

Abstract
Importance of the field
Many chronic diseases of various etiologies universally lead to fibrosis and organ dysfunction. Despite many advances in medicine in recent years, options to slow the progression of fibrotic diseases have remained limited. The recent availability of pirfenidone, an anti-fibrotic and anti-inflammatory investigational agent, thus offers a new hope for treating progressive fibrotic diseases.
Areas covered in this review
This review provides concise review of the available data regarding mechanism and pharmacokinetics of pirfenidone and preclinical and clinical data regarding efficacy and safety in fibrotic diseases of the kidney. It also reviews results of clinical trials involving pirfenidone in other fibrotic diseases.
What the reader will gain
The review will provide in-depth review of pirfenidone with a renal focus.
Take home message
Because many of the available clinical trials have been small and/or uncontrolled, conclusive evidence regarding efficacy and safety of pirfenidone is lacking, particularly in patients with renal or hepatic dysfunction. Larger studies are needed both to better understand long-term efficacy and safety of this medication in various patient populations.
Keywords: fibrosis, inflammation, renal failure, glomerulosclerosis, proteinuria
1. General Overview
Pirfenidone is an orally available pyridone derivative that has anti-fibrotic and anti-inflammatory effects. Since its discovery as an antifibrotic agent in a hamster model of bleomycin-induced pulmonary fibrosis [1], pirfenidone has been clinically evaluated for its safety and efficacy in numerous disorders such as idiopathic pulmonary fibrosis, multiple sclerosis, primary sclerosing cholangitis, chronic hepatitis C, myelofibrosis, neurofibromatosis, and fibrotic renal disorders. Most of the studies have been small with mixed results, though many suggest a possible benefit and the need for larger randomized controlled trials. The largest clinical data obtained pertain to idiopathic pulmonary fibrosis, in which pirfenidone may help to stabilize pulmonary function [2, 3]. The efficacy of pirfenidone in idiopathic pulmonary fibrosis has been reviewed else where in detail [4–6]. The goal of this review is to provide a more in-depth review of the available preclinical and clinical data regarding the scientific rationale and the experience of pirfenidone in fibrotic renal diseases.
2. Introduction
Renal fibrosis is the common pathway underlying the progression of chronic renal injury to end stage renal disease, regardless of the initiating event. It universally involves progressive glomerulosclerosis, tubulointerstitial atrophy and fibrosis, and loss of glomerular and peritubular microvasculature. Mechanisms contributing to renal fibrosis are complex and incompletely understood. It is a dynamic, interactive process initiated by cell injury and characterized by the twin processes of loss of differentiated cellular phenotype and accumulation of extracellular matrix proteins. Cell injury of any type results in release of proinflammatory cytokines and inflammation is intrinsically linked to the fibrotic process in the kidney [7]. Inadequate repair or prolonged tissue injury may lead to cell injury, loss of differentiated functions, and death. The complex web of pathways activated in the process eventually leads to matrix production, inhibition of matrix degradation, mesangial and fibroblast activation, lymphocyte/macrophage infiltration, and epithelial-to-mesenchymal transition (EMT) of tubular epithelial cells (Figure 1).
Figure 1. Mechanisms of progressive kidney injury.
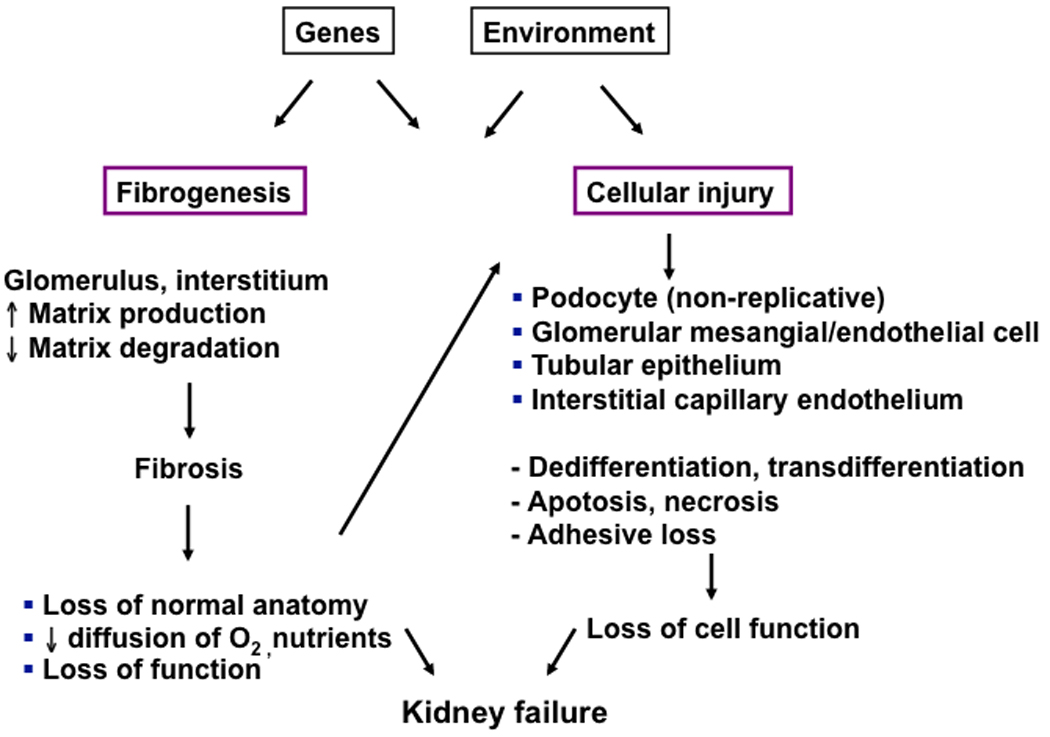
Tissue injury is initiated by genetic factors (e.g sickle cell anemia) or environmental factors (e.g. viral infection) or most frequently by diseases arising out of a combination of both processes (e.g hypertension, diabetes, coronary heart disease). The figure illustrates some pathways for kidney injury but similar processes affect every tissue. Tissue injury, often involving inflammation, leads to the twin processes of fibrogenesis and cellular injury, which interact as shown. Ultimately, organs fail because cells and the tissue as a whole can no longer fulfill critical organ functions. It is likely that successful therapeutic strategies to slow and perhaps reverse organ failure will be those that limit and reverse fibrogenesis and stabilize differentiate cell phenotypes.
Over the years, transforming growth factor (TGF) -β and its downstream signaling pathways, in addition to various proinflammatory cytokines such as tumor necrosis factor (TNF), have emerged as one of the central players in activating and mediating cellular pathomechanisms responsible for progression of renal diseases [8–11]. Specifically, the role of TGF-β and its downstream Smad pathways in mediating apoptosis and EMT has been discussed in recent years [12–16].
Given its critical importance in the renal fibrotic process, targeting TGF-β for clinical intervention has been attractive. Various attempts to antagonize the TGF-β system, however, have been limited. The widely available angiotensin pathway antagonists can slow the progression of various chronic renal diseases, but most patients continue to have progressive loss of renal function. Other possibilities such as neutralizing antibodies of TGF-β have not been clinically available. It is thus not surprising that pirfenidone, a relatively new compound with good bioavailability, has garnered much interest in investigators studying various forms of fibrotic diseases.
3. Pirfenidone: mechanism of action and pharmacokinetics
3.1 mechanism of action
While the precise the mechanism of pirfenidone is incompletely understood, it exhibits both anti-inflammatory and antifibrotic effects. Much attention has been focused the antifibrotic effect of the drug. An important aspect of the antifibrotic mechanism of pirfenidone is associated with its inhibition of both production and activity of TGF-β. In bleomycin-induced lung injury hamster model, pirfenidone treatment significantly suppresses the TGF-β gene transcription by 33% [17] and suppresses bleomycin-induced overexpression of procollagen I and III genes [18]. In animal models of hepatic fibrosis, pirfenidone also decreases TGF-β and collagen type 1 mRNA expression [19]. In addition, pirfenidone inhibits pulmonary fibroblast expression of heat shock protein 47, a collagen-specific molecular chaperone thought to play an important role in extracellular matrix synthesis and remodeling [20]. In cultured renal cortical fibroblasts isolated from rats with ureteric obstruction, pirfenidone has been shown to inhibit fibroblast mitogenesis and expression of connective tissue growth factor [21].
Various animal models of fibrosis demonstrate that pirfenidone exerts anti-inflammatory effects. It suppresses bleomycin-induced increased pulmonary vascular permeability and increased influx of inflammatory cells, such as neutrophils, macrophages, and lymphocytes [22]. In a superantigen-induced shock model, mice treated with pirfenidone had substantially improved survival, with the outcome closely correlating with the reduction in TNF-α and interleukin-1 expression [23]. Pirfenidone similarly ameliorates the tissue damage, hypotension, and completely blocks the increase in TNF-α in a ischemia reperfusion injury model of rat intestine [24]. The mechanism underlying the anti-inflammatory effect of pirfenidone in part involves its ability to inhibit the expression of potent pro-inflammatory cytokines, as described above. In the murine endotoxin shock model, pirfenidone potently suppresses production of TNF, interleukin-6, and interferon gamma at the translation level, but not at the transcription level [25]. In cultured hepatocytes treated with interleukin-1β, moreover, pirfenidone suppresses the induction of inducible nitric oxide synthase mRNA and protein partly through inhibition of the activation of NF-kappaβ [26]. Additionally, pirfenidone may further ameliorate oxidative stress through its ability to scavenge hydroxyl radicals in a dose-dependent manner, with the concentration of pirfenidone required to inhibit 50% signal height being about 2.5 mM [27]. Given that this represents a much higher concentration by several logs compared to clinically-relevant drug levels, however, it is unclear if pirfenidone may indeed have antioxidant effect in vivo.
3.2 Metabolism and pharmacokinetics
There are limited clinical data on metabolism and pharmacokinetics of pirfenidone. Pirfenidone is thought to be primarily metabolized through liver, though the specific mode of metabolism remains unknown. Furthermore, pharmacokinetics and metabolism of pirfenidone have not been formally evaluated in patients with reduced glomerular filtration rate (GFR) or with end stage renal disease. The long-term efficacy and safety in patients with decreasing or minimal GFR, therefore, are unknown. In a phase I study involving 48 healthy and young Chinese adult volunteers, investigators assessed for both single- and multiple-dose pharmacokinetics of pirfenidone, including the effect of food and gender [28]. In the first part of the study, the participants took a single dose of oral pirfenidone at 200, 400, or 600 mg while fasting. To examine the effect of food, 12 subjects of the 400-mg dose group were randomized to fasted and fed conditions and received pirfenidone following a high-fat meal in a crossover design with a washout period of 1 week. In a second part of the study, 12 subjects (6 women and 6 men) received 400 mg of pirfenidone 3 times daily for 5 consecutive days in a fed state (low-fat diet) to prevent potential side effects. Pirfenidone was shown to be rapidly absorbed following single oral doses, with maximum plasma concentrations occurring 0.33 to 1 hour after administration. The drug was cleared from plasma in a dose-dependent manner with terminal elimination t1/2 of approximately 2 to 2.5 hours after single oral doses of pirfenidone. The presence of food significantly decreased absorption of pirfenidone (AUC0− = 37.4 ± 15.4 mg·h/L, fed state vs 46.6 ± 16.8 mg·h/L, fasted state; p < 0.01). Ingestion of food also considerably delayed the rate of absorption: tmax = 1.5 ± 0.4 hours (fed) versus 0.7 ± 0.2 hours (fasted). Correspondingly, peak plasma concentrations were significantly (P < .001) lower with food ingestion (Cmax= 9.2 ± 2.9 mg/L) compared to fasted state (Cmax =13.0 ± 1.8 mg/L). In the second part of the study evaluating the pharamcokinetic of multiple dosing, steady-state conditions were achieved by the third day with 400 mg three times daily. Pirfenidone was again rapidly absorbed within 1 to 2 hours after oral administration and cleared from plasma with a half-life of 2.1 ± 0.4 hours. The pharmacokinetic parameters after multiple dosing were similar to those obtained after single doses under fed conditions, indicating no significant accumulation of PF with repeated dosing. There was no effect of gender on pharmacokinetics.
In another study involving older healthy volunteers (ages between 50–66 years), similar effect of food was seen on the pharmacokinetics of the drug [29]. Approximately 80% of the dose of pirfenidone was recovered in the urine as parent drug or metabolites over the 72-h collection period. One study involving three hemodialysis patients with peritoneal sclerosis suggests that pirfenidone is dialyzable to some extent, though adjustment of dosage in dialysis patients is likely unnecessary [30]. Pediatric pharmacokinetics of pirfenidone appears to be comparable to adults, when two doses (250 mg/m2 or 500 mg/m2) were compared [31]. Within the pediatric population between the ages 3–19 years, the study did not reveal any evidence of age dependence in drug disposition.
4. Preclinical studies in animal models of renal fibrosis
Pirfenidone has been evaluated in various experimental models of renal fibrosis. The available data can be divided largely into two groups: (1) studies examining the effect of pirfenidone in experimental settings or models for native fibrotic glomerular disorders and (2) studies evaluating the effect of pirfenidone in calcineurin-inhibitor induced renal fibrosis.
4.1 Use of pirfenidone in models of native fibrotic renal disorders
Pirfenidone has been studied in experimental models of glomerulosclerosis (remnant kidney [32], unilateral ureteral obstruction [33], and FGS/Kist mouse [34]), diabetes (streptozotocin rats [35] and db/db mice [36]), and anti-glomerular basement membrane glomerulonephritis [37]. In the remnant kidney rat model, Shimizu et al. noted that the treatment with pirfenidone was significantly more effective at stabilizing renal function at 12 weeks and reducing renal cortical collagen accumulation compared to control. While pirfenidone also suppressed the increase in TGF-β mRNA in the cortex of the remnant kidney, pirfenidone therapy did not result in significant reduction of proteinuria. Shimizu et al. also found similar results in the unilateral ureteral obstruction model [33]. Administration of pirfenidone was again associated with significant suppression of collagen content, collagen I and IV mRNA expression, matrix metalloproteinase-2, and TGF-β mRNA in the renal cortex at 21 days. More significantly, pirfenidone therapy for five weeks led to significant improvement of inulin clearance, while the untreated control group continued to have poor renal function without any recovery. In parallel with this functional improvement, the renal histologic examination following five weeks of pirfenidone therapy also revealed striking attenuation of fibrotic lesions. Likewise, Park et al. described similar beneficial effect of pirfenidone to reduce sclerosis and preserve GFR after 3 months of administration in a separate model of focal segmental glomerulosclerosis (FSGS) [34].
In streptozotocin-diabetic rats, Miric et al. described that 4 weeks of pirfenidone administration attenuated fibrotic changes both in the heart and kidney and decreased renal and plasma concentration of fibronectin [35]. More recently, RamachandraRao et al. have evaluated the therapeutic potential of pirfenidone in a different diabetic model with db/db mice [36]. Four weeks of pirfenidone administration significantly reduced mesangial matrix expansion and expression of renal matrix genes in the mice. As previously seen, however, pirfenidone did not improve albuminuria in this study. In addition, a separate part of the study using murine mesangial cell line again demonstrated that pirfenidone inhibits TGF-β production at the transcriptional and protein levels and blocks TGF-β-induced Smad phosphorylation, thereby suppressing the pathways felt to be critical for apoptosis and fibrosis. Pirfenidone also led to therapeutic benefit in a rat model of anti-glomerular basement membrane glomerulonephritis [37]. Pirfenidone administration for eight weeks in this model was associated with significant improvement of renal architecture, including attenuated podocyte injury and tubular degeneration, compared to untreated controls. Unlike other studies, pirfenidone therapy led to significant reduction of proteinuria by week 7, though this antiproteinuric effect was noted earlier and to a greater degree in the group that was randomized to candesartan. Combination of pirfenidone and candesartan resulted in the greatest reduction of proteinuria.
4.2 Experimental models to evaluate drug-induced renal fibrosis
Some medications, while required to treat specific conditions, may have the undesirable effect of nephrotoxicity through progressive fibrosis. Calcineurin inhibitors, the mainstay of immunosuppressive regimen for organ transplantation, ironically pose one of the greatest long-term threats to renal allograft outcome. Calcineurin inhibitors cause glomerulosclerosis, tubulointerstitial and mesangial fibrosis associated with increased collagen production and insufficient extracellular matrix degradation [38]. It has been well established that calcineurin inhibitors also induce TGF-β both in vitro and vivo and this is thought to be an important pathomechanism underlying the profibrotic effect of the drugs. Cyclosporine causes TGF-β gene and protein induction in activated human T cells [39] and proximal tubular cells and tubulointerstitial fibroblast cell lines [40]. Cyclosporine has also been shown to significantly increase circulating levels of TGF-β protein in the serum of mice, in addition to inducing the intra-renal expression of TGF-β gene [41]. Given the abundant data to suggest that pirfenidone opposes the induction of TGF-β and its downstream pathways, this novel antifibrotic agent has been considered as a potential therapeutic agent in management of chronic allograft dysfunction. The topic has been recently reviewed more extensively by Dosanjh [42, 43].
The available data from experimental models support the rationale for pirfenidone use in clinical conditions that would require long-term use with calcineurin inhibitors or other profibrotic agents. In salt-depleted rats treated with cyclosporine, pirfenidone administration led to significant reduction in renal cortical TGF-β1 mRNA and still greater reduction of TGF-β1 protein expression by 80% [44]. Concurrently, pirfenidone also significantly reduced renal cortical expression of plasminogen activator inhibitor-1, a protease inhibitor that is directly stimulated by TGF-β and blocks matrix degradation. While pirfenidone did not improve renal function in cyclosporine-treated rats, it was associated with striking improvement of histologic findings, ameliorating cyclosporine-induced fibrosis by 50%. In another study also using salt-depleted rats, pirfenidone attenuated cyclosporine-induced increase in mRNA expression of profibrotic genes and increased expression of antifibrotic genes, while improving serum creatinine [45]. The beneficial effect of pirfenidone may in part be associated with inhibition of the pro-apoptotic effect of TGF-β The significant renal protection noted with pirfenidone in rats was associated with down-regulation of cyclosporine-induced pro-apoptotic genes and significant reduction of apoptosis in the renal tubules and interstitium [46]. Moreover, pirfenidone may have immune modulating properties, including inhibition of T-cell activation, proliferation and attenuation of associated induction of multiple cytokines [47].
5. Clinical studies in renal fibrosis and other fibrotic diseases
To date, only one small clinical trial has been completed to assess safety and efficacy of pirfenidone in fibrotic kidney disease [48]. It was a single-center, open-label pilot study to evaluate if pirfenidone can slow the GFR decline in adult patients with biopsy-proven idiopathic and post-adaptive FSGS. Out of 21 patients enrolled, 18 completed a median of 13 months of pirfenidone treatment in the study with a median dose of 800 mg three times daily. In the months preceding the study, patients had well-controlled blood pressure, with most of them receiving angiotensin pathway antagonists. The participants also had advanced chronic kidney disease at baseline, with mean MDRD-estimated eGFR of 26±9.4 ml/min/1.73m2 and median baseline proteinuria of 2.8 g/d. The monthly eGFR decline rate improved from a median of −0.61 ml/min/1.73m2 during the baseline period to −0.45 ml/min/1.73m2 during the 12-months of pirfenidone therapy (P<0.01). Pirfenidone had no effect on proteinuria.
Two significant problems with the study were the lack of placebo control and the choice of the GFR decline rate as the study primary outcome variable. Calculating eGFR change rate based on multiple serum creatinine values can be problematic as one outlier can cause significant shift in the overall slope. Thus comparing the GFR decline slope during the treatment phase to the pre-study entry period may or may not accurately illustrate the effect of the drug on this endpoint.
The study of FSGS, nonetheless, provided sufficient rationale to evaluate pirfenidone in a larger, randomized, placebo-controlled trial to investigate if it can have similar benefit in patients with diabetic nephropathy. This phase II, dose-ranging, multi-center trial had the main objective of assessing if pirfenidone can slow the loss of GFR in diabetic patients after 54 weeks of therapy. The inclusion criteria included adult type 1 or 2 diabetic patients with established nephropathy (eGFR between 20–75 ml/min and proteinuria >300 mg/24h) while being treated with angiotensin pathway antagonists. Patients with unstable cardiovascular complications or requiring immunosuppressive medications were excluded. The patients were randomized to take 1200 mg, 2400 mg of pirfenidone or respective placebo doses. The investigators were blinded to the randomization. The primary outcome of the study was change in GFR from baseline to 54 weeks. The study has been completed recently, with results available in the near future (Kumar Sharma, personal communication).
Pirfenidone has been evaluated in clinical trials for various other fibrotic diseases. It has been most extensively studied in patients with idiopathic pulmonary fibrosis, in two open-label studies (n=10, n=54) [2, 3] and one double-blind, placebo-controlled trial (Azuma et al., n=107) [49]. The maximal targeted doses of pirfenidone in these trials ranged between 1800 mg/d to 3600 mg/d with variable duration of treatment, from 1 to 2 years. The two larger studies suggested that pirfenidone stabilizes or improves pulmonary function. Azuma et al. found that pirfenidone therapy significantly improved exercise-induced hypoxemia and that it was associated with decreased episodes of acute exacerbation compared to the placebo group. The randomized trial was terminated at 9 month (3 months prior to the planned date) on ethical grounds, to allow the placebo group to receive pirfenidone. Pirfenidone was also shown to slow the pulmonary functional decline in 21 patients with Hermansky-Pudlak syndrome in a separate randomized, placebo-controlled trial [50].
A randomized, controlled trial (n=43) of pirfenidone has been conducted to assess its efficacy in patients with advanced secondary progressive multiple sclerosis [51]. Twelve-month therapy with pirfenidone was associated with significant reduction in incidence of relapse (28% on placebo vs. 8% on pirfenidone) and marked improvement of bladder function. The investigators concluded that an expanded, multi-center trial of pirfenidone is warranted. Other small, uncontrolled studies have suggested that pirfenidone may also be beneficial in advanced liver fibrosis [52], neurofibromatosis type 1 [53], and radiation-induced fibrosis [54]. Pirfenidone, however, has not been uniformly beneficial in all clinical trials. It was not found to have clinical or histologic benefits in patients with myelofibrosis [55] or primary sclerosing cholangitis, while being associated with increased adverse events [56].
6. Pirfenidone: safety in clinical trials
Most clinical trials describe that pirfenidone is generally well tolerated in doses up to 2400 mg daily (800 mg three times daily). The most common adverse effects include gastrointestinal (nausea, dyspepsia, diarrhea, abdominal discomfort, and vomiting), anorexia, fatigue, sedation, and photosensitivity rash (Table 1). All of these symptoms can be severe enough to cause dose reduction or termination, though many of the symptoms seem to improve with time. The adverse effects of pirfenidone appear to be dose-related and typically resolve completely once the drug is withdrawn. Patients with advanced hepatic dysfunction receiving higher doses of the drug appear to have the greatest difficulty tolerating the medication. The renal function does not appear to correlate with the incidence or severity of adverse events. In addition, many of the gastrointestinal symptoms can be ameliorated by taking the drug with food. The risk of photosensitivity dermatitis can be lessened by avoiding contact with direct sun and wearing protective sunscreen. Pediatric patients with neurofibromatosis, receiving doses comparable to those in adults, also demonstrate similar tolerance. Overall, pirfenidone appears to be reasonably safe in various patient populations with chronic fibrotic disorders, though it is premature to understand the long-term safety or the full range of the adverse effects of the drug.
Table 1.
Pirfenidone safety data from clinical trials
| Author | N | Target Dose (mg/d) |
Study duration (months) |
Mean Age (years) |
Adverse events |
|---|---|---|---|---|---|
| Renal fibrosis | Dyspepsia: 8 (38%), 3 requiring dose reduction, 4 requiring | ||||
| • Cho et al. (2007) [48] |
21 | 2400 | 12 | 47* | anti-reflux therapy; Sedation: 6 (29%), 1 exited protocol; Photosensitive dermatitis: 2 (10%); Transaminitis: 1 (5%) |
| Pulmonary fibrosis | |||||
| • Raghu et al. (1999) [3] |
54 | 3600 | > 12 | 62 | GI: 35 (64%), 2 stopping drug; dermatologic 20 (37%), 4 stopping drug; fatigue 23 (42%) |
| • Nagai et al. (2002) [2] |
10 | 40 mg/Kg |
12 | 65* | Anorexia: 5 (50%); photosensitivity: 3 (30%); fatigue 2 (20%) |
| • Gahl et al. (2002) [50] |
11 | 2400 | 19 (mean) | 42 | GI: 7 (64%); fatigue: 4 (36%); dizziness: 3 (27%), photosensitivity: 1 (9%) |
| • Azuma et al. (2005) [49] |
72 | 1800 | 9 | 64 | GI: 50 (69%) anorexia: 23 (32%); fatigue/sedation: 33 (46%); photosensitivity: 32 (44%); transaminitis: 24 (33%) |
| Hepatic fibrosis | |||||
| • Angulo et al (2002)= [56] |
24 | 2400 | 12 | 42 | GI: 18 (75%); fatigue 11 (46%); photosensitivity 10 (42%) dose reduction or discontinuation in 50% of patients |
| • Armendariz- Borunda et al. 2006) [52] |
15 | 1200 | 12 | 57 | Combined GI and photosensitivity: 15%. Symptoms resolved after 2–3 months of therapy. |
| Neurofibromatosis | |||||
| • Babovic- Vuksanovic et al. (2006) [53] |
24 | 2400 | 24 | 30 | GI: 10 (42%, Treatment terminating toxicity in 3/10); fatigue 2 (8%); dizziness 3 (13%); photosensitivity 1 (4%) |
| • Babovic- Vuksanovic et al. (2007) [31] |
16 | 1500 mg/m2/d | 1 | 11* | GI: Nausea 5 (31%), emesis 9 (56%), diarrhea 3 (19%), abdominal pain 3 (19%), dyspepsia 1 (6%); fatigue 4 (25%); pruritis 1 (6%); palpitation 1 (6%) |
| Multiple Sclerosis | |||||
| • Bowen et al. (2003) [57] |
20 | 2400 | 12 | 48 | GI: Nausea 7 (35%), diarrhea 1 (5%); fatigue/sedation: 4 (20%), rash 1 (5%); irritability 1 (5%); dyspnea: 1 (5%) |
| • Walker et al. (2005) [51] |
25 | 2400 | 12 | 49 | 3 (12%) exited the study due to adverse effects: nausea 1, anorexia 1, and skin rash 1 |
| Myelofibrosis | |||||
| • Mesa et al. (2001) [55] |
28 | 2400 | 12 | 60 | GI: Nausea 8 (29%), diarrhea 3 (11%); rash: 1 (4%); neuropathy 1 (4%) 3 withdrew from study due to drug intolerance. |
median value; = Adverse events were more severe in patients with more severe hepatic dysfunction; GI: gastrointestinal, combined adverse events including nausea, vomiting, abdominal discomfort, dyspepsia, and diarrhea. The number of study size includes only those who were assigned to receive pirfenidone and does not include the placebo arm, when available.
7. Conclusion
Treatment for chronic fibrotic diseases is limited and only partially successful. The currently available data from multiple pilot studies of pirfenidone provide a significant rationale to pursue further studies to better understand this novel antifibrotic agent. Further investigation is needed to better define its role, indications, and contraindications in appropriate patient populations. It may also possibly serve as an adjunctive therapy to slow the progression of disease.
8. Expert Opinion
Identifying specific target of intervention or developing effective means to slow the progression of the fibrotic process has been elusive. Translating laboratory success into clinical practice has been challenging, partly because no experimental models can adequately reproduce the medical environment or account for the extraordinarily complex interdependent regulatory pathways and their redundancy in clinical settings. Pirfenidone, with its anti-inflammatory and antifibrotic effect, offers a new hope in management of chronic and debilitating fibrotic diseases which currently have only limited treatment options. While preclinical data show possible mechanisms which may partially explain the clinical effect of the drug, the clinical trials thus far have been small and often uncontrolled. The trials have encompassed various diseases, endpoints, and duration of follow up. The available data, therefore, are insufficient and inconsistent, making it difficult to draw a meaningful overall conclusion.
Much work remains to better understand the potential therapeutic role of pirfenidone in various patient populations. Some of the remaining research questions include identification of the target diseases, the minimal effective dose, and if pirfenidone can positively impact clinically relevant endpoints such as organ or patient survival. Large, randomized controlled phase II or phase III clinical trials with adequate power are required to answer many of these questions. Given that many of the adverse effects of the drug are dose related, it would be of clinical benefit to know the smallest dose needed to exert therapeutic effect. It is also unclear if the dosing needs to be adjusted in patients with hepatic and renal dysfunction. More pharmacokinetic studies are required to better answer this question, and to have better understanding of the metabolism of the drug and its metabolites. The exact mechanism underlying the antifibrotic and anti-inflammatory effects also needs to be further investigated. Long-term safety data regarding pirfenidone also remain unknown.
Acknowledgements
This work was supported by the Intramural Research Program, NIDDK and NIH. The authors appreciate the critical review of the manuscript by Meryl Waldman.
Footnotes
Declaration of interest:
The authors declare no conflicts of interest.
REFERENCES
- 1.Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125:779–785. [PubMed] [Google Scholar]
- 2.Nagai S, Hamada K, Shigematsu M, Taniyama M, Yamauchi S, Izumi T. Open-label compassionate use one year-treatment with pirfenidone to patients with chronic pulmonary fibrosis. Intern Med. 2002;41:1118–1123. doi: 10.2169/internalmedicine.41.1118. [DOI] [PubMed] [Google Scholar]
- 3.Raghu G, Johnson WC, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label Phase II study. Am J Respir Crit Care Med. 1999;159:1061–1069. doi: 10.1164/ajrccm.159.4.9805017. [DOI] [PubMed] [Google Scholar]
- 4. Antoniu SA. Pirfenidone for the treatment of idiopathic pulmonary fibrosis: therapeutic potential prompts further investigation. Expert Opin Investig Drugs. 2005;14:1443–1447. doi: 10.1517/13543784.14.11.1443. • • Very good and comprehensive review of pirfenidone in idiopathic pulmonary fibrosis.
- 5. Antoniu SA. Pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert Opin Investig Drugs. 2006;15:823–828. doi: 10.1517/13543784.15.7.823. • • Very good review.
- 6. Lasky J. Pirfenidone. IDrugs. 2004;7:166–172. • • Excellent overall review of pirfenidone.
- 7. Schlondorff DO. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int. 2008;74:860–866. doi: 10.1038/ki.2008.351. • Good review to better understand pathophysiology of chronic renal disease.
- 8.Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature. 1990;346:371–374. doi: 10.1038/346371a0. [DOI] [PubMed] [Google Scholar]
- 9.Border WA, Ruoslahti E. Transforming growth factor-beta 1 induces extracellular matrix formation in glomerulonephritis. Cell Differ Dev. 1990;32:425–431. doi: 10.1016/0922-3371(90)90059-6. [DOI] [PubMed] [Google Scholar]
- 10.Sharma K, Ziyadeh FN, Alzahabi B, McGowan TA, Kapoor S, Kurnik BR, et al. Increased renal production of transforming growth factor-beta1 in patients with type II diabetes. Diabetes. 1997;46:854–859. doi: 10.2337/diab.46.5.854. [DOI] [PubMed] [Google Scholar]
- 11.Ziyadeh FN. Role of transforming growth factor beta in diabetic nephropathy. Exp Nephrol. 1994;2:137. [PubMed] [Google Scholar]
- 12. Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. • • Very good review of pathomechanism of fibrosis in the kidney.
- 13.Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P. TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112(Pt 24):4557–4568. doi: 10.1242/jcs.112.24.4557. [DOI] [PubMed] [Google Scholar]
- 14.Schiffer M, Schiffer LE, Gupta A, Shaw AS, Roberts IS, Mundel P, et al. Inhibitory smads and tgf-Beta signaling in glomerular cells. J Am Soc Nephrol. 2002;13:2657–2666. doi: 10.1097/01.asn.0000033276.06451.50. [DOI] [PubMed] [Google Scholar]
- 15.Schnaper HW, Hayashida T, Poncelet AC. It's a Smad world: regulation of TGF-beta signaling in the kidney. J Am Soc Nephrol. 2002;13:1126–1128. doi: 10.1681/ASN.V1341126. [DOI] [PubMed] [Google Scholar]
- 16.Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci U S A. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;291:367–373. [PubMed] [Google Scholar]
- 18.Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;289:211–218. [PubMed] [Google Scholar]
- 19.Tada S, Nakamuta M, Enjoji M, Sugimoto R, Iwamoto H, Kato M, et al. Pirfenidone inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Clin Exp Pharmacol Physiol. 2001;28:522–527. doi: 10.1046/j.1440-1681.2001.03481.x. [DOI] [PubMed] [Google Scholar]
- 20.Nakayama S, Mukae H, Sakamoto N, Kakugawa T, Yoshioka S, Soda H, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008;82:210–217. doi: 10.1016/j.lfs.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Hewitson TD, Kelynack KJ, Tait MG, Martic M, Jones CL, Margolin SB, et al. Pirfenidone reduces in vitro rat renal fibroblast activation and mitogenesis. J Nephrol. 2001;14:453–460. [PubMed] [Google Scholar]
- 22.Iyer SN, Hyde DM, Giri SN. Anti-inflammatory effect of pirfenidone in the bleomycin-hamster model of lung inflammation. Inflammation. 2000;24:477–491. doi: 10.1023/a:1007068313370. [DOI] [PubMed] [Google Scholar]
- 23.Hale ML, Margolin SB, Krakauer T, Roy CJ, Stiles BG. Pirfenidone blocks the in vitro and in vivo effects of staphylococcal enterotoxin B. Infect Immun. 2002;70:2989–2994. doi: 10.1128/IAI.70.6.2989-2994.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arumugam TV, Shiels IA, Margolin SB, Taylor SM, Brown L. Pirfenidone attenuates ischaemia-reperfusion injury in the rat small intestine. Clin Exp Pharmacol Physiol. 2002;29:996–1000. doi: 10.1046/j.1440-1681.2002.03766.x. [DOI] [PubMed] [Google Scholar]
- 25.Nakazato H, Oku H, Yamane S, Tsuruta Y, Suzuki R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-alpha at the translational level. Eur J Pharmacol. 2002;446:177–185. doi: 10.1016/s0014-2999(02)01758-2. [DOI] [PubMed] [Google Scholar]
- 26.Nakanishi H, Kaibori M, Teshima S, Yoshida H, Kwon AH, Kamiyama Y, et al. Pirfenidone inhibits the induction of iNOS stimulated by interleukin-1beta at a step of NF-kappaB DNA binding in hepatocytes. J Hepatol. 2004;41:730–736. doi: 10.1016/j.jhep.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Misra HP, Rabideau C. Pirfenidone inhibits NADPH-dependent microsomal lipid peroxidation and scavenges hydroxyl radicals. Mol Cell Biochem. 2000;204:119–126. doi: 10.1023/a:1007023532508. [DOI] [PubMed] [Google Scholar]
- 28.Shi S, Wu J, Chen H, Chen H, Wu J, Zeng F. Single- and multiple-dose pharmacokinetics of pirfenidone, an antifibrotic agent, in healthy Chinese volunteers. J Clin Pharmacol. 2007;47:1268–1276. doi: 10.1177/0091270007304104. [DOI] [PubMed] [Google Scholar]
- 29.Rubino CM, Bhavnani SM, Ambrose PG, Forrest A, Loutit JS. Effect of food and antacids on the pharmacokinetics of pirfenidone in older healthy adults. Pulm Pharmacol Ther. 2009;22:279–285. doi: 10.1016/j.pupt.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Taniyama M, Ohbayashi S, Narita M, Nakazawa R, Hasegawa S, Azuma N, et al. Pharmacokinetics of an antifibrotic agent, pirfenidone, in haemodialysis patients. Eur J Clin Pharmacol. 1997;52:77–78. doi: 10.1007/s002280050252. [DOI] [PubMed] [Google Scholar]
- 31.Babovic-Vuksanovic D, Widemann BC, Dombi E, Gillespie A, Wolters PL, Toledo-Tamula MA, et al. Phase I trial of pirfenidone in children with neurofibromatosis 1 and plexiform neurofibromas. Pediatr Neurol. 2007;36:293–300. doi: 10.1016/j.pediatrneurol.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu T, Fukagawa M, Kuroda T, Hata S, Iwasaki Y, Nemoto M, et al. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int Suppl. 1997;63:S239–S243. [PubMed] [Google Scholar]
- 33.Shimizu T, Kuroda T, Hata S, Fukagawa M, Margolin SB, Kurokawa K. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int. 1998;54:99–109. doi: 10.1046/j.1523-1755.1998.00962.x. [DOI] [PubMed] [Google Scholar]
- 34.Park HS, Bao L, Kim YJ, Cho IH, Lee CH, Hyun BH, et al. Pirfenidone suppressed the development of glomerulosclerosis in the FGS/Kist mouse. J Korean Med Sci. 2003;18:527–533. doi: 10.3346/jkms.2003.18.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol. 2001;133:687–694. doi: 10.1038/sj.bjp.0704131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.RamachandraRao SP, Zhu Y, Ravasi T, McGowan TA, Toh I, Dunn SR, et al. Pirfenidone is renoprotective in diabetic kidney disease. J Am Soc Nephrol. 2009;20:1765–1775. doi: 10.1681/ASN.2008090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leh S, Vaagnes O, Margolin SB, Iversen BM, Forslund T. Pirfenidone and candesartan ameliorate morphological damage in mild chronic anti-GBM nephritis in rats. Nephrol Dial Transplant. 2005;20:71–82. doi: 10.1093/ndt/gfh562. [DOI] [PubMed] [Google Scholar]
- 38.Duymelinck C, Deng JT, Dauwe SE, De Broe ME, Verpooten GA. Inhibition of the matrix metalloproteinase system in a rat model of chronic cyclosporine nephropathy. Kidney Int. 1998;54:804–818. doi: 10.1046/j.1523-1755.1998.00050.x. [DOI] [PubMed] [Google Scholar]
- 39.Li B, Sehajpal PK, Khanna A, Vlassara H, Cerami A, Stenzel KH, et al. Differential regulation of transforming growth factor beta and interleukin 2 genes in human T cells: demonstration by usage of novel competitor DNA constructs in the quantitative polymerase chain reaction. J Exp Med. 1991;174:1259–1262. doi: 10.1084/jem.174.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolf G, Thaiss F, Stahl RA. Cyclosporine stimulates expression of transforming growth factor-beta in renal cells. Possible mechanism of cyclosporines antiproliferative effects. Transplantation. 1995;60:237–241. doi: 10.1097/00007890-199508000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Khanna A, Kapur S, Sharma V, Li B, Suthanthiran M. In vivo hyperexpression of transforming growth factor-beta1 in mice: stimulation by cyclosporine. Transplantation. 1997;63:1037–1039. doi: 10.1097/00007890-199704150-00026. [DOI] [PubMed] [Google Scholar]
- 42.Dosanjh A. Pirfenidone: anti-fibrotic agent with a potential therapeutic role in the management of transplantation patients. Eur J Pharmacol. 2006;536:219–222. doi: 10.1016/j.ejphar.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 43.Dosanjh A. Pirfenidone: a novel potential therapeutic agent in the management of chronic allograft rejection. Transplant Proc. 2007;39:2153–2156. doi: 10.1016/j.transproceed.2007.07.078. [DOI] [PubMed] [Google Scholar]
- 44.Shihab FS, Bennett WM, Yi H, Andoh TF. Pirfenidone treatment decreases transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am J Transplant. 2002;2:111–119. doi: 10.1034/j.1600-6143.2002.020201.x. [DOI] [PubMed] [Google Scholar]
- 45.Brook NR, Waller JR, Bicknell GR, Nicholson ML. The experimental agent pirfenidone reduces pro-fibrotic gene expression in a model of tacrolimus-induced nephrotoxicity. J Surg Res. 2005;125:137–143. doi: 10.1016/j.jss.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Shihab FS, Bennett WM, Yi H, Andoh TF. Effect of pirfenidone on apoptosis-regulatory genes in chronic cyclosporine nephrotoxicity. Transplantation. 2005;79:419–426. doi: 10.1097/01.tp.0000151721.99418.48. [DOI] [PubMed] [Google Scholar]
- 47.Visner GA, Liu F, Bizargity P, Liu H, Liu K, Yang J, et al. Pirfenidone inhibits T-cell activation, proliferation, cytokine and chemokine production, and host alloresponses. Transplantation. 2009;88:330–338. doi: 10.1097/TP.0b013e3181ae3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho ME, Smith DC, Branton MH, Penzak SR, Kopp JB. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2007;2:906–913. doi: 10.2215/CJN.01050207. [DOI] [PubMed] [Google Scholar]
- 49.Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 50.Gahl WA, Brantly M, Troendle J, Avila NA, Padua A, Montalvo C, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2002;76:234–242. doi: 10.1016/s1096-7192(02)00044-6. [DOI] [PubMed] [Google Scholar]
- 51.Walker JE, Giri SN, Margolin SB. A double-blind, randomized, controlled study of oral pirfenidone for treatment of secondary progressive multiple sclerosis. Mult Scler. 2005;11:149–158. doi: 10.1191/1352458505ms1134oa. [DOI] [PubMed] [Google Scholar]
- 52.Armendariz-Borunda J, Islas-Carbajal MC, Meza-Garcia E, Rincon AR, Lucano S, Sandoval AS, et al. A pilot study in patients with established advanced liver fibrosis using pirfenidone. Gut. 2006;55:1663–1665. doi: 10.1136/gut.2006.107136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Babovic-Vuksanovic D, Ballman K, Michels V, McGrann P, Lindor N, King B, et al. Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology. 2006;67:1860–1862. doi: 10.1212/01.wnl.0000243231.12248.67. [DOI] [PubMed] [Google Scholar]
- 54.Simone NL, Soule BP, Gerber L, Augustine E, Smith S, Altemus RM, et al. Oral Pirfenidone in patients with chronic fibrosis resulting from radiotherapy: a pilot study. Radiat Oncol. 2007;2:19. doi: 10.1186/1748-717X-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mesa RA, Tefferi A, Elliott MA, Hoagland HC, Call TG, Schroeder GS, et al. A phase II trial of pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone), a novel anti-fibrosing agent, in myelofibrosis with myeloid metaplasia. Br J Haematol. 2001;114:111–113. doi: 10.1046/j.1365-2141.2001.02883.x. [DOI] [PubMed] [Google Scholar]
- 56.Angulo P, MacCarty RL, Sylvestre PB, Jorgensen RA, Wiesner RH, LaRusso NA, et al. Pirfenidone in the treatment of primary sclerosing cholangitis. Dig Dis Sci. 2002;47:157–161. doi: 10.1023/a:1013240225965. [DOI] [PubMed] [Google Scholar]
- 57.Bowen JD, Maravilla K, Margolin SB. Open-label study of pirfenidone in patients with progressive forms of multiple sclerosis. Mult Scler. 2003;9:280–283. doi: 10.1191/1352458503ms907oa. [DOI] [PubMed] [Google Scholar]