (Figure 7C, bar 4) but not by PML3S– (Figure 7C, bar 5). Similar results were also obtained in SaOS-2 cells, which express low levels of PML (not shown).

Fig. 7. Sumolation of PML3 and recruitment of p53 into NBs are necessary for enhancing p53 transcriptional activity. (A) PML–/– MEFs were microinjected with 10 ng/µl p53 wt or 1–355 together with 30 ng/µl PML3S– and analysed for p53 and PML3 staining as described in Figure 1B. (B) Lysates from SaOS-2 cells transfected with PML3S– and precipitated with GST–p53 or GST, as indicated, were analysed by western blotting with an anti-PML polyclonal antibody. (C) Luciferase activity assay in PML–/– MEFs transfected with the PIG3-LUC reporter and pcDNA3p53 1–355, either alone or together with PML3 or PML3S–, as indicated. pRL-CMV (50 ng) was cotransfected in each case to normalize transfection efficiency. The graph represents the mean of at least three independent experiments. An aliquot of each lysate was analysed by western blotting with DO-1 and anti-PML antibodies to monitor the efficiency of transfection (lower panels).
Taken together, these results demonstrate that binding to PML3 is not sufficient to affect p53 activity, since in addition an efficient relocalization into NBs is required.
Discussion
A possible link between NBs and p53 has been suggested recently (Gostissa et al., 1999) by the finding that similarly to PML and Sp100, known components of NBs, p53 is covalently conjugated to the small ubiquitin-related modifier SUMO-1. In this study we demonstrated that PML3 mediates the recruitment of p53 into these structures (Figure 1B). Overexpression of SUMO-1 and its conjugating enzyme, hUbc9, induced a similar relocalization of p53 that, however, was not dependent on the direct sumolation of the protein, since the conjugation-deficient mutant K386R was efficiently targeted to NBs as well (Figure 1A). A recent report demonstrating that conjugation of SUMO-1 to PML is a prerequisite for its ability to form NBs and consequently to recruit other proteins into these structures (Zhong et al., 2000a) let us hypothesize that enforced expression of SUMO-1 and hUbc9 results in the augmented assembly of NBs where p53 is also targeted. That this is likely to be the case is further supported by the finding that treatment of UV-irradiated cells with As2O3, a known inducer of PML sumolation and used for therapy of APL patients (Chen et al., 1997; Muller et al., 1998a), led to recruitment of endogenous p53 into PML3-containing NBs (Figure 1C).
A detailed microinjection and coimmunoprecipitation analysis with a set of p53 deletions demonstrated that the observed relocalization of p53 into NBs is dependent on a direct association through its core domain with PML3 (Figure 2). Interestingly, amino acids from 355 to 363 of p53 play a negative role in NB targeting (Figure 2B). The region comprising the last 40 amino acids of p53 is a well known target for various post-translational modifications and serves as a surface for intense protein–protein interactions that may modulate p53 functions (Ko and Prives, 1996; Giaccia and Kastan, 1998). Therefore, it is tempting to speculate that the segment between residues 355 and 363 is a binding site for a factor that removes or, more probably, keeps p53 out from the NBs. Alternatively, the presence of a serine at position 362 raises the possibility that phosphorylation of this residue may have the same effect.
PML exists in numerous alternatively spliced variants that mostly differ in their C-terminal sequences (de The et al., 1991; Fagioli et al., 1992). All of them described so far contain the RING finger, B-box and coiled-coil motifs (RBCC) and a nuclear localization signal, which together have been shown to be required and sufficient to target PML into NBs. Nevertheless, the available data do not exclude the possibility that the different PML proteins may interact with diverse cellular partners, thus affecting NB composition and functions. Supporting this interpretation, here we provided evidence that the interaction between p53 and PML is specific for the PML3 variant, since no significant binding was detected with the PML-L isoform and consequently p53 was not found in PML-L NBs (Figure 3). These findings are, to our knowledge, the first data on functional differences between the various PML proteins and raise the possibility that the complex splicing pattern of PML represents a cellular mechanism generating alternative binding interfaces for a variety of factors. In addition to splicing, enhanced SUMO-1 modification of PML may provide another level of complexity either by directly affecting these interactions or by enhancing the ability of PML to form NBs.
The function of the NBs is not yet fully understood; however, their possible involvement in growth suppression has been postulated since the discovery that leukaemia cells from APL patients have an aberrant nuclear dot organization (Dyck et al., 1994; Koken et al., 1994; Weis et al., 1994). Moreover, fibroblasts derived from PML–/– mice show an increased proportion of cells in S phase (Wang et al., 1998a) and these knockout mice are less sensitive to lethal doses of gamma irradiation or Fas antibody treatment, proposing a pro-apoptotic role for PML (Wang et al., 1998b). Several additional correlative results suggested that PML is a critical component for death induction, probably due to its activity in recruiting apoptotic proteins into NBs (Quignon et al., 1998). Since the role of p53 in induction of apoptosis and growth suppression is well established (Sionov and Haupt, 1999), we hypothesized that recruitment of p53 into NBs may contribute to PML3-dependent growth inhibition. In line with this idea, we consistently observed a significant reduction of cell survival when PML3 was introduced into the p53-expressing U2OS cells but not in the p53-null MG63 cell line (Figure 4A). Moreover, the effect observed with the p53 1–355 deletion mutant that shows complete relocalization into NBs suggests that recruitment of p53 into NBs is instrumental for PML3-mediated reduction of cell survival (Figure 4B).
The apoptotic function of p53 has been shown to involve transcription-dependent activities as well as its capability to associate with other cellular factors (Sionov and Haupt, 1999). Here we demonstrated that PML3 selectively increases the p53-dependent activation of the PIG3 promoter (Figure 5). PIG3 was originally isolated as one of several p53-regulated genes with the potential to induce or mimic oxidative stress (Polyak et al., 1997). The evidence that reactive oxygen species are involved in apoptosis and in cell aging (Migliaccio et al., 1999) suggests that PML3-dependent relocalization of transcriptionally active p53 into NBs could contribute to these physiological processes. Furthermore, the lack of p53 activation observed with PML3S–, which still binds to p53 but does not organize the NB structures, definitively proves the importance of p53 recruitment into NBs for modulation of its functions.
Which mechanisms control this promoter specificity? The interaction of p53 with PML3 and other NB-targeted factors involved in transcriptional control, like p300/CBP, which is also a p53 coactivator (Avantaggiati et al., 1997), can regulate the recognition of p53 target genes.
In addition, distinct post-translational modifications taking place in NBs could also contribute to activate p53 transcriptional functions in a promoter-specific manner. Interestingly, it was recently shown that expression of PML3 induces phosphorylation of p53 on serine 15 and enhances its acetylation (Ferbeyre et al., 2000; Pearson et al., 2000). In this context the p53 1–355 protein, which exhibits an increased PML3-dependent ability to activate the PIG3 promoter, could mimic a special conformational change of the full-length protein that is a result of a post-translational modification upon relocalization to NBs. Our subcellular fractionation experiments as well as our immunofluorescence data strongly argue for the functional modification of p53 in the NBs and not in the nucleoplasm. Whether this is true for all types of cells remains to be investigated, since NB composition may vary among the various cell types and may depend on the differentiation state of the cells. The latter is consistent with very recent findings demonstrating that PML is induced by oncogenic ras and promotes premature senescence in fibroblasts in a p53-dependent manner (Ferbeyre et al., 2000; Pearson et al., 2000), while in transformed cells PML may activate p53 apoptotic pathways.
Finally, since it has been suggested that NBs are also involved in chromatin remodelling (Seeler et al., 1998), access to a particular promoter region may depend on the association between transcription factors and non-histone chromosomal proteins.
Of note, hDaxx, a protein involved in Fas-mediated apoptosis, has recently been found to bind to PML and exert its apoptotic function in NBs (Ishov et al., 1999; Torii et al., 1999). The physical interaction between hDaxx and p53 (M.Gostissa and G.Del Sal, unpublished results) may add another level of complexity in the role of cell death control of NBs.
Approximately 5–15% of p53 mutations occur in the C-terminal domain and result in truncated proteins that, although transcriptionally active, are defective in apoptosis induction (Zhou et al., 1999). It is tempting to speculate that in cells expressing such p53 mutants, apoptosis can still be induced by stimulating the relocalization of p53 into PML-containing NBs following treatment with agents that modulate the expression (or the sumolation) of PML3.
Control of cell death and differentiation may proceed through pathways involving either PML or p53 and, as demonstrated in this work, at least some of them are converging. Relocalization of various factors involved in transcriptional and growth control into NBs may allow the formation of specific protein–protein interactions and lead to the transactivation of particular promoters (Figure 8). The knowledge of integration and cross-talks between different apoptotic pathways could therefore allow the implementation of methods for blocking the transformation process and the design of novel therapeutic strategies.

Fig. 8. A model for PML3-mediated recruitment of p53 into NBs and the postulated interactions with other NB-resident factors. Conjugation of SUMO-1 (S) on PML3 results in the assembly of NBs either by direct interactions between PML and various proteins like p53, p300/CBP and hDaxx or by the relocalization of other elements (Sp100) through still unknown mechanisms. Protein–protein interactions as well as post-translational modifications in NBs may regulate specific biological functions.
Materials and methods
Cell lines, plasmids and antibodies
U2OS, MG63 and SaOS-2 cells are human osteosarcoma cell lines, respectively, wt for p53 and pRb, wt for pRb but null for p53 and null for both p53 and pRb. LOVO is a colon carcinoma cell line containing wt p53. PML–/– MEFs have been described previously (Wang et al., 1998a).
pcDNA3p53wt, pcDNA3HAp53 294–393, pGFPSUMO-1, pcDNA3HAhUbc9, pRcCMVp53 K386R (Gostissa et al., 1999), pSG5PML-L and pSG5PMLRARα (de The et al., 1991) have been described previously. pCMVp53Δ63–91, pCMVp53Δ12–69, pcDNA3PML3 and pNLS-βgal were kindly provided by A.J.Levine, J.Jenkins, P.G.Pelicci and C.Kuhne, respectively. pcDNA3 plasmids containing the different p53 deletions and pGEXPML3Ct were constructed by PCR. pcDNA3PML3S– has been generated by site-directed mutagenesis. All PCR-amplified products were fully sequenced to exclude the possibility of second site mutations.
The following primary antibodies were used: rabbit polyclonal anti-p53 (Gostissa et al., 1999); DO-1 (monoclonal anti-p53, Santa Cruz); 1801 (monoclonal anti-p53, gift from L.Banks); PG-M3 (monoclonal anti-PML, Santa Cruz); rat polyclonal anti-PML (Sternsdorf et al., 1997); rabbit polyclonal anti-PML3 (kindly provided by P.G.Pelicci); monoclonal anti-β-galactosidase (Promega); and 12CA5 (monoclonal anti-HA, Roche Molecular Biochemicals).
Microinjection, immunofluorescence analysis and survival assay
Cells grown on coverslips in 35-mm Petri dishes were microinjected using the Automated Injection System (Zeiss, Oberkochen, Germany) as described previously (Del Sal et al., 1992).
For immunofluorescence analysis, cells were fixed with 3% paraformaldehyde in phosphate-buffered saline (PBS) at room temperature for 20 min, then incubated for 5 min in 0.1 M glycine–PBS and permeabilized with 0.1% Triton X-100 in PBS for 5 min. The coverslips were incubated for 1 h at 37°C with the different primary antibodies. Primary antibodies were revealed by a 30 min incubation with goat anti-mouse or anti-rabbit FITC- or TRITC-conjugated secondary antibodies (Sigma). GFP–SUMO-1 and GFP staining were revealed by means of the intrinsic green fluorescence of GFP. For UV and/or As2O3 treatment cells were irradiated with 30 J/m2 UV light and subsequently incubated for 18 h in the presence of 1 µM As2O3 prior to fixation. The pre-extraction protocol on UV- and/or AS2O3-treated cells was performed as previously described (Lombard and Guarente, 2000). Images were obtained with a Zeiss laser scan microscope (LSM 410).
Cell survival was analysed by microinjecting 200 cells for each experiment with the gene of interest and GFP as marker, and calculated as the percentage of recovered cells expressing GFP, as described previously (Brancolini et al., 1999).
Immunoprecipitation, in vitro binding assay and western blot analysis
Transfections were performed with the standard calcium phosphate method. Immunoprecipitations were performed in 150 mM NaCl-containing lysis buffer (Sandy et al., 2000) with 1 µg of PG-M3 antibody, covalently crosslinked to 20 µl of protein A–Sepharose CL-4B beads (Amersham Pharmacia Biotech). In vitro binding assays were performed as previously described (Sandy et al., 2000). For in vivo binding assays, transfected cells were lysed in 300 mM NaCl-containing lysis buffer (300 mM NaCl, 50 mM Tris–HCl pH 7.5, 0.5% NP-40, 10% glycerol). The lysates were then diluted twice and incubated with 4 µg of GST–p53 or GST alone. Western blotting was performed according to standard procedures. Primary antibodies were revealed by HRPO-conjugated secondary antibodies (Sigma), followed by enhanced chemiluminescence (Pierce).
Nuclear fractionation
MG63 cells were plated in 6-cm Petri dishes and transfected with 3 µg of pcDNA3PML3 and 1.5 µg of the different p53 constructs. Twenty-four hours after transfection, cells were collected in PBS and nuclei were separated by lysis in buffer 1 (50 mM Tris–HCl pH 7.9, 10 mM KCl, 1 mM EDTA, 0.2% NP-40, 10% glycerol) and centrifugation at 6000 r.p.m. for 3 min at 4°C. Pellet was washed with buffer 1 without detergent and lysed with buffer 2 (400 mM NaCl, 1% NP-40, 20% glycerol, 20 mM HEPES pH 7.9, 10 mM KCl, 1 mM EDTA) for 20 min at 4°C. The insoluble and soluble nuclear fractions were separated by centrifugation at 14 000 r.p.m. for 10 min.
Transactivation assays
Petri dishes (35 mm) were transfected with 500 ng of p21-LUC (El-Deiry et al., 1993) or PIG3-LUC (Polyak et al., 1997) reporters, 100 ng of different p53 constructs and 300 ng of PML3 or PML-L, as indicated. In each transfection, 50 ng of pRL-CMV (Promega), expressing the Renilla luciferase, were added and luciferase activity was measured and normalized using the Dual Luciferase Reporter Assay System (Promega).
MG63 cells were transfected with the standard calcium phosphate method, while PML–/– MEFs were transfected by lipofection using the Lipofectamine Plus kit (Life Technologies).
Acknowledgments
Acknowledgements
We thank P.G.Pelicci for PML3 expression vector and antibodies, M.Oren for p21-Luc and B.Vogelstein for PIG3-Luc reporters. This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) and MURST (Cofin) to G.D.S. M.G. and P.S. are FIRC and ICGEB fellows, respectively. The work of H.W., T.S. and K.J. was supported by grants from the Deutsche Forschungsgemeinschaft and the Deutsche Krebshilfe. The HPI is supported by the Ministry of Health and the Freie and Hansestadt Hamburg.
Note added in proof
While this manuscript was under revision, Guo et al. (Nature Cell Biol., 2000, 730–736) have shown that PML is involved in p53-dependent apoptosis.
References
- Alcalay M., Tomassoni,L., Colombo,E., Stoldt,S., Grignani,F., Fagioli,M., Szekely,L., Helin,K. and Pelicci,P.G. (1998) The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol. Cell. Biol., 18, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avantaggiati M.L., Ogryzko,V., Gardner,K., Giordano,A., Levine,A.S. and Kelly,K. (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell, 89, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Brancolini C., Marzinotto,S., Edomi,P., Agostoni,E., Fiorentini,C., Muller,H.W. and Schneider,C. (1999) Rho-dependent regulation of cell spreading by the tetraspan membrane protein Gas3/PMP22. Mol. Biol. Cell, 10, 2441–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.Q. et al. (1997) Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): As2O3 exerts dose-dependent dual effects on APL cells. Blood, 89, 3345–3353. [PubMed] [Google Scholar]
- Del Sal G., Ruaro,M.E., Philipson,L. and Schneider,C. (1992) The growth arrest-specific gene, gas1, is involved in growth suppression. Cell, 70, 595–607. [DOI] [PubMed] [Google Scholar]
- de The H., Lavau,C., Marchio,A., Chomienne,C., Degos,L. and Dejean,A. (1991) The PML–RARα fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell, 66, 675–684. [DOI] [PubMed] [Google Scholar]
- Dyck J.A., Maul,G.G., Miller,W.H.J., Chen,J.D., Kakizuka,A. and Evans,R.M. (1994) A novel macromolecular structure is a target of the promyelocyte–retinoic acid receptor oncoprotein. Cell, 76, 333–343. [DOI] [PubMed] [Google Scholar]
- El Deiry W.S. et al. (1993) WAF-1, a potential mediator of p53 tumor suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Fagioli M., Alcalay,M., Pandolfi,P.P., Venturini,L., Mencarelli,A., Simeone,A., Acampora,D., Grignani,F. and Pelicci,P.G. (1992) Alternative splicing of PML transcripts predicts coexpression of several carboxy-terminally different protein isoforms. Oncogene, 7, 1083–1091. [PubMed] [Google Scholar]
- Ferbeyre G., de Stanchina,E., Querido,E., Baptiste,N., Prives,C. and Lowe,S.W. (2000) PML is induced by oncogenic ras and promotes premature senescence. Genes Dev., 14, 2015–2027. [PMC free article] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Gostissa M., Hengstermann,A., Fogal,V., Sandy,P., Schwarz,E., Scheffner,M. and Del Sal,G. (1999) Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J., 18, 6462–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges M., Tissot,C., Howe,K., Grimwade,D. and Freemont,P.S. (1998) Structure, organization and dynamics of promyelocytic leukemia protein nuclear bodies. Am. J. Hum. Genet., 63, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Floss,S. and Stamminger,T. (2000) Covalent modification of the transactivator protein IE2-p86 of human cytomegalovirus by conjugation to the ubiquitin-homologous proteins SUMO-1 and hSMT3b. J. Virol., 74, 2510–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov A.M., Sotnikov,A.G., Negorev,D., Vladimirova,O.V., Neff,N., Kamitani,T., Yeh,E.T., Strauss,J.F.,III and Maul,G.G. (1999) PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol., 147, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizuka A., Miller,W.H.J., Umesono,K., Warrell,R.P.J., Frankel,S.R., Murty,V.V.V.S., Dmitrovsky,E. and Evans,R.M. (1991) Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RARα with a novel putative transcription factor, PML. Cell, 66, 663–674. [DOI] [PubMed] [Google Scholar]
- Kamitani T., Kito,K., Nguyen,H.P., Wada,H., Fukuda-Kamitani,T. and Yeh,E.T. (1998) Identification of three major sentrinization sites in PML. J. Biol. Chem., 273, 26675–26682. [DOI] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Koken M.H. et al. (1994) The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J., 13, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lain S., Midgley,C., Sparks,A., Lane,E.B. and Lane,D.P. (1999) An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp. Cell Res., 248, 457–472. [DOI] [PubMed] [Google Scholar]
- LaMorte V.J., Dyck,J.A., Ochs,R.L. and Evans,R.M. (1998) Localization of nascent RNA and CREB binding protein with the PML-containing nuclear body. Proc. Natl Acad. Sci. USA, 95, 4991–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavau C. et al. (1995) The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene, 11, 871–876. [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Lin R.J., Egan,D.A. and Evans,R.M. (1999) Molecular genetics of acute promyelocytic leukemia. Trends Genet., 15, 179–184. [DOI] [PubMed] [Google Scholar]
- Lombard D.B. and Guarente,L. (2000) Nijmegen breakage syndrome disease protein and MRE11 at PML nuclear bodies and meiotic telomeres. Cancer Res., 60, 2331–2334. [PubMed] [Google Scholar]
- Mahajan R., Delphin,C., Guan,T., Gerace,L. and Melchior,F. (1997) A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell, 88, 97–107. [DOI] [PubMed] [Google Scholar]
- Maul G.G. (1998) Nuclear domain 10, the site of DNA virus transcription and replication. BioEssays, 20, 660–667. [DOI] [PubMed] [Google Scholar]
- Migliaccio E., Giorgio,M., Mele,S., Pelicci,G., Reboldi,P., Pandolfi,P.P., Lanfrancone,L. and Pelicci,P.G. (1999) The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature, 402, 309–313. [DOI] [PubMed] [Google Scholar]
- Moll U.M., Ostermayer,A.G., Haladay,R., Winkfield,B., Frazier,M. and Zambetti,G. (1996) Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol. Cell. Biol., 16, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S., Matunis,M.J. and Dejean,A. (1998a) Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J., 17, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S., Miller,W.H.,Jr and Dejean,A. (1998b) Trivalent antimonials induce degradation of the PML–RAR oncoprotein and reorganization of the promyelocytic leukemia nuclear bodies in acute promyelocytic leukemia NB4 cells. Blood, 92, 4308–4316. [PubMed] [Google Scholar]
- Muller S., Berger,M., Lehembre,F., Seeler,J.-S., Haupt,Y. and Dejean,A. (2000) c-Jun and p53 activity is modulated by SUMO-1 modification. J. Biol. Chem., 275, 13321–13329. [DOI] [PubMed] [Google Scholar]
- Ostermayer A.G., Runko,E., Winkfield,B., Ahn,B. and Moll,U.M. (1996) Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide. Proc. Natl Acad. Sci. USA, 93, 15190–15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M. et al. (2000) PML regulates p53 acetylation and premature senescence by oncogenic Ras. Nature, 406, 207–210. [DOI] [PubMed] [Google Scholar]
- Polyak K., Xia,Y., Zweier,J.L., Kinzler,K.W. and Vogelstein,B. (1997) A model for p53-induced apoptosis. Nature, 389, 300–305. [DOI] [PubMed] [Google Scholar]
- Quignon F., De Bels,F., Koken,M., Feunteun,J., Ameisen,J.C. and de The,H. (1998) PML induces a novel caspase-independent death process. Nature Genet., 20, 259–265. [DOI] [PubMed] [Google Scholar]
- Rodriguez M.S., Desterro,J.M.P., Lain,S., Midgley,C.A., Lane,D.P. and Hay,R.T. (1999) SUMO-1 modification activates the transcriptional response of p53. EMBO J., 18, 6455–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandy P., Gostissa,M., Fogal,V., De Cecco,L., Szalay,K., Rooney,R.J., Schneider,C. and Del Sal,G. (2000) p53 is involved in the p120E4F-mediated growth arrest. Oncogene, 19, 188–199. [DOI] [PubMed] [Google Scholar]
- Seeler J.S. and Dejean,A. (1999) The PML nuclear bodies: actors or extras? Curr. Opin. Genet. Dev., 9, 362–367. [DOI] [PubMed] [Google Scholar]
- Seeler J.S., Marchio,A., Sitterlin,D., Transy,C. and Dejean,A. (1998) Interaction of SP100 with HP1 proteins: a link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc. Natl Acad. Sci. USA, 95, 7316–7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sionov R. and Haupt,Y. (1999) The cellular response to p53: the decision between life and death. Oncogene, 18, 6145–6157. [DOI] [PubMed] [Google Scholar]
- Sternsdorf T., Jensen,K. and Will,H. (1997) Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol., 139, 1621–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternsdorf T., Jensen,K., Reich,B. and Will,H. (1999) The nuclear dot protein Sp100, characterization of domains necessary for dimerization, subcellular localization and modification by small ubiquitin-like modifiers. J. Biol. Chem., 274, 12555–12566. [DOI] [PubMed] [Google Scholar]
- Tarunina M. and Jenkins,J.R. (1993) Human p53 binds DNA as a protein homodimer but monomeric variants retain full transcription transactivation activity. Oncogene, 8, 3165–3173. [PubMed] [Google Scholar]
- Torii S., Egan,D.A., Evans,R.A. and Reed,J.C. (1999) Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). EMBO J., 18, 6037–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.G., Delva,L., Gaboli,M., Rivi,R., Giorgio,M., Cordon-Cardo,C., Grosveld,F. and Pandolfi,P.P. (1998a) Role of PML in cell growth and the retinoic acid pathway. Science, 279, 1547–1551. [DOI] [PubMed] [Google Scholar]
- Wang Z.G., Ruggero,D., Ronchetti,S., Zhong,S., Gaboli,M., Rivi,R. and Pandolfi,P.P. (1998b) PML is essential for multiple apoptotic pathways. Nature Genet., 20, 266–271. [DOI] [PubMed] [Google Scholar]
- Weis K., Rambaud,S., Lavau,C., Jansen,J., Carvalho,T., Carmo-Fonseca,M., Lamond,A. and Dejean,A. (1994) Retinoic acid regulates aberrant nuclear localization of PML–RARα in acute promyelocytic leukemia cells. Cell, 76, 345–356. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Xiong,Y. (1999) Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol. Cell, 3, 579–591. [DOI] [PubMed] [Google Scholar]
- Zhong S., Muller,S., Ronchetti,S., Freemont,P.S., Dejean,A. and Pandolfi,P.P. (2000a) Role of SUMO-1-modified PML in nuclear body formation. Blood, 95, 2748–2753. [PubMed] [Google Scholar]
- Zhong S., Salomoni,P. and Pandolfi,P.P. (2000b) The transcriptional role of PML and the nuclear body. Nature Cell Biol., 2, E85–E90. [DOI] [PubMed] [Google Scholar]
- Zhou X., Wang,X.W., Xu,L., Hagiwara,K., Nagashima,M., Wolkowicz,R., Zurer,I., Rotter,V. and Harris,C.C. (1999) COOH-terminal domain of p53 modulates p53-mediated transcriptional transactivation, cell growth and apoptosis. Cancer Res., 59, 843–848. [PubMed] [Google Scholar]