

Abstract
The pathogenic bacterium Helicobacter pylori produces the cytotoxin VacA, which is implicated in the genesis of gastric epithelial lesions. By transfect ing HEp-2 cells with DNAs encoding either the N-terminal (p34) or the C-terminal (p58) fragment of VacA, p34 was found localized specifically to mitochondria, whereas p58 was cytosolic. Incubated in vitro with purified mitochondria, VacA and p34 but not p58 translocated into the mitochondria. Microinjection of DNAs encoding VacA–GFP and p34–GFP, but not GFP–VacA or GFP–p34, induced cell death by apoptosis. Transient transfection of HeLa cells with p34–GFP or VacA–GFP induced the release of cytochrome c from mitochondria and activated the executioner caspase 3, as determined by the cleavage of poly(ADP–ribose) polymerase (PARP). PARP cleavage was antagonized specifically by co-transfection of DNA encoding Bcl-2, known to block mitochondria-dependent apoptotic signals. The relevance of these observations to the in vivo mechanism of VacA action was supported by the fact that purified activated VacA applied externally to cells induced cytochrome c release into the cytosol.
Keywords: apoptosis/Bcl-2/green fluorescent protein/Helicobacter pylori vacuolating cytotoxin
Introduction
Several studies have provided evidence that the pathogenic bacterium Helicobacter pylori is the major risk factor for gastroduodenal diseases (Atherton, 1997). The vacuolating cytotoxin VacA has been reported to be an important pathogenic factor, although its precise role in H.pylori virulence is still unknown (Cover, 1996; Reyrat et al., 1999). VacA-induced vacuolation has been observed in most epithelial cell types studied, including gastric epithelial cells (Cover, 1996; Reyrat et al., 1999). Vacuoles have been suggested to originate from an intermediate compartment between lysosomes and late endosomes (Molinari et al., 1997). They express the late endosomal marker Rab7 (Papini et al., 1994), and their formation depends on the presence of sub-vacuolating concentrations of weak bases such as ammonia (Cover et al., 1992; Ricci et al., 1997; Sommi et al., 1998). Recently, it has been reported that purified VacA forms pores with a relative selectivity for anions both across planar lipid bilayers (Czajkowski et al., 1999; Iwamoto et al., 1999; Tombola et al., 1999) and in HeLa cell plasma membranes (Szabò et al., 1999). According to Szabò et al. (1999), endocytosed VacA channels might stimulate the turnover of the endosomal V-ATPase by increasing the membrane permeability to anions, leading to accumulation of osmotically active species such as NH4Cl. VacA also decreases the trans-epithelial electrical resistance of polarized epithelial monolayers (Papini et al., 1998; Pelicic et al., 1999). This effect, probably due to the modulation of tight junctions, takes place in the absence of weak bases (Papini et al., 1998; Pelicic et al., 1999), indicating that VacA may exhibit activities other than induction of vacuolation.
In its mature form, VacA has a molecular weight of ∼90 kDa (Cover, 1996). VacA released by H.pylori is often cleaved into two subunits of 34 kDa (the N-terminal fragment ‘p34’) and 58 kDa (the C-terminal fragment ‘p58’), which remain associated by non-covalent interactions (Cover, 1996; Reyrat et al., 1999). Purified VacA toxin forms oligomeric structures of ∼1000 kDa (Lupetti et al., 1996; Cover et al., 1997). Recent work suggests that the toxin is able to bind a cell receptor (Massari et al., 1998) through a segment of the p58 domain (Garner and Cover, 1996; Pagliaccia et al., 1998). Upon binding, VacA could either be endocytosed and accumulated in late endosomes where vacuoles originate (Ricci et al., 1997; Sommi et al., 1998), or translocated across the plasma membrane into the cytosol where it could exert its action (Molinari et al., 1998). Studies addressing the latter mechanism have revealed that vacuolation could be obtained by cytosolic expression of the toxin molecule in HeLa cells (de Bernard et al., 1997, 1998; Vinion-Dubiel et al., 1999; Ye et al., 1999). In these studies the minimal intracellular vacuolating domain of VacA was found to encompass p34 and <200 residues belonging to the C-terminal moiety of p58 (de Bernard et al., 1998; Ye et al., 1999). Very recently a protein (VIP54) co-localizing with the intermediate filament protein vimentin was found, by the yeast two-hybrid screening method, to be a possible cytosolic target of VacA (de Bernard et al., 2000).
To gain further insights into the molecular mechanism of VacA action, we performed cell transfections of DNAs encoding either VacA, p34 or p58 linked to the green fluorescent protein (GFP). Since all data available to date concerning a possible mechanism of action of VacA in the cytosol have been obtained by expression of transfected DNAs using the vaccinia virus system (de Bernard et al., 1997, 1998; Vinion-Dubiel et al., 1999; Ye et al., 1999), we wondered whether one or several modifications induced by infection with the vaccinia virus could have influenced the activity of the toxin in the cytosol. Therefore, we expressed DNA constructs under the control of the cytomegalovirus (CMV) promoter. This technique allowed us to show that p34 is targeted selectively to mitochondria. Using an in vitro mitochondrial import assay, we studied the association of VacA or its fragments with this organelle. Finally, we found that by targeting mitochondria, VacA p34 induces cytochrome c release and apoptosis.
Results
GFP–p34 expressed by transfection localizes to mitochondria
Transfections of the various DNA constructs shown in Figure 1 were performed using a eukaryotic expression vector in which transcription is under the control of the CMV promoter. Expression of the DNAs encoding p34, p58 or VacA, fused at either the N-terminus or the C-terminus to GFP, was performed and the fluorescence was observed by confocal microscopy. No expression of VacA–GFP was observed, and only very low expression of p34–GFP could be detected, exhibiting a dot-like distribution (Table I). In contrast, p58–GFP expression was observed in a large number of cells, with a cytosolic distribution (Table I). Constructs encoding GFP–p34 and GFP–VacA were expressed at higher levels than their C-terminally fused counterparts (Table I). Whereas cells transfected with GFP–p34 displayed GFP-labeled dot-like structures (Figure 2A), as in the rare cells that expressed p34–GFP (Table I), GFP–p58 and GFP–VacA-transfected cells exhibited a cytosolic expression pattern (Figure 2C and E). GFP–p34, GFP–p58 and GFP–VacA specifically reacted with antibodies directed against VacA (Figure 2B, D and F). Immunofluorescence staining for various intracellular compartment markers revealed that the GFP–p34-labeled structures were not early or late endosomes, and they were not derived from the lysosomal compartment, the Golgi apparatus or the endoplasmic reticulum (not shown). When GFP–p34-transfected cells were incubated with purified activated VacA, vacuoles induced by the cytotoxin (in the presence of ammonium chloride) did not co-localize with GFP–p34-positive structures (data not shown). Rather, the GFP–p34 dot-like structures co-localized with the mitochondrial compartment, as revealed by an antibody specifically directed against this organelle (Figure 3). Furthermore, by ultrastructural immunocytochemistry, GFP–p34 was found to be located inside mitochondria (Figure 4). Both single grains and small groups of grains were observed in ∼30% of the cell population, similar to the yield of transfected cells, whereas virtually no labeling could be observed in the remaining 70% of cells. Quantitative evaluation of immunogold deposits (performed on 20 gold-positive cells) revealed the presence of 3.67 ± 0.41 gold particles per square micrometer of mitochondrial area, compared with 0.29 ± 0.04 gold particles per square micrometer of surrounding cytoplasm. Localization of GFP–p34 to mitochondria was also observed in other cell lines such as HeLa or Vero cells (not shown).

Fig. 1. VacA chimeric proteins expressed in HEp-2 cells by DNA transfection. DNA constructs, fused with GFP, encompassing the VacA cytotoxin or various fragments of the toxin were constructed in the pEGFP-C1 and -N1 vectors under the control of the CMV promoter. Numbers refer to mature VacA toxin amino acids (the first 33 amino acids, corresponding to the signal sequence, are not included). p34 and its fragments are represented by empty boxes and p58 by shaded boxes.
Table I. Expression levels and subcellular localization of the different constructs.
| Constructs | Expression levels (% of detectable GFP-positive cells) | Subcellular localization |
|---|---|---|
| p34–GFP | <1 | mitochondria |
| p58–GFP | 17 ± 3 | cytosol |
| VacA–GFP | 0 | – |
| GFP–p34 | 17 ± 3 | mitochondria |
| GFP–p58 | 17 ± 3 | cytosol |
| GFP–VacA | 2 ± 1 | cytosol |

Fig. 2. Intracytoplasmic expression of p34 and p58. Confocal immunofluorescence microscopy of HEp-2 cells transfected with DNA encoding GFP–p34 (A), GFP–p58 (C) or GFP–VacA (E). Expressed proteins were stained with antibodies directed against p34 (B) and the C-terminal part of VacA (D and F). Arrows indicate the presence of non-transfected cells not reacting with specific antibodies. Bar, 10 µm.

Fig. 3. Transfected GFP–p34 localizes to mitochondria. HEp-2 cells, transfected with GFP–p34 (green, A), were stained with the anti-mitochondria mAb 1273 (red, B). Merged images show yellow fluorescence indicating colocalization of the markers (C). Bar, 10 µm.

Fig. 4. Immunolocalization of GFP–p34 by transmission electron microscopy. Immunolocalization was performed on HEp-2 cells transfected with GFP–p34, using anti-GFP antibodies and gold colloidal particles. Arrows indicate the location of gold particles.
In order to delineate sequences involved in mitochondrial localization of GFP–p34, the first 30 codons were deleted from the DNA coding for this VacA fragment (GFP–Δp34). Alternatively, stop codons were introduced after the first 100 or 200 codons of p34 in GFP–p34 DNA (Figure 1) to generate C-terminally truncated proteins. None of the truncated proteins encoded by these DNA constructs was targeted to mitochondria (Table II). These findings suggest that the entire sequence of p34 is essential for the targeting of GFP–p34 to mitochondria.
Table II. Sequence requirements for GFP–p34 localization to mitochondria.
| Constructs | Subcellular localization |
|---|---|
| GFP–p34 | mitochondria |
| GFP–Δp34 | cytosol |
| GFP–Stop100 | cytosol |
| GFP–Stop200 | cytosol |
VacA and p34, but not p58, are imported in vitro into purified mitochondria
The possible mitochondrial targeting of VacA and its fragments, p34 and p58, was studied by a procedure that had previously been used for the import of intrinsic mitochondrial proteins (Rassow, 1999). VacA and its fragments were produced and radiolabeled in vitro with [35S]methionine using a rabbit reticulocyte lysate system. Denaturing polyacrylamide gel electrophoresis demonstrated that VacA and its subunits were produced with the correct molecular weight (Figure 5A). Slow-migrating forms of VacA and p58 were observed under blue native gel electrophoresis conditions (Schägger and von Jagow, 1991), indicating that a sizeable proportion of p58 and VacA, but not of p34, was produced in an oligomeric form in the rabbit reticulocyte lysate (Figure 5B). These proteins were incubated with isolated yeast mitochondria to test their ability to interact with this organelle in vitro. After an incubation period of 5 or 10 min at 25°C, a proteolytic treatment was performed to remove proteins that were not imported. Prior experiments had demonstrated that VacA, p34 and p58 were equally sensitive to treatment with proteinase K, since complete degradation was observed with 10 µg/ml proteinase K (Figure 6A). Under the conditions of our mitochondrial import assay, similar amounts of VacA, p34 and p58 were found to associate with purified mitochondria (Figure 6B). However, after a short treatment with 30 µg/ml proteinase K, only p34 and VacA were resistant to proteinase K treatment (Figure 6B). Protection from proteolysis was specific, as we observed no residual p34 or VacA proteins when reactions were performed in the absence of mitochondria (Figure 6C). A quantification of the import efficiency showed that ∼20% of the initial amounts of both p34 and VacA added to this assay were imported under the experimental conditions (Figure 6C). Similar efficiencies were found with the intrinsic mitochondrial proteins porin or the ADP/ATP nucleotide transporter (AAC) studied under the same conditions (data not shown). Compared with porin and AAC, p34 and VacA penetrated into mitochondria more quickly (Figure 6D).
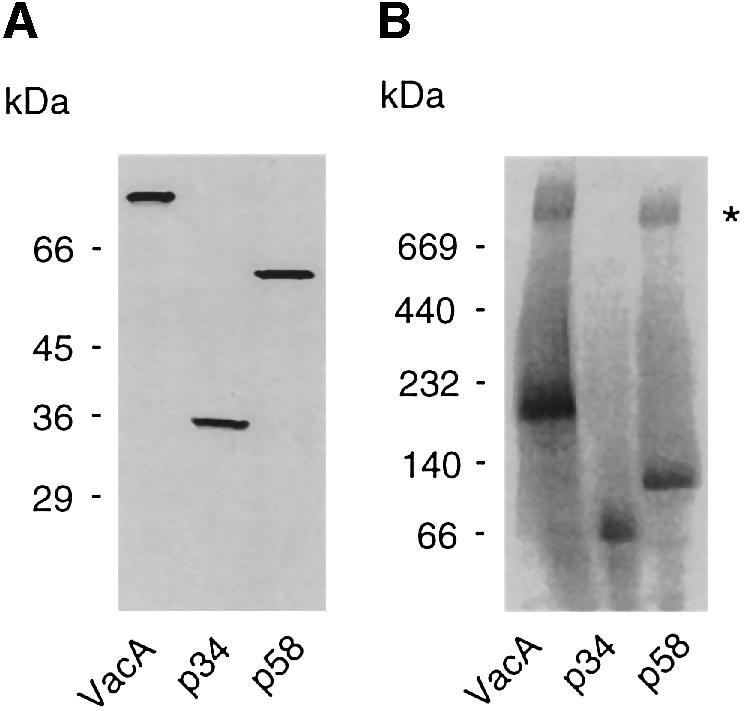
Fig. 5. Denaturing/native blue gel electrophoresis of VacA, p34 and p58 produced in the rabbit reticulocyte system. VacA, p34 and p58 were electrophoretically analyzed upon in vitro synthesis in a rabbit reticulocyte lysate. (A) Under denaturing conditions (12% SDS–PAGE), proteins were observed at their expected molecular weight. (B) The same proteins were also resolved under native conditions (using the blue native–PAGE technique, with a 3–16% acrylamide gradient). This technique allowed the observation of large oligomers formed by VacA and p58 (asterisk).

Fig. 6. Import of p34, p58 and VacA into isolated mitochondria. (A) Sensitivity of p34, p58 and VacA to proteinase K. 35S-labeled p34, p58 or VacA synthesized in reticulocyte lysates was diluted with 10 mM MOPS pH 7.4, and incubated with increasing amounts of proteinase K for 10 min at 0°C. Proteolysis was stopped by addition of PMSF, and the samples were mixed with Laemmli buffer, separated on SDS–PAGE and analyzed using a PhosphorImager. (B) Binding and import of p34, p58 and VacA to mitochondria. Reticulocyte lysates containing p34, p58 or VacA were incubated with yeast mitochondria at 25°C for 5 or 10 min. For the determination of mitochondrial import (right panel), samples were subsequently treated with proteinase K (30 µg/ml) for 10 min at 0°C. The reaction was stopped by the addition of 1 mM PMSF. After re-isolation of mitochondria, samples were separated on SDS–PAGE and analyzed on a PhosphorImager. (C) Specificity of import. Quantification of the efficiency of the mitochondrial import was performed taking into account total amounts of initially added p34, p58 or VacA. The import reaction was performed for 10 min with the same conditions as in (B), in the presence or in the absence of mitochondria. (D) Kinetics of import.The reticulocyte lysates containing p34 (empty circles), VacA (filled circles), the intrinsic mitochondrial proteins porin (empty squares) and AAC (filled squares) were incubated together with mitochondria in import buffer for 1, 2, 5 or 10 min. After a 10 min proteinase K digestion, the samples were analyzed on SDS–PAGE and arelative targeting efficiency was calculated, taking the 10 min import as 100%.
In order to determine the localization of p34 and VacA after their import into mitochondria, we used two different classical procedures for disruption of mitochondrial outer membrane (i.e. to form mitoplasts), swelling and incubation with low doses of digitonin. The marker proteins AAC (a mitochondrial inner membrane protein partially exposed to the mitochondrial intermembrane space) and Mge1 (a marker for the mitochondrial matrix) were used to control the fractionation procedure. In the presence of proteinase K, opening of the outer membrane resulted in a mobility shift of the AAC, due to partial proteolysis of this protein (Figure 7). Swelling experiments and digitonin extraction showed that both imported p34 and VacA were present either free in the mitochondrial matrix or associated with the inner membrane. Indeed, conditions that allowed a near complete rupture of the mitochondrial outer membrane in the absence of matrix opening did not render p34 or VacA accessible to proteinase K (Figure 7).

Fig. 7. Localization of the imported proteins into mitochondria. To localize imported proteins in mitochondria, their outer membranes were disrupted using either swelling, or a digitonin extraction protocol (as described in Materials and methods). (A) Swelling. The amounts of imported p34/VacA remaining protected after proteinase K treatment were determined after formation of mitoplasts. Mitoplasts were obtained with an osmotic shock of 15 min in 1 mM EDTA, 10 mM MOPS pH 7.2. Immunodecoration of marker proteins AAC and Mge1 was performed in order to follow the degree of opening of mitochondrial outer membrane. The accessibility of the inner membrane protein AAC was detected by monitoring the formation of the proteolytic fragment (asterisk). Integrity of the inner membrane was determined using the matrix protein Mge1 as a marker. Amounts of proteinase K-protected proteins were quantified, and the values obtained without swelling were set as 100% (control). (B) Digitonin treatment. Graded concentrations of digitonin (from 0 to 0.5%) were applied for 2 min at 0°C. Percentages of p34, VacA, AAC and porin that were inaccessible to proteinase K were plotted for each digitonin concentration.
VacA–GFP and p34–GFP, but not GFP–VacA or GFP–p34, induce apoptosis
Transfection of VacA–GFP, p34–GFP and GFP–VacA DNAs cloned in the CMV vector resulted in expression of the cytotoxin by very few or no cells (Table I). To determine whether expression of VacA or p34 was deleterious to cells, DNA encoding either VacA–GFP, p34–GFP, GFP–VacA or GFP–p34 was microinjected into HEp-2 cells and the morphology of microinjected cells was observed at different times. A clear cytopathic effect was evident soon after VacA–GFP and p34–GFP microinjection: 6 h after microinjection, 30–40% of microinjected cells exhibited cytoplasmic shrinkage, cell rounding, membrane blebbing (Figures 8D and 9D) and chromatin condensation (Figures 8F and 9F). After overnight expression, VacA–GFP and p34–GFP microinjected cells could not be found, whereas all cells that had been microinjected with DNA encoding GFP expressed the protein, suggesting that cell death and detachment had occurred. In contrast, no apparent toxic effect was observed after GFP–VacA or GFP–p34 microinjection (Figures 8A and 9A). Whereas GFP–p34 expression was detected in all microinjected cells with its fluorescence localized to mitochondria, GFP–VacA was only detectable in a very low percentage of microinjected cells. As observed after transfection (see Figure 2), injection of DNA encoding GFP–VacA resulted in cytosolic expression at very low levels. Given the fact that no toxicity or mitochondrial targeting of this protein could be detected, we examined the possibility that GFP–VacA might be degraded upon synthesis. To do so, HEp-2 cells were treated with either a broad spectrum calpain/proteasome inhibitor (N-acetyl-leucine-leucine-norleucine, MG101) (20 µM) or a specific proteasome inhibitor (lactacystin) (100 µM), prior to their microinjection with GFP–VacA or VacA–GFP plasmids. MG101 and to a lesser extent lactacystin allowed expression of GFP–VacA, but not that of VacA–GFP (data not shown). This strongly suggests that low GFP–VacA expression levels most likely resulted from a degradation of the protein.

Fig. 8. VacA DNA microinjections. HEp-2 cells were microinjected with DNAs encoding GFP–VacA (A–C) or VacA–GFP (D–F). Microinjected cells (white arrows) were observed 6 h after microinjection. The same microinjected preparations were used for phase-contrast observation (A and D) and determination of annexin V binding (B and E). A separate coverslip was used for chromatin staining with DAPI (C and F). Bar, 10 µm.

Fig. 9. p34 DNA microinjections. HEp-2 cells were microinjected with DNAs encoding GFP–p34 (A–C) or p34–GFP (D–F). Microinjected cells (white arrows) were observed 6 h after microinjection. The same microinjected preparations were used for phase-contrast observation (A and D) and determination of annexin V cell binding (B and E). A separate coverslip was used for chromatin staining with DAPI (C and F). Bar, 10 µm.
The cytopathogenic effect observed with p34–GFP or VacA–GFP was clearly reminiscent of an apoptotic process (cell rounding, blebbing and chromatin condensation). Thus, we examined the appearance of apoptotic cells in a population of cells microinjected with GFP–VacA or VacA–GFP constructs, using annexin V binding as a marker (Bossy-Wetzel and Green, 2000). As shown in Figures 8B and 9B, no apoptotic cells could be detected in cells microinjected with GFP–VacA (Figure 8B) or GFP–p34 (Figure 9B). In contrast, a sizeable proportion of cells microinjected with VacA–GFP (Figure 8E) or p34–GFP (Figure 9E) bound annexin V. From these results we concluded that intracellularly expressed VacA induces apoptosis, and that p34 is the active moiety of the toxin in this process.
VacA and p34 induce cytochrome c release and activation of procaspase 3
Since mitochondria play an essential role in the onset of apoptosis (Kroemer and Reed, 2000), we examined whether p34 or VacA might elicit cell death by inserting into mitochondria. Two biochemical events known to accompany the apoptotic process were studied: (i) the release of cytochrome c from mitochondria into the cytosol; and (ii) activation of the executioner caspase 3. HeLa clone X1 cells (Gossen and Bujard, 1992) were used in these experiments for their high yield of transfection. The cytochrome c content of cytosol from HeLa cells transfected with DNA constructs encoding GFP, GFP–p34, GFP–VacA, p34–GFP or VacA–GFP was determined by immunoblotting using a specific antibody directed against this protein. As shown in Figure 10A, cytochrome c was released only into the cytosol of cells transfected with p34–GFP or VacA–GFP. In order to monitor the activation of caspase 3 in cells expressing p34–GFP, we studied the cleavage of the poly(ADP– ribose) polymerase (PARP), known to be a specific target for this caspase (Lazebnik et al., 1994). A construct encoding a fragment of PARP containing the caspase 3 cleavage site was tagged with the c-myc epitope and co-transfected with DNA coding for GFP, p34–GFP or GFP–p34. This technique allowed us to monitor selectively the activation of the executioner caspase in transfected cells. As demonstrated in Figure 10B, p34–GFP, but not GFP or GFP–p34, induced a marked cleavage of PARP, identical to that elicited by UV-B irradiation or staurosporin treatment of cells. Similarly, VacA–GFP (but not GFP–VacA) induced PARP cleavage (data not shown). Transfection of HeLa cells with increasing quantities of p34 DNA revealed that the extent of caspase 3 activation correlated well with the amount of p34 present in mitochondria (data not shown). Activation of terminal procaspases by a mitochondrial pathway is known to be regulated by Bcl-2 family members (Vander Heiden and Thompson, 1999). Indeed, Bcl-2 overexpression efficiently inhibited p34–GFP-induced cleavage of PARP (Figure 10C). In contrast, overexpression of a dominant-negative form of FADD, which blocks apoptosis induced by death receptors (reviewed by Nagata, 1997), failed to block this cleavage (Figure 10C). These observations indicate that p34-induced caspase activation involves a mitochondrial step.

Fig. 10. VacA and p34 induce cytochrome c release and PARP cleavage. (A) Cytochrome c release in the cytosol. HeLa cells were transfected with different VacA constructs. After overnight expression, cells were collected and cytosolic fractions were prepared as described in Materials and methods. Cytochrome c was detected by western blotting. (B) PARP cleavage. HeLa cells were co-transfected with a DNA encoding a c-myc-tagged PARP fragment and either GFP or p34 constructs. After overnight expression, cells were lysed and cytosolic extracts were analyzed for PARP cleavage by immunodetection with the c-myc (9E10) antibody. The arrowhead indicates the size of the intact PARP fragment, whereas the arrow indicates the cleaved PARP fragment. Other bands are non-specific cross-reactants, recognized by the anti-myc 9E10 antibody in control non-transfected cells. (C) Effects of the overexpression, by DNA transfection into HeLa cells, of Bcl-2 or a dominant-negative FADD protein on PARP cleavage induced by p34–GFP transfection. (D) Effect of extracellularly applied purified activated VacA (5 µg/ml for 24 h) on the cytochrome c content of HeLa cell cytosol. For positive controls, apoptosis was induced with either UV irradiation or 1 µg/ml staurosporine as described in Materials and methods.
Finally, to extend our findings, we analyzed the action of extracellularly applied toxin. Purified toxin, activated by a brief acidic pulse (de Bernard et al., 1995; McClain et al., 2000), was applied to HeLa cell cultures at a final concentration of 5 µg/ml. As can be seen in Figure 10D, a clear release of cytochrome c was observed in the cytosol of cells after 24 h of toxin treatment.
Discussion
VacA is the only protein toxin known to be produced and released by the bacterium H.pylori. When administered to mice, this cytotoxin induces gastric epithelial lesions reminiscent of those observed in H.pylori-colonized patients (Telford et al., 1994). In the present study we made the new and unexpected observation that p34, the N-terminal fragment of VacA, targets mitochondria when it is expressed in the cytosol. Localization of p34 into mitochondria was demonstrated to be highly specific, both by confocal immunofluorescence microscopy and by ultrastructural immunochemistry. Despite the fact that we can not completely eliminate the possibility that p34 may interact with other cell compartments, these interactions clearly appear to be very minor. Localization, by immunofluorescence or by ultrastructural microscopy, of VacA subunit p34 in mitochondria upon extracellular application of the cytotoxin is most likely to be technically impossible, since bacterial toxins that act intracellularly introduce a very low number of their active subunits into the cytosol (Yamaizumi et al., 1978; Falnes et al., 2000).
Using an in vitro assay, widely used to study the import of intrinsic mitochondrial proteins, we found that p34 and VacA are translocated into this organelle. Most proteins addressed to the mitochondria contain an N-terminal cleavable polypeptide that is positively charged and comprised of ∼20 amino acids (Roise and Schatz, 1988). However, several other proteins are specifically targeted to mitochondria although they lack this typical signal sequence (Kurz et al., 1999; Rapaport and Neupert, 1999; Schleiff et al., 1999). No classical mitochondrial targeting sequence could be found in p34. Further evidence that no single peptidic sequence was sufficient to promote mitochondrial localization of p34 was brought by the lack of mitochondrial localization of truncated constructs, deleted either at the N- or the C-terminus of p34. Therefore, interaction of p34 with mitochondria requires different polypeptidic segments throughout the molecule. In most cases, the precise elements that confer the ability of mitochondrial proteins to be imported without signal sequences are unknown (Neupert, 1997). Recruitment of p34 to mitochondria indicates that this toxin fragment has an affinity for a component present in this organelle. We have recently observed that p34 and VacA were still imported into purified mitochondria after these organelles had been pretreated with trypsin (data not shown). We have also observed that p34 and VacA attain the internal part of mitochondria, and remain either free in the matrix or associated with the inner membrane.
The mitochondria has been shown to be an essential organelle for apoptosis (Kroemer and Reed, 2000). It is worth noting that previous attempts to express VacA in eukaryotic cells have all relied on the use of the vaccinia virus expression system (de Bernard et al., 1997, 1998; Ye et al., 1999). Vaccinia virus is known to express potent anti-apoptotic activities in infected cells (Gagliardini et al., 1994; Garcia-Calvo et al., 1998), which might play a permissive role in the expression of the cytotoxin. We have observed that microinjection of the DNA encoding p34 or VacA fused at their C-termini to GFP induced the hallmarks of apoptotic cell death, including cell shrinkage and blebbing, chromatin condensation and annexin V staining. The apoptosis-inducing activity of p34 and VacA was found to be strictly dependent on their ability to insert into mitochondria and on the fact that their N-termini were free. After transfection, VacA and p34 were found to induce cytochrome c release from mitochondria and to activate the executioner procaspase 3, as determined by the cleavage of PARP. Release of cytochrome c from mitochondria induces oligomerization of the cytosolic protein Apaf-1, which allows activation of procaspase 9 (Budihardjo et al., 1999). Caspase 9 in turn activates ‘terminal’ procaspases 3, 6 and 7, which are the effectors of cell death (reviewed by Thornberry and Lazebnik, 1998). The fact that expression of Bcl-2 but not a dominantnegative FADD inhibited p34-induced caspase activation further supports our conclusion that mitochondrial localization is responsible for p34-induced apoptosis. It is interesting to compare the sequence requirements on VacA for vacuolation and for promotion of apoptosis following transient expression of the toxin; similar to induction of apoptosis, vacuolation seems to be critically dependent on the N-terminal end of VacA (Vinion-Dubiel et al., 1999; Letley and Atherton, 2000; Ye and Blanke, 2000). However, in contrast to the effect of p34 on induction of apoptosis, vacuolation elicited by intracellular expression of VacA strictly requires a portion of p58 (de Bernard et al., 1998; Ye et al., 1999).
Pro-apoptotic activities of a growing number of bacterial virulence factors are currently being described (reviewed by Weinrauch and Zychlinsky, 1999; Gao and Abu Kaik, 2000). The finding that extracellularly applied VacA is able to cause gastric epithelial cell death has been shown in different model systems (Rudi et al., 1998; Peek et al., 1999). Our present data showing that externally applied VacA induces cytochrome c release appear to be consistent with these reports. However, it is noteworthy that externally applied toxin failed to induce a significant PARP cleavage (data not shown). Accordingly, mitochondrial dysfunctions without overt induction of apoptosis have recently been reported after the application of purified VacA on cultured gastric epithelial cells (Kimura et al., 1999). It is likely that a threshold level of mitochondrial damage is required for cell death (Vander Heiden and Thompson, 1999). Intracellular expression of p34 (or VacA) by transfection, as compared with external application of VacA, results in a higher cytosolic accumulation of this toxin fragment. Rather than being directly fatal for the cell, limited mitochondrial damage induced by VacA released by the bacteria may account for the observed sensitization of gastric epithelial cells to apoptotic stimuli such as CD95 (Rudi et al., 1998). Increased levels of apoptosis have been well documented in the gastric epithelium of patients colonized by H.pylori (reviewed by Shirin and Moss, 1998). This increased and sustained occurrence of cell death probably plays a causal role in the appearance of atrophic gastritis, an established pre-neoplastic condition (Shirin and Moss, 1998). Our data therefore suggest that the cytotoxin might play a role early during the process of gastric carcinogenesis.
VacA applied extracellularly apparently targets mitochondria, since it induces the release of cytochrome c. This indicates that VacA or its p34 moiety is released into the cytosol. Such a translocation could be accomplished in an endosomal compartment, by a mechanism similar to those described for A-B toxins such as diphtheria toxin or the Bacillus anthracis toxic factors (reviewed by Falnes and Sandvig, 2000). In this case, the channel-forming ability of the toxin would be merely a side effect of the membrane translocating activity of the toxin.
Materials and methods
Antibodies and reagents
Anti-mitochondria antibodies mAb 1273 were from Chemicon (Temecula, CA). Polyclonal antibodies against the yeast mitochondrial proteins AAC and Mge1 were raised against the whole proteins after expression and purification from Escherichia coli. Monoclonal anti-cytochrome c antibody (clone 7H8.2C12) was from PharMingen. Purified annexin V and the anti-annexin V rabbit antibody were gifts from Dr Françoise Russo-Marie (INSERM U332, Paris, France). Living Color anti-GFP antibody was from Clontech (Palo Alto, CA) and Texas Red-labeled secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Rabbit polyclonal anti-VacA antibodies (serum 958) directed against the VacA p58 C-terminal portion, and purified VacA toxin were a gift from Dr Timothy L.Cover (Vanderbilt University, TN). The anti-VacA p34 antibody (serum AC2) was a gift from Dr Rino Rappuoli (IRIS Chiron, Siena, Italy). Lactacystin was from Calbiochem (La Jolla, CA). All other reagents were of the highest analytical grade available and were purchased from Sigma (St Quentin Fallavier, France) or Boehringer Mannheim (Mannheim, Germany).
Eukaryotic cell cultures
HeLa clone X1 cells (Gossen and Bujard, 1992) and HEp-2 cells were cultured in DMEM (Bio-Wittaker, Verviers, Belgium) with 7% fetal calf serum (Bio-Media, Boussens, France) and 2 mM l-glutamine (Gibco-BRL, Paisley, UK) at 37°C in an incubator with 5% CO2.
Construction and transfection into HEp-2 cells of DNA encoding GFP–VacA fusion proteins
The eukaryotic expression vectors pEGFP-C1 and pEGFP-N1 (Clontech), encoding GFP under the CMV promoter, were used for easy identification of transfected cells. Starting from the VacA gene cloned into the pQE30 vector (a kind gift from Dr Rino Rappuoli) (Telford et al., 1994), VacA and its fragments (as shown in Figure 1) were amplified with the following primers adding BamHI and XhoI sites: p34–5′, AAAAAAGGATCCGCCTTTTTCACAACCGTGATCATTCCAGCC; p34–3′, AAAAAACTCGAGAGCGCTTTCATTTTTGTCGTTTTTAGCACC; p58–5′, AAAAAAGGATCCGAGAGCAGTCAAAATAATAGTAACACTCAG; p58–3′, AAAAAACTCGAGAGAACGTGCATTGCTAGTGGTGTTTGTGGG; Δp34–5′, AAAAAAGGATCCGGGCTCAAACAAGCCGAAGAAGCCAATAAA.
The amplified DNAs were digested and inserted into the pEGFP-C1 and -N1 vectors. Premature stop codons were introduced at positions 100 and 200 of the GFP–p34 construct using the QuickChange kit (Stratagene, La Jolla, CA), with the following primers: Stop100–5′, GGGCAACAGAATAAGCTTGAATGAGATATGAAAGACGCTGTAGGG; Stop100–3′, CCCTACAGCGTCTTTCATATCTCATTCAAGCTTATTCTGTTGCCC; Stop200–5′, GGGATCACTAGCGATAAAAA CTGAGAAATTTCTCTTTATGATGGT; Stop200–3′, ACCATCATAAAGAGAAATTTCTCAGTTTTTATCGCTAGTGATCCC.
Plasmid DNAs were produced, purified and then transfected into HEp-2 cells using the transfecting reagent ExGen500 (Euromedex, Souffelweyersheim, France) according to the manufacturer’s recommendations, or into HeLa X1 cells using the calcium phosphate precipitation technique (Chen and Okayama, 1987).
Immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde for 20 min, treated with 50 mM NH4Cl for 1 min and permeabilized with 0.5% saponin or 0.1% Triton X-100. The primary antibodies, diluted in phosphate-buffered saline (PBS), were incubated for 30 min with cells. After several washes, Texas Red-labeled secondary antibodies were diluted in PBS and incubated with cells for 30 min. For annexin V staining, cells grown on glass coverslips were incubated for 20 min at room temperature with a concentration of 10 µg/ml of the protein in DMEM containing 5 mM CaCl2, 140 mM NaCl, 25 mM HEPES pH 7.4. The samples were fixed with 4% paraformaldehyde, and an anti-annexin V immunofluorescence was performed. For cell nuclei observation, samples were fixed, permeabilized and stained for 10 min in 50 ng/ml DAPI (4,6-diamidino-2-phenylindole dihydrochloride) (Boehringer Mannheim). Samples were mounted in Mowiol (Calbiochem) and observed with a confocal microscope (Leica TCS-SP, Heidelberg, Germany).
Ultrastructural immunocytochemistry
Ultrastructural immunocytochemistry was performed as described previously (Sommi et al., 1998). Briefly, 24 h after transfection, cell monolayers were washed twice with cacodylate buffer, and fixed with freshly prepared mixture of one part 2.5% glutaraldehyde and two parts 1% osmium tetroxide in cacodylate buffer for 40 min at 4°C. Fixed monolayers were scraped and collected in cacodylate buffer, centrifuged at 10 000 g for 10 min, and embedded in Epon–Araldite mixture. Immunolocalization of GFP–p34 was performed by means of the colloidal gold-labeling technique, using the Living Color anti-GFP antibody as a primary antibody.
Mitochondrial import of VacA and its fragments, p34 and p58
DNAs encoding VacA or the toxin fragments p34 or p58 were cloned into the pET28a plasmid (Novagen). Transcription and translation of these plasmids were performed in a rabbit reticulocyte lysate kit (T7 TNT Quick coupled transcription/translation, Promega) in the presence of [35S]methionine (Redivue, Amersham, UK). To study the mitochondrial targeting of VacA, p34 and p58, we used the classical procedure described for the study of the import of mitochondrial intrinsic preproteins (Rassow, 1999). Briefly, mitochondria, isolated from Sacchromyces cerevisiae as described in Daum et al. (1982) were incubated (20 µg protein/100 µl assay) with reticulocyte lysate containing the toxin in import buffer (3% w/v bovine serum albumin, 250 mM sucrose, 80 mM KCl, 5 mM MgCl2, 2 mM ATP, 2 mM NADH, 10 mM MOPS–KOH pH 7.2) at 25°C for 5–10 min. For protease protection experiments, proteinase K (30 µg/ml) was added to import reactions for 10 min at 0°C. The reaction was stopped by addition of 1 mM phenylmethylsulfonyl fluoride (PMSF). Mitochondria were then pelleted by centrifugation at 16 000 g for 10 min at 2°C, rinsed with SEM (250 mM sucrose, 1 mM EDTA, 10 mM MOPS pH 7.2) and resuspended in sample buffer and submitted to SDS–PAGE separation. Gels were analyzed for radioactivity with a PhosphorImager (Molecular Dynamics).
Mitochondrial subfractionation
Mitochondrial swelling was performed according to Rassow and Pfanner (1991); after import, mitochondria were diluted with five times their volume SEM buffer or EM (i.e. the same buffer without sucrose, in order to induce a hypo-osmotic shock of the mitochondria) in the presence of 30 µg/ml proteinase K. After a 15 min incubation at 0°C, PMSF was added and the mitochondria were pelleted, rinsed and resuspended in sample buffer. Digitonin treatment was performed according to Wienhues et al. (1992); after import, mitochondria were resuspended in SEMK buffer (SEM plus 100 mM KCl). Graded concentrations of digitonin (from 0 to 0.5%) were applied for 2 min at 0°C. A 20-fold dilution in SEMK buffer was performed in order to stop the action of digitonin, and proteinase K was added to all the samples for 10 min at 0°C. PMSF was then added and the mitochondria were pelleted, rinsed and resuspended in sample buffer. After SDS–PAGE, proteins were transferred to nitrocellulose membrane. Quantitation of radiolabeled proteins was performed in parallel with immunodetection of marker proteins AAC and Mge1, allowing an estimation of the degree of mitoplasting.
Microinjection of DNAs into cells
DNAs were microinjected into the nucleus of HEp-2 cells growing on coverslips using an Eppendorf microinjecting system 5171 (Hamburg, Germany). Each DNA was diluted at a final concentration of 40 ng/µl in a 130 mM KCl, 2 mM MgCl2, 50 mM Tris pH 7.4 solution, together with 5 mg/ml rhodamine-labeled dextran (Sigma) to identify microinjected cells.
Assays for cytochrome c release and PARP cleavage
For the determination of cytochrome c release from mitochondria, HeLa cells were transfected with the DNAs encoding different parts of the toxin, and expression was allowed for 24 h. Once collected, cells were washed twice in PBS and incubated for 30 min on ice in lysis buffer [68 mM sucrose, 200 mM mannitol, 50 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol with 1× complete protease inhibitor (Boehringer Mannheim)]. Cells were then lysed with 40 passages through a 25G 5/8 needle, and centrifuged at 1500 g for 10 min. Cytosolic extracts were recovered after centrifugation at 22 000 g for 15 min. For each condition, 20 µg of mitochondrial protein were loaded on a 15% SDS–polyacrylamide gel, and an anti-cytochrome c western blot was performed. For the determination of procaspase 3 activation after transfection with the DNAs encoding VacA p34 fragments, cells were co-transfected with a construct encoding a fragment of the PARP tagged with the myc epitope (J.C.Chambard, personal communication) and allowed to express the proteins for 24 h. Control cells were triggered for apoptosis with UV irradiation (7.5 mJ/cm2) or staurosporin (1 µg/ml), and maintained in culture for 6 h. After cell lysis, cytosolic extracts were prepared and analyzed by 10% SDS–PAGE. PARP cleavage was analyzed by western blotting using the c-myc antibody (Mab 9E10).
Miscellaneous
Standard techniques were used for SDS–PAGE. Immunoblots were performed on nitrocellulose membranes (Pharmacia Biotech). Secondary antibodies coupled to horseradish peroxidase (Sigma) were used and immune complexes were revealed by enhanced chemiluminescence reaction. Blue native gel electrophoresis was performed as described in Schägger and von Jagow (1991). Samples were resuspended in a loading buffer containing 1% (w/v) digitonin, 20 mM Tris–HCl pH 7.4, 50 mM NaCl, 0.1 mM EDTA, 10% (w/v) glycerol, 1 mM PMSF.
Acknowledgments
Acknowledgements
We would like to thank Professor N.Pfanner and the members of his laboratory (Institut für Biochemie und Molekularbiologie, Freiburg, Germany), especially Andreas Geissler and Birgit Schonfish, for their continuous interest and support of the project. We would also like to thank V.Necchi (University of Pavia) for his assistance in ultrastructural immunocytochemistry, and Drs T.L.Cover (Vanderbilt University), R.Rappuoli (IRIS Chiron, Siena, Italy) and F.Russo-Marie (INSERM U332, Paris, France) for providing reagents used throughout this work.
References
- Atherton J.C. (1997) The clinical relevance of strain types of Helicobacter pylori. Gut, 40, 701–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E. and Green,D.R. (2000) Detection of apoptosis by annexin V labelling. Methods Enzymol., 322, 15–18. [DOI] [PubMed] [Google Scholar]
- Budihardjo I., Oliver,H., Lutter,M., Luo,X. and Wang,X. (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol., 15, 269–290. [DOI] [PubMed] [Google Scholar]
- Chen C. and Okayama,H. (1987) High efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol., 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover T.J. (1996) The vacuolating cytotoxin of Helicobacter pylori. Mol. Microbiol., 20, 241–246. [DOI] [PubMed] [Google Scholar]
- Cover T.J., Hanson,P.I. and Heuser J.E. (1997) Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J. Cell Biol., 138, 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover T.L., Vaughn,S.G., Cao,P. and Blaser,M.J. (1992) Potentiation of Helicobacter pylori vacuolating toxin activity by nicotine and other weak bases. J. Infect. Dis., 166, 1073–1078. [DOI] [PubMed] [Google Scholar]
- Czajkowski D.M., Iwamoto,H., Cover,T.L. and Shao,Z. (1999) The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc. Natl Acad. Sci. USA, 96, 2001–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G., Bohni,P.C. and Schatz,G. (1982) Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem., 257, 13028–13033. [PubMed] [Google Scholar]
- de Bernard M., Papini,E., de Filipis,V., Gottardi,E., Telford,J.L., Manetti,R., Fontana,A., Rappuoli,R. and Montecucco,C. (1995) Low pH activates the vacuolating toxin of Helicobacter pylori which becomes acid and pepsin resistant. J. Biol. Chem., 270, 23937–23940. [DOI] [PubMed] [Google Scholar]
- de Bernard M., Aricò,B., Papini,E., Rizzuto,R., Grandi,G., Rappuoli,R. and Montecucco,C. (1997) Helicobacter pylori toxin VacA induces vacuole formation by acting in the cell cytosol. Mol. Microbiol., 26, 665–674. [DOI] [PubMed] [Google Scholar]
- de Bernard M., Burroni,D., Papini,E., Rappuoli,R., Telford,J.L. and Montecucco,C. (1998) Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect. Immun., 66, 6014–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bernard M., Moschioni,M., Napolitani,G., Rappuoli,R. and Montecucco,C. (2000) The VacA toxin of Helicobacter pylori identifies a new intermediate filament-interacting protein. EMBO J., 19, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnes P.O. and Sandvig,K. (2000) Penetration of protein toxins into cells Curr. Opin. Cell Biol., 12, 407–413. [DOI] [PubMed] [Google Scholar]
- Falnes P.O., Ariansen,S., Sandvig,K. and Olsnes,S. (2000) Requirement for prolonged action in the cytosol for optimal protein synthesis inhibition by diphtheria toxin. J. Biol. Chem., 275, 4363–4368. [DOI] [PubMed] [Google Scholar]
- Gagliardini V., Fernadez,P.A., Lee,R.K., Drexler,H.C., Rotello,R.J., Fishman,M.C. and Yuan,J. (1994) Prevention of vertebrate neuronal death by the CrmA gene. Science, 263, 826–828. [DOI] [PubMed] [Google Scholar]
- Gao L.-Y. and Abu Kaik,Y. (2000) The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol., 8, 306–313. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson,E.P., Leiting,B., Ruel,R., Nicholson,D.W. and Thornberry,N.A. (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem., 273, 32608–32613. [DOI] [PubMed] [Google Scholar]
- Garner J.A. and Cover T.L. (1996) Binding and internalization of the Helicobacter pylori vacuolating cytotoxin by epithelial cells. Infect. Immun., 64, 4197–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M. and Bujard,H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto H., Czajkowsky,D.M., Cover,T.L., Szabo,G. and Shao,Z. (1999) VacA from Helicobacter pylori: a hexameric chloride channel. FEBS Lett., 450, 101–104. [DOI] [PubMed] [Google Scholar]
- Kimura M., Goto,S., Wada,A., Yahiro,K., Niidome,T., Hatakeyama,T., Aoyagi,H., Hirayama,T. and Kondo,T. (1999) Vacuolating cytotoxin purified from Helicobacter pylori causes mitochondrial damage in human gastric cells. Microb. Pathog., 26, 45–52. [DOI] [PubMed] [Google Scholar]
- Kroemer G. and Reed,J.C. (2000) Mitochondrial control of cell death. Nature Med., 6, 513–519. [DOI] [PubMed] [Google Scholar]
- Kurz M., Martin,H., Rassow,J., Pfanner,N. and Ryan,M.T. (1999) Biogenesis of Tim proteins of the mitochondrial carrier import pathway: differential targeting mechanisms and crossing over with the main import pathway. Mol. Biol. Cell., 10, 2461–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik Y.A., Kaufmann,S.H., Desnoyer,S., Poirier,G.G. and Earnshaw,W.C. (1994) Cleavage of poly(ADP–ribose) polymerase by a proteinase with properties like ICE. Nature, 371, 346–347. [DOI] [PubMed] [Google Scholar]
- Letley D.P. and Atherton,J.C. (2000) Natural diversity in the N-terminus of the mature vacuolating cytotoxin of Helicobacter pylori determines cytotoxin activity. J. Bacteriol., 182, 3278–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupetti P., Heuser,J.E., Manetti,R., Massari,P., Lanzavecchia,S., Bellon,P.L., Dallai,R., Rappuoli,R. and Telford,J.L. (1996) Oligomeric and subunit structure of the Helicobacter pylori vacuolating cytotoxin. J. Cell Biol., 133, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari P., Manetti,R., Burroni,D., Nuti,S., Norais,N., Rappuoli,R. and Telford,J.L. (1998) Binding of the Helicobacter pylori vacuolating cytotoxin to target cells. Infect. Immun., 66, 3981–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain M., Schraw,W., Ricci,V., Boquet,P. and Cover,T. (2000) Acid activation of Helicobacter pylori vacuolating cytotoxin (VacA) results in toxin internalization by eukaryotic cells. Mol. Microbiol., 37, 433–442. [DOI] [PubMed] [Google Scholar]
- Molinari M., Galli,C., Norais,N., Telford,J., Rappuoli,R., Luzio,J.P. and Montecucco,C. (1997) Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J. Biol. Chem., 272, 25339–25344. [DOI] [PubMed] [Google Scholar]
- Molinari M., Galli,C., de Bernard,M., Norais,N., Ruysschaert,J.M., Rappuoli,R. and Montecucco,C. (1998) The acid activation of Helicobacter pylori toxin VacA: structural and membrane binding studies. Biochem. Biophys. Res. Commun., 248, 334–340. [DOI] [PubMed] [Google Scholar]
- Nagata S. (1997) Apoptosis by death factor. Cell, 88, 355–365. [DOI] [PubMed] [Google Scholar]
- Neupert W. (1997) Protein import into mitochondria. Annu. Rev. Biochem., 66, 863–917. [DOI] [PubMed] [Google Scholar]
- Pagliaccia C. et al. (1998) The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc. Natl Acad. Sci. USA, 95, 10212–10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papini E., de Bernard,M., Milia,E., Bugnoli,M., Zerial,M., Rappuoli,R. and Montecucco,C. (1994) Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc. Natl Acad. Sci. USA, 91, 9720–9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papini E., Satin,B., Norais,N., de Bernard,M., Telford,J.L., Rappuoli,R. and Montecucco,C. (1998) Selective increase of the permeability of polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin. J. Clin. Invest., 102, 813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek R.M.,Jr, Blaser,M.J., Mays,D.J., Forsyth,M.H., Cover,T.L., Song,S.Y., Krishna,U. and Pietenpol,J.A. (1999) Helicobacter pylori strain-specific genotypes and modulation of the gastric epithelial cell cycle. Cancer Res., 59, 6124–6131. [PubMed] [Google Scholar]
- Pelicic V., Reyrat,J.M., Sartori,L., Pagliaccia,C., Rappuoli,R., Telford,J.L., Montecucco,C. and Papini,E. (1999) Helicobacter pylori VacA cytotoxin associated with the bacteria increases epithelial permeability independently of its vacuolating activity. Microbiology, 145, 2043–2050. [DOI] [PubMed] [Google Scholar]
- Rapaport D. and Neupert,W. (1999) Biogenesis of Tom40, core component of the TOM complex of mitochondria. J. Cell Biol., 146, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassow J. (1999) Protein folding and import into organelles. In Higgins,S.J. and Hames,B.D. (eds), Post-translational Processing: A Practical Approach. Oxford University Press, Oxford, UK, pp. 43–94. [Google Scholar]
- Rassow J. and Pfanner,N. (1991) Mitochondrial preproteins en route from the outer membrane to the inner membrane are exposed to the intermembrane space. FEBS Lett., 293, 85–88. [DOI] [PubMed] [Google Scholar]
- Reyrat J.M., Pelicic,V., Papini,E., Montecucco,C., Rappuoli,R. and Telford,J.L. (1999) Towards deciphering the Helicobacter pylori cytotoxin. Mol. Microbiol., 34, 197–204. [DOI] [PubMed] [Google Scholar]
- Ricci V., Sommi,P., Fiocca,R., Romano,M., Solcia,E. and Ventura,U. (1997) Helicobacter pylori vacuolating toxin accumulates within the endosomal-vacuolar compartment of cultured gastric cells and potentiates the vacuolating activity of ammonia. J. Pathol., 183, 453–459. [DOI] [PubMed] [Google Scholar]
- Roise D. and Schatz,G. (1988) Mitochondrial presequences. J. Biol. Chem., 263, 4509–4511. [PubMed] [Google Scholar]
- Rudi J., Kuck,D., Strand,S., Von Herbay,A., Mariani,S.M., Krammer,P.H., Galle,P.R. and Stremmel,W. (1998) Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J. Clin. Invest., 102, 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H. and von Jagow,G. (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem., 199, 223–231. [DOI] [PubMed] [Google Scholar]
- Schleiff E., Silvius,J.R. and Shore,G.C. (1999) Direct membrane insertion of voltage-dependent anion-selective channel protein catalyzed by mitochondrial Tom20. J. Cell Biol., 145, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirin H. and Moss,S.F. (1998) Helicobacter pylori induced apoptosis. Gut, 43, 592–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommi P., Ricci,V., Fiocca,R., Necchi,V., Romano,M., Telford,J.L., Solcia,E. and Ventura,U. (1998) Persistence of Helicobacter pylori VacA toxin and vacuolating potential in cultured gastric epithelial cells. Am. J. Physiol., 275, G681–G688. [DOI] [PubMed] [Google Scholar]
- Szabò I. et al. (1999) Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J., 18, 5517–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford J.L. et al. (1994) Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med., 179, 1653–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry N.A. and Lazebnik,Y. (1998) Caspases: enemies within. Science, 281, 1312–1316. [DOI] [PubMed] [Google Scholar]
- Tombola F. et al. (1999) Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers: possible implications for the mechanism of cellular vacuolation. Biophys. J., 76, 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G. and Thompson,C.B. (1999) Bcl2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nature Cell Biol., 1, E209–E216. [DOI] [PubMed] [Google Scholar]
- Vinion-Dubiel A.D. et al. (1999) A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem., 274, 37736–37742. [DOI] [PubMed] [Google Scholar]
- Weinrauch Y. and Zychlinsky,A. (1999) The induction of apoptosis by bacterial pathogens. Annu. Rev. Microbiol., 53, 155–187. [DOI] [PubMed] [Google Scholar]
- Wienhues U., Koll,H., Becker,K., Guiard,B. and Hartl,F.U. (1992) Protein targeting to mitochondria. In Magee,A.I. and Wileman,T. (eds), Protein Targeting: A Practical Approach. Oxford University Press, Oxford, UK, pp. 135. [Google Scholar]
- Yamaizumi M., Mekada,E., Uchida,T. and Okada,Y. (1978) One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell, 15, 245–250. [DOI] [PubMed] [Google Scholar]
- Ye D. and Blanke,S.R. (2000) Mutational analysis of the Helicobacter pylori vacuolating cytotoxin amino terminus: identification of amino acids essential for cellular vacuolation. Infect. Immun., 68, 4354–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D., Willhite,D.C. and Blanke,S.R. (1999) Identification of the minimal intracellular vacuolating domain of the Helicobacter pylori vacuolating toxin. J. Biol. Chem., 274, 9277–9282. [DOI] [PubMed] [Google Scholar]