

Abstract
The stress-responsive p38 MAPK, when activated by genotoxic stresses such as UV radiation, enhances p53 activity by phosphorylation and leads to cell cycle arrest or apoptosis. Here we report that a member of the protein phosphatase type 2C family, Wip1, has a role in down-regulating p38-p53 signaling during the recovery phase of the damaged cells. Wip1 was originally identified as a gene whose expression is induced following γ or UV radiation in a p53-dependent manner. We found that Wip1 is also inducible by other environmental stresses, such as anisomycin, H2O2 and methyl methane sulfonate. UV-induction of Wip1 requires p38 activity in addition to the wild-type p53. Wip1 selectively inactivates p38 by specific dephosphorylation of its conserved threonine residue. Furthermore, Wip1 expression attenuates UV-induced p53 phosphorylation at Ser33 and Ser46, residues previously reported to be phosphorylated by p38. Wip1 expression also suppresses both p53-mediated transcription and apoptosis in response to UV radiation. These results suggest that p53-dependent expression of Wip1 mediates a negative feedback regulation of p38-p53 signaling and contributes to suppression of the UV-induced apoptosis.
Keywords: dephosphorylation/DNA damage/p38/p53/Wip1
Introduction
Eukaryotic cells respond to DNA damage by activating signal transduction pathways that lead to cell cycle arrest, DNA repair and/or apoptosis. These choices can maximize cellular survival while minimizing the chance of carcinogenesis. One prominent event in the early responses induced by DNA damage is the activation of the stress responsive p38 and JNK MAPK (mitogen-activated protein kinase) cascades (Kyriakis and Avruch, 1996), and another is the activation of the tumor suppressor p53 (Ko and Prives, 1996).
The central core of each MAPK pathway is conserved in all eukaryotic cells, from yeast and plants to mammals, and consists of three protein kinases that are commonly referred to as MAPK, MAPK kinase (MAPKK) and MAPKK kinase (MAPKKK) (Marshall, 1994). An activated MAPKKK phosphorylates and activates a specific MAPKK, which in turn activates its cognate MAPK through phosphorylation of conserved threonine and tyrosine residues. In mammalian cells, at least three distinct MAPK cascades have been identified. While the prototypic MAPKs, ERKs, are activated by mitogenic stimuli, two other types of MAPKs, p38 and JNK, are activated by environmental stresses such as γ-radiation, UV radiation, DNA damaging reagents, osmotic shock and oxidant stresses (Waskiewicz and Cooper, 1995; Kyriakis and Avruch, 1996; Ip and Davis, 1998). Persistent activation of p38/JNK, especially in the absence of mitogenic stimuli, has been shown to induce apoptosis (Chen et al., 1996). Inhibition of p38 and/or JNK activation, either by genetic inactivation, the use of dominant inhibitory mutants or by treatment with their specific inhibitors, confers resistance to cell death induced by diverse stimuli including DNA damage (Zanke et al., 1996; Yang et al., 1997; Tournier et al., 2000).
The tumor suppressor p53 plays a central role in preserving genomic integrity by arresting cell cycle progression or inducing apoptosis after DNA damage and certain other cellular stresses (Levine, 1997). In response to DNA damage, the p53 protein is transiently stabilized and accumulates in the nucleus. DNA damage also activates p53 as a transcription factor, which induces the transcription of several genes, including WAF1, Bax, GADD45 and MDM2 (Gottlieb and Oren, 1996). Both stabilization and functional activation of the p53 protein are regulated by post-transcriptional modifications such as phosphorylation and acetylation (Gu and Roeder, 1997; Giaccia and Kastan, 1998). Phosphorylation at several different serine and threonine residues in p53 occurs after cells are exposed to DNA damaging agents including UV radiation. Several protein kinases are reported to phosphorylate p53, including ATM kinase, ATR kinase, DNA-PK, cyclin dependent kinases, cdk-activating kinase and Chk1/Chk2 (reviewed in Meek, 1998; Prives and Hall, 1999; Caspari, 2000).
In addition to these kinases, p38 MAPK has recently been shown to be a prominent activator of p53 in response to UV radiation (Bulavin et al., 1999; Huang et al., 1999; Keller et al., 1999) and certain anticancer drugs (Sanchez-Prieto et al., 2000). It has been reported that p38 is physically associated with p53 in vivo, and directly phosphorylates p53 on Ser33 and Ser46. Phosphorylation of p53 by p38 at these two sites is crucial for the subsequent p53 phosphorylation at other N-terminal residues including Ser37. Furthermore, UV-induced phosphorylation of p53 by p38 is associated with both activation of p53-mediated transcription and p53-dependent apoptosis (Bulavin et al., 1999). These studies thus indicate that the p38 and p53 cascades cooperate to induce apoptosis in cells exposed to UV. Despite their importance, however, the regulatory mechanism(s) of these two signaling cascades is incompletely understood. In particular, it still remains to be elucidated how p53 and p38 are negatively regulated in the process of cellular recovery from DNA damage. Since both p38 and p53 are activated by phosphorylation, protein phosphatases are likely to play a key role in down-regulation of UV-induced p38-p53 signaling.
The protein phosphatase Wip1 was initially identified as a gene whose expression is induced in response to γ or UV radiation in a p53-dependent manner (Fiscella et al., 1997). Wip1 is a nuclear protein and a member of the serine/threonine specific protein phosphatase type 2C (PP2C) family. Interestingly, induction of Wip1 was observed only in cells with an intact p53, suggesting that the Wip1 gene is a potential downstream target of p53. However, the roles of the Wip1 phosphatase in DNA damage-induced responses remain obscure.
In this study, we demonstrate that not only DNA damage but also other environmental stresses induce the expression of Wip1 mRNA, and that UV-induced Wip1 expression is dependent on both p38 MAPK activity and the wild-type p53 gene. Furthermore, in vivo and in vitro studies indicate that Wip1 selectively dephosphorylates and inactivates p38 in the nucleus, but not JNK, ERK or MAPKKs. The inhibition of p38 by Wip1 attenuates UV-induced phosphorylation of p53 at Ser33 and Ser46, resulting in suppression of p53-mediated transcription and apoptosis. We propose that the p53-inducible protein phosphatase Wip1 mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation.
Results
Wip1 mRNA is inducible by various stresses, and UV induction of Wip1 is regulated by p53 and the p38 MAPK
Initially, we performed northern blot analyses of Wip1 gene expression under various stress conditions. The expression of Wip1 mRNA is known to be induced by γ or UV radiation, but the effects of other environmental stresses have not been examined. For this analysis, we used the p53-intact A549 lung carcinoma cells because radiation-mediated induction of Wip1 is p53 dependent. Consistent with previous reports (Fiscella et al., 1997), expression of Wip1 mRNA was highly inducible by γ and UV radiation in A549 cells (Figure 1A). In addition, we found that Wip1 mRNA was also induced by other stress stimuli including methyl methane sulfonate (MMS), anisomycin and H2O2, whereas high osmolarity had little inducing potential (Figure 1B). Similar results were observed in ML-1, a p53-positive human myeloid leukaemia cell line (data not shown). Thus, Wip1 mRNA is inducible not only by γ or UV radiation but also by a certain subset of environmental stresses.

Fig. 1. Northern blot analysis of Wip1 expression. (A and B) Induction of Wip1 mRNA in the p53-positive A549 cells was monitored by northern blot analysis following: (A) γ-ray (20 Gly) or UV (30 J/m2) radiation, or (B) various stress conditions as described in Materials and methods. (C and D) Absence of Wip1 mRNA induction in the p53-deficient H1299 cells following (C) UV (30 J/m2) radiation or (D) various stress conditions as indicated. Ethidium bromide (EtBr)-stained gels of the same samples are shown at the bottom to indicate the amounts of RNA.
In order to test whether the stress-mediated induction of Wip1 also depends on the wild-type p53, we analyzed Wip1 mRNA levels in H1299, a p53-null lung adenocarcinoma cell line. In H1299 cells, Wip1 was not significantly induced in response to UV radiation (Figure 1C), MMS and H2O2 (Figure 1D). However, apparent Wip1 induction was observed when H1299 cells were stimulated with anisomycin (Figure 1D). These findings suggest that transcription of the Wip1 gene is regulated by both p53-dependent and -independent mechanisms depending on individual stress stimuli.
Since previous studies have reported that p38 MAPK has a pivotal role in UV-induced p53 activation (Bulavin et al., 1999; Huang et al., 1999; Keller et al., 1999), we examined whether the p38 activity is required for the induction of Wip1 by UV radiation, using the specific inhibitor of p38, SB203580 (Hazzalin et al., 1996). A549 cells were exposed to UV in the presence of various concentrations of SB203580. As shown in Figure 2A, the p38 inhibitor significantly reduced the expression of Wip1 mRNA in a dose-dependent manner. The inhibition of p38 by SB203580 was confirmed by monitoring the activity of the endogenous MAPKAP-kinase2 in an in vitro kinase assay (Figure 2B). Because MAPKAP-K2 is directly phosphorylated and activated by p38 in response to UV radiation, MAPKAP-K2 activity reflects UV-induced activation of p38 (Eyers et al., 1999).
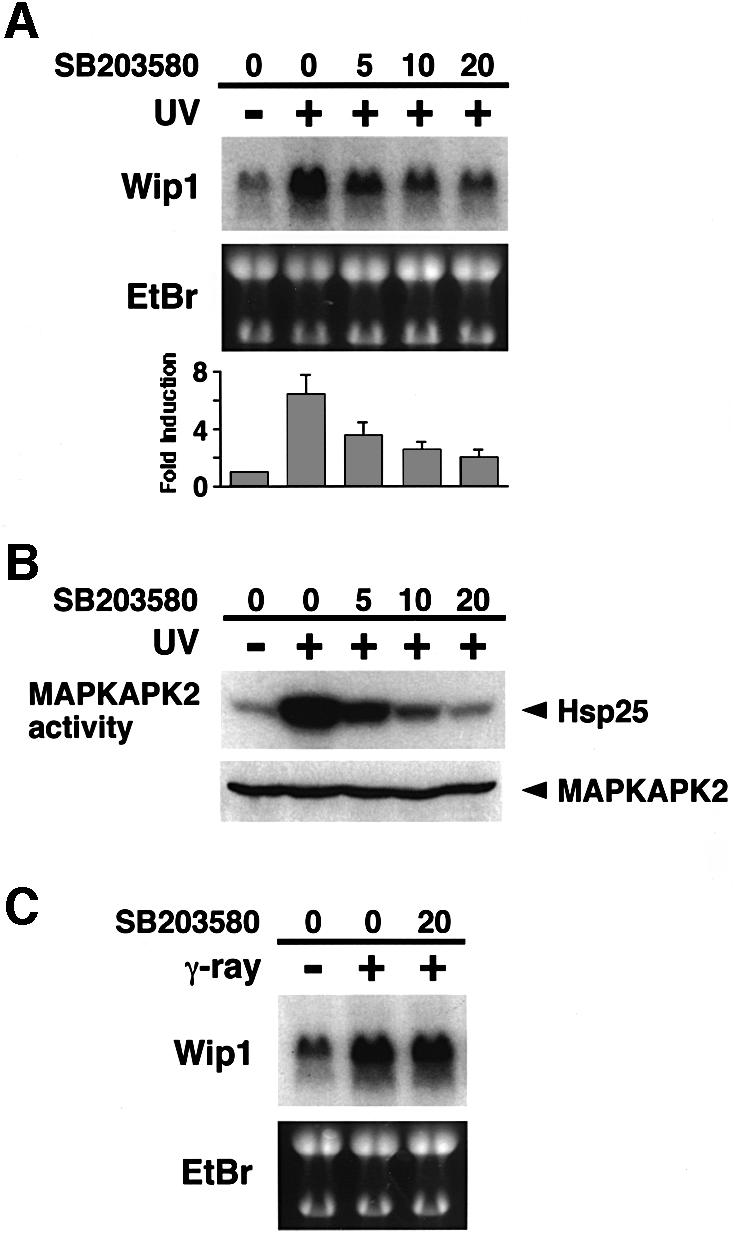
Fig. 2. Effect of the p38-specific inhibitor SB203580 on Wip1 mRNA induction. (A) Inhibition of Wip1 mRNA induction by SB203580. The inhibitor was added to A549 cells at the concentrations indicated (0, 5, 10 or 20 µM) 1 h before exposure to 30 J/m2 UV radiation, and total RNA was prepared 12 h later. The fold induction of Wip1 mRNA was determined by densitometry from three independent experiments and is indicated below each lane. Error bars indicate standard error of the mean. (B) Inhibition of UV-induced MAPKAP-K2 activity by SB203580. A549 cells were treated with SB203580 1 h before irradiation with UV (30 J/m2), and extracts were prepared 1 h later. Endogenous MAPKAP-K2 was immunoprecipitated, and its kinase activity was assessed in an in vitro kinase assay using Hsp25 as a substrate. (C) SB203580 has little effect on γ radiation-induced Wip1 expression. A549 cells were exposed to γ-ray (20 Gly) in the presence or absence of SB203580, and total RNA was prepared 4 h later.
It was demonstrated that, unlike UV radiation, γ-radiation-mediated p53 phosphorylation and transcription is not inhibited by SB203580 (Bulavin et al., 1999; Sanchez-Prieto et al., 2000). If so, SB203580 should not affect γ-radiation-induced Wip1 expression. Consistent with this prediction, SB203580 did not significantly reduce the levels of Wip1 mRNA induced by γ-radiation (Figure 2C). Thus, the inhibition of UV-induced Wip1 expression by SB203580 is not a non-specific effect of the drug. These data strongly suggest that both p53 and p38 are prerequisite for expression of Wip1 in response to UV radiation, and support the notion that UV-induced activation of p53 is, at least partly, mediated by p38.
Wip1 selectively inhibits p38 activation in response to environmental stresses
We tested whether expression of Wip1 has any effect on activation of the stress-responsive p38 and JNK MAPK cascades or on the mitogen-activated ERK MAPK cascade. COS-7 cells were transfected with various amounts of the Wip1 cDNA expression plasmid together with a constant amount of the epitope-tagged MAPK constructs, Flag–p38, HA–JNK1 or HA–ERK2. Trans fected cells were stimulated with either UV radiation or with mitogen, phorbol-12-myristate-13-acetate (PMA). The tagged MAPKs were immunoprecipitated and their kinase activities were determined in an in vitro kinase assay using specific substrates (GST–ATF2 for p38, GST–jun for JNK1 and MBP for ERK2). As shown in Figure 3A, activation of the p38 induced by UV radiation was suppressed in a dose-dependent manner by increasing amounts of the Wip1 expression plasmid. In clear contrast, Wip1 did not have any inhibitory effect on activation of JNK1 and ERK2. We then examined whether Wip1 also inhibits activation of the p38 pathway by any other stress stimuli. As shown in Figure 3B, expression of Wip1 inhibited stimulation of the p38 pathway by anisomycin, sorbitol and MMS, whereas activation of the JNK pathway was not inhibited by Wip1 under any of the conditions used. Thus, the expression of Wip1 selectively inhibits activation of the p38 pathway irrespective of the inducing agent, but not that of the JNK or ERK pathways.

Fig. 3. Selective inhibition of the p38 MAPK pathway by Wip1 in vivo. (A) Wip1 selectively inhibits the p38 MAPK cascade. COS-7 cells were co-transfected with 0.1 µg of expression plasmids encoding Flag–p38, HA–JNK1 or HA–ERK2, with increasing amounts of the pWip1 expression vector as indicated. The total amount of transfected DNA was kept constant by including appropriate amounts of the empty vector pcDNA3. Transfected cells were serum starved for 12 h and stimulated either with UV (60 J/m2 followed by a 1 h incubation at 37°C) or with PMA (100 nM for 20 min). The activities of the epitope-tagged MAPKs were measured in immunocomplex kinase assays using specific exogenous substrates (GST–ATF2, GST–jun or MBP). The expression levels of the MAPKs in cell lysates were monitored by immunoblotting. These experiments were repeated at least three times with similar results. (B) Wip1 inhibits the activation of the p38 pathway caused by distinct stimuli. COS-7 cells were transfected with 0.3 µg/plate of either pWip1 (+) or the empty vector pcDNA3 (–), together with Flag–p38 or HA–JNK1 expression plasmid (0.1 µg). After 24 h, the cells were treated with MMS (100 µg/ml for 3 h), anisomycin (10 µg/ml for 30 min), sorbitol (0.4 M for 20 min) or left untreated (–). The activities and expression levels of Flag–p38 and HA–JNK1 were measured as described in (A). (C) Kinetics of p38 activation in response to UV radiation. A549 cells were treated with UV (30 J/m2) and cell extracts were prepared at various points in time as indicated. The phosphorylation status of endogenous p38 was analyzed by immunoblotting using antibodies specific to phosphorylated p38 (upper panel). The levels of p38 in cell lysates were indicated (lower panel).
In order to explore roles of Wip1 in regulating p38 activity after UV radiation, we examined the kinetics of p38 activation in UV-irradiated A549 cells using anti-phospho-p38 immunoblot analysis. As shown in Figure 3C, p38 was rapidly phosphorylated in response to UV radiation (within 1 h), but p38 phosphorylation was apparently attenuated at 8 h post-treatment. A decline in p38 activity roughly coincided with Wip1 mRNA expression, consistent with the notion that Wip1 plays a role in down-regulation of p38.
Effect of Wip1 on nuclear accumulation of the activated p38 MAPK
Members of the MAPK family are translocated to the nucleus upon activation (Su and Karin, 1996). Thus, we examined whether Wip1 can influence the cellular localization of p38. In these experiments, HeLa or COS-7 cells were transiently transfected with an expression plasmid encoding Flag-tagged Wip1. Cells were then treated with UV radiation and stained with phospho-specific p38 antibodies to monitor the subcellular localization of the endogenous activated p38 protein. The transfected cells that express Flag–Wip1 were specifically detected by indirect-immunofluorescent staining using the anti-Flag antibody M2. As reported previously (Fiscella et al., 1997), Flag–Wip1 was exclusively expressed in the nucleus in HeLa cells (Figure 4A) and Cos-7 cells (data not shown). Essentially the same result was obtained when the green fluorescent protein (GFP)–Wip1 fusion protein was used (data not shown). In response to UV radiation, the activated p38 was primarily localized in the nucleus (Figure 4B). Nuclear accumulation of the activated p38, however, was strongly attenuated in the cells that express the Flag–Wip1 protein (Figure 4B, arrows).

Fig. 4. Wip1 inhibits nuclear accumulation of activated p38 kinase. HeLa cells were transfected with 0.4 µg of an expression plasmid encoding Flag–Wip1. One hour after treatment with UV (60 J/m2), the cells were fixed and stained as follows: (A) Flag–Wip1 was detected with the anti-Flag M2 antibody (red). Some cells are unstained because they do not express the Flag–Wip1 protein. (B) The activated endogenous p38 MAPK was detected by the anti-phospho-specific p38 antisera (green). Cells transfected with Wip1 are indicated by arrows. (C) Flag–Wip1 and activated p38 MAPK are shown simultaneously.
Wip1 interacts with, and inactivates p38 MAPK in vivo
In principle, Wip1 could inhibit the p38 pathway either by directly inactivating p38 or by inactivating upstream kinase(s). To determine the target(s) of Wip1 in vivo, we tested whether Wip1 can specifically inhibit the p38 MAPK or its upstream kinases, such as MAPKKs. To test the effect of Wip1 expression on the activity of MAPKKs, COS-7 cells were co-transfected with a Wip1 expression plasmid together with the GST-tagged MAPKK constructs, GST–MKK6, GST–SEK1 or GST–MEK1. Together, these MAPKKs represent the three major MAP kinase pathways (i.e. the p38, JNK and ERK pathways) (Waskiewicz and Cooper, 1995; Kyriakis and Avruch, 1996). After stimulation of the transfected cells with either UV radiation (in the cases of MKK6 and SEK1) or PMA (for MEK1), the phosphorylation status of the GST–MAPKKs was probed with antibodies specific to the phosphorylated form of each MAPKK. Expression of Wip1 did not significantly dephosphorylate any of the MAPKKs (Figure 5A). Thus, while Wip1 strongly suppressed p38 activation in vivo, no significant inactivation of its direct activator, MKK6, was observed, suggesting that Wip1 inhibits neither the MKK6 MAPKK nor its more upstream elements.

Fig. 5. Direct inactivation of the p38 MAPK by Wip1 in vivo. (A) Wip1 does not inhibit any MAPKK tested in vivo. COS-7 cells were transfected with 0.1 µg/plate of expression plasmids encoding GST–MKK6, GST–SEK1 or GST–MEK1, together with the indicated amounts of pWip1. After 18 h, the cells were serum starved for an additional 12 h and then stimulated with UV or PMA. The phosphorylation status of the affinity-purified GST–MAPKKs was analyzed by immunoblotting using antibodies specific to phosphorylated MKK6, SEK1 or MEK1. The expression level of each GST–MAPKK was also monitored using an anti-GST antibody. (B) Wip1 inhibits p38 activation induced by a constitutively active MKK6 mutant in vivo. COS-7 cells were co-transfected with Flag–p38 (0.1 µg/plate), MKK6DD (0.07 µg/plate) and Wip1 (0.2 or 0.4 µg/plate) expression plasmids as indicated. After 24 h, the kinase activity and the expression level of Flag–p38 were determined as described in Figure 2. The fold activation of kinase activity was determined by densitometrical quantitation and is indicated below each lane. (C) Co-immunoprecipitation of p38 and Wip1 expressed in vivo. COS-7 cells were transfected with 0.2 µg/plate of an expression plasmid encoding HA–p38, together with 0.2 µg of an expression plasmid encoding Flag-tagged Wip1 or vector control (pcDNAI-Flag). Cells were harvested before (–) or 1 h after (+) treatment with UV radiation. Flag–Wip1 was immunoprecipitated, and the presence of HA–p38 in the precipitates was detected by anti-HA antibody 12CA5 immunoblotting (top panel). The levels of HA–p38 (middle panel) and Flag–Wip1 (lower panel) in the total lysates are shown.
The MKK6DD mutant, in which the two activating phosphorylation sites are replaced by aspartate residues, is constitutively active independent of phosphorylation (Takekawa et al., 1998). Using this constitutively active MKK6 mutant, we examined further whether Wip1 directly inactivates p38 in vivo. To test whether Wip1 inhibits the activation of p38 by the constitutively active MKK6DD, COS-7 cells were co-transfected with the MKK6DD and p38 expression plasmids with or without the Wip1 expression plasmid. As seen in Figure 5B, co-expression of Wip1 suppressed MKK6DD-induced p38 activity. Because this suppression of p38 activation cannot be attributed to reduced MKK6DD activity, it is concluded that Wip1 directly affects the activity of p38, most likely through dephosphorylation of the activated p38.
We then tested whether Wip1 forms a specific physical complex with p38. For this purpose, Flag-tagged Wip1 and HA-tagged p38 were co-expressed in COS-7 cells, and Flag–Wip1 was immunoprecipitated before or after UV radiation. Immunoprecipitates were then probed with an anti-HA antibody to detect co-precipitated HA–p38. As shown in Figure 5C, HA–p38 was associated with Flag–Wip1 only after UV radiation, suggesting that Wip1 preferentially binds phosphorylated p38 with high affinity. Together, these findings indicate that Wip1 selectively inactivates the p38 pathway at the level of MAPK.
Wip1 dephosphorylates the p38 MAPK at a threonine residue in vitro
We then examined whether p38 can serve as a direct substrate for the Wip1 phosphatase using an in vitro phosphatase assay. Initially, we tested whether purified GST–Wip1 can inactivate MAPKs (p38, JNK1 and ERK2) in vitro, since MAPK activity reflects their phosphorylation status. In order to prepare phosphorylated MAPKs, COS-7 cells were transfected with HA–p38, HA–JNK1 or HA–ERK2 expression plasmids, and stimulated with UV radiation (in the case of p38 and JNK1) or PMA (for ERK2). The activated HA–MAPKs were immunopurified, and incubated with GST–Wip1 in vitro in the presence or absence of Mg2+ because, like other PP2C enzymes, Wip1 requires Mg2+ or Mn2+ for its activity (Fiscella et al., 1997). After incubation, the remaining kinase activities were measured in vitro using specific substrates. As seen in Figure 6A, Wip1 efficiently inactivated p38 in a Mg2+-dependent manner. In contrast, JNK1 and ERK2 activities were resistant to the action of the Wip1 phosphatase. These results are consistent with the in vivo results demonstrating that Wip1 directly dephosphorylates p38 with a high specificity.
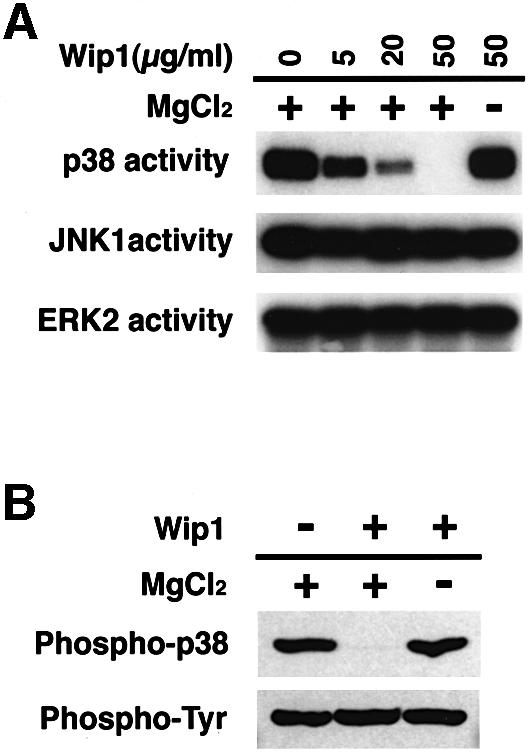
Fig. 6. In vitro dephosphorylation of the p38 MAPK by Wip1. (A) p38 can serve as a direct substrate of Wip1. Activated MAPKs were isolated from UV- or PMA-treated COS-7 cells and incubated with the indicated concentrations of purified GST–Wip1 in the presence or absence of Mg2+. Residual kinase activities were assessed in an in vitro kinase assay using specific substrates (GST–ATF2 for p38, GST–jun for JNK1, MBP for ERK2). (B) Threonine-specific dephosphorylation of p38 by Wip1 in vitro. Phosphorylated GST–p38 was affinity-purified from UV-treated COS-7 cells and incubated with purified GST–Wip1 in the presence or absence of Mg2+. The phosphorylation state of GST–p38 was examined by immunoblotting with either anti-phospho-p38 antibodies (upper panel) or anti-phosphotyrosine antibody 4G10 (lower panel).
MAPK activity is dependent on phosphorylation at both a threonine and a tyrosine residue (Anderson et al., 1990; Gómez and Cohen, 1991). Because dephosphorylation of either site alone is sufficient to inactivate MAPKs, the inactivation of p38 by Wip1 could be mediated by dephosphorylation of either the threonine or the tyrosine residue. To determine which residue on the p38 is dephosphorylated by Wip1, we used anti-dual phospho-p38 and anti-phosphotyrosine antibodies in an immunoblot analysis. Dual-phosphorylated GST–p38 reacted with both antibodies as anticipated (Figure 6B, left lane). After incubation with GST–Wip1 in vitro, however, GST–p38 only reacted with the anti-phosphotyrosine antibody, indicating that the Wip1 phosphatase dephosphorylated p38 at the threonine residue, but not at tyrosine. Thus, these data strongly suggest that Wip1 dephosphorylates p38 at the essential threonine residue.
Wip1 expression down-regulates phosphorylation of the p53 tumor suppressor protein in response to UV radiation
It has been reported recently that p38 directly phosphorylates and thereby functionally activates the tumor suppressor p53 protein at Ser33 and Ser46 after exposure of cells to UV (Bulavin et al., 1999). Because the expression of Wip1 inhibited UV-induced activation of p38, we sought to determine whether Wip1 expression also leads to inhibition of the p53 activity. Initially, COS-7 cells were transfected with the Wip1 cDNA expression plasmid together with the Flag-tagged p38 construct, and stimulated with UV radiation. p38 activity in immunoprecipitates was then assayed in vitro with GST–p53, instead of GST–ATF2, as a substrate. Consistent with our result shown in Figure 3A, co-expression of Wip1 strongly inhibits phosphorylation of GST–p53 by p38 (Figure 7A).

Fig. 7. Wip1 inhibits phosphorylation of p53 induced by UV radiation. (A) In vitro phosphorylation of p53 by p38. COS-7 cells were cotransfected with 0.1 µg of the expression plasmid encoding Flag–p38, with 0.3 µg of the empty vector or the pWip1 expression vector. Transfected cells were stimulated with UV, and kinase activity of Flag–p38 was assayed in an immunocomplex kinase assay as in Figure 3A, except that GST–p53 was used as a substrate. (B) Wip1 blocks phosphorylation of p53 at Ser33 and Ser46 induced by UV radiation. The p53-deficient H1299 cells were transiently transfected with 0.1 µg of an expression vector encoding wild-type p53, together with 0.3 µg of either the pWip1 expression vector or the empty vector pcDNA3 (Vector). After 18 h, cells were either treated with UV (25 J/m2) (+) or left untreated (–). Seven hours after UV treatment, cell extracts were prepared and analyzed for p53 phosphorylation by immunoblot analysis using anti-p53 antibodies specific for phosphorylation at Ser33, Ser46 or Ser392 as indicated. The levels of p53 expression in the samples were monitored using a p53-specific mAb, DO-1. (C) Wip1 does not dephosphorylate p53 at Ser33 and Ser46 in vitro. Phosphorylated p53 was immunopurified from UV-treated H1299 cells transfected with wild-type p53 expression vector. Following incubation with purified GST–Wip1, in the presence or absence of Mg2+, the phosphorylation state of p53 was examined by immunoblotting using antibodies specific for the phosphorylated Ser33 or Ser46 sites of p53. (D) Time-course of p53 phosphorylation at Ser33 and Ser46 in A549 cells. Phosphorylation status of p53 was determined in cell extracts from A549 cells treated with UV (30 J/m2) by immunoblotting using antibodies specific for p53 Ser33 (upper panel) and Ser46 (middle panel) phosphorylation sites, respectively. The levels of p53 expression in the samples were indicated (lower panel). (E) Effect of SB203580 on p53 phosphorylation at Ser33 and Ser46 in response to UV radiation. p38-specific inhibitor SB203580 were added to A549 cells 1 h before irradiation with UV (30 J/m2), and extracts were prepared 12 h later. Endogenous wt-p53 was analyzed for phosphorylation by immunoblotting as in (D). The amount of p53 in cell lysates was indicated (lower panel).
Next, we analyzed the effect of Wip1 on UV-induced phosphorylation of p53 at Ser33, Ser46 and Ser392 using site- and phosphorylation-specific antibodies; these residues were previously reported to be phosphorylated after UV radiation (Lu et al., 1998). p53-null H1299 cells were transiently co-transfected with a plasmid carrying wild-type human p53 together with the Wip1 expression plasmid or an empty plasmid pcDNA3. As shown in Figure 7B, co-expression of Wip1 significantly reduced UV-induced phosphorylation of p53 at Ser33 and Ser46, but not at Ser392.
The Wip1 phosphatase may suppress p53 phosphorylation in vivo, either directly by dephosphorylating p53 or indirectly by inhibiting the p38 pathway. To distinguish these possibilities, we examined the ability of Wip1 to dephosphorylate p53. The phosphorylated p53 was immunopurified from UV-irradiated H1299 cells transfected with the p53 expression vector, and incubated in vitro with GST–Wip1. The phosphorylation status of p53 was then monitored by immunoblotting using the phospho-specific antibodies. GST–Wip1 did not dephosphorylate p53 at either Ser33 or at Ser46 in vitro (Figure 7C). Thus, it is likely that Wip1 indirectly attenuates UV-induced phosphorylation of p53 on Ser33 and Ser46.
We found that phosphorylation of p53 at Ser33 and Ser46 occurred within 4 h of UV treatment of A549 cells, reaching a maximum at 8–12 h (Figure 7D), almost at the same time as Wip1 induction (Figure 1A), and then decreased. Next, we analyzed the effect of p38 inhibitor SB203580 on p53 phosphorylation at Ser33 and Ser46 in UV-treated A549 cells. Phosphorylation of p53 on Ser33 was shown to be suppressed by treatment of the cells with SB203580 (Bulavin et al., 1999; Sanchez-Prieto et al., 2000). However, the effects of SB203580 on UV-induced p53 phosphorylation on Ser46 have not been examined. As shown in Figure 7E, SB203580 significantly reduced p53 phosphorylation on Ser33, whereas the inhibitor had little effect on UV-induced p53 phosphorylation at Ser46 (see Discussion). Taken together with our results that Wip1 expression inhibited p38 activation and p53 phosphorylation, these data suggest that p38 participates in the phosphorylation of p53, at least on Ser33, in response to UV radiation, and that Wip1 reduces p53 phosphorylation, probably through inhibition of p38.
Wip1 expression reduced p53-mediated transcription and apoptosis in response to UV radiation
Next, we asked whether Wip1 expression affects p53-mediated transcription and apoptosis after UV induction, because p53 phosphorylation on Ser33 or Ser46 is known to be essential for functional activation of p53 (Bulavin et al., 1999; Oda et al., 2000; Sanchez-Prieto et al., 2000). To generate a negative control for the following experiments, we first constructed a catalytically inactive Wip1 mutant by altering the conserved Asp314 to an Ala, because a corresponding substitution mutation in murine PP2Cβ inactivated its phosphatase activity (Hanada et al., 1998). Indeed, expression of the Wip1(D/A) mutant did not inhibit the activation of p38 in vivo under the same conditions whereby expression of the wild-type Wip1 robustly blocked p38 activation (Figure 8A).
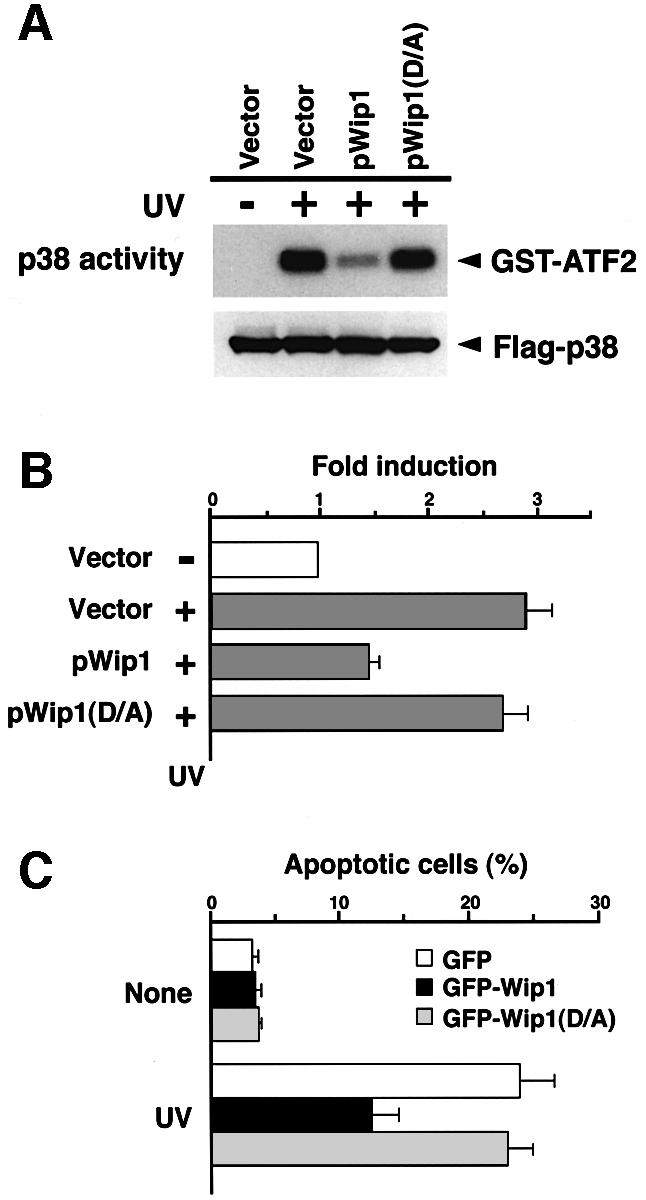
Fig. 8. Wip1 prevents p53-mediated transcription and apoptotic cell death after exposure to UV radiation. (A) p38 activation is inhibited by the wild-type Wip1 phosphatase, but not by the catalytically inactive Wip1(D/A) mutant. COS-7 cells were co-transfected with 0.1 µg/plate of the Flag–p38 plasmid and 0.3 µg of an expression plasmid encoding Wip1 or Wip1(D/A), or their vector control pcDNA3 (Vector). After 24 h, the transfected cells were treated with UV (60 J/m2), and p38 activity was assayed as in Figure 3. The level of Flag–p38 in the total extracts is shown in the lower panel. (B) Suppression of p53-mediated transcription by Wip1. MCF7 cells, which express intact p53, were transiently co-transfected with a p53–luciferase reporter plasmid pGL3-p53RE (0.1 µg) and pCMV β-galactosidase expression vector (0.07 µg), with 0.3 µg of either the control pcDNA3 (Vector), the pWip1 expression vector or the pWip1(D/A) expression vector. After 18 h, the cells were treated with UV (25 J/m2) (+) or left untreated (–). Seven hours after irradiation, luciferase activity was determined. The data represents the average fold-induction of luciferase activity from three independent experiments, in which each assay was performed in duplicate. Error bars indicate standard errors of the mean. (C) Wip1 prevents p53-mediated apoptotic cell death induced by exposure to UV radiation. A549 cells were transfected with either the pGFP expression vector, pGFP-Wip1 or pGFP-Wip1(D/A). Eighteen hours after transfection, the transfected cells were treated with UV (15 J/m2) or left untreated (None). Sixteen hours after UV treatment, the cells were fixed and their nuclei were stained with DAPI. The fraction of apoptotic cells was determined by dividing the number of GFP-positive cells that exhibit apoptotic morphology (round cell shape with condensed nucleus) by the total number of GFP-positive cells. At least 500 GFP-positive cells from five randomly chosen fields were counted. The averages of three independent experiments are shown.
Initially, to ascertain a role of Wip1 in p53-dependent transcription, a luciferase reporter plasmid driven by the p53-responsive element pGL-p53RE was co-transfected with either an empty vector (pcDNA3), wild-type Wip1 (pWip1) or mutant Wip1 [pWip1(D/A)] into MCF7 cells, which harbor the wild-type p53. A CMV-driven β-galactosidase construct was utilized as an internal control for normalization of luciferase activities. Seven hours after exposure to UV light, cells were harvested for luciferase assays. As shown in Figure 8B, UV radiation stimulated the p53-dependent luciferase activity only when the empty vector was present. In contrast, expression of the wild-type Wip1 resulted in a marked decrease in the UV-induced luciferase activity. This inhibitory effect of Wip1 is dependent on its phosphatase activity, because expression of a catalytically inactive mutant Wip1(D/A) did not significantly suppress the p53-dependent luciferase activity. Similar results were also observed when another p53-positive cell, A549, was used (data not shown).
To examine the role of Wip1 in regulating p53-mediated apoptosis, A549 cells were transiently transfected with expression plasmids for GFP–Wip1 or GFP–Wip1(D/A) fusion protein. GFP allows for specific detection of transfected cells by its green fluorescence. Twelve hours after treatment of the cells with UV radiation, we examined cell death as determined from nuclear morphology after 4′-6-diamidine-2-phenylindole (DAPI) staining. UV-induced apoptosis was significantly reduced by expression of GFP–Wip1, compared with cells transfected with the control GFP vector or the catalytically inactive Wip1(D/A) expression vector (Figure 8C). These data indicate that Wip1 may play an important role in the negative regulation of p53-mediated apoptosis as well as p53-dependent transcription in response to UV radiation.
Discussion
The activity of MAPK reflects a balance between the upstream activating kinases and inactivating protein phosphatases. Since phosphorylation at both a specific threonine and a specific tyrosine residue is required for MAPK activity, MAPK inactivation can be achieved by either dual-specificity phosphatases, tyrosine-specific phosphatases (PTPases) or serine/threonine-specific protein phosphatases. Both dual-specificity phosphatases and PTPases were shown to inactivate various members of the MAPK family (reviewed in Keyse, 2000). Among the Ser/Thr-specific protein phosphatases, the type 2A protein phosphatase (PP2A) was shown to dephosphorylate and inactivate ERK1/ERK2 MAPKs and MEK1/MEK2 MAPKKs (Gómez and Cohen, 1991; Alessi et al., 1995).
In addition to these phosphatases, recent work has shown that another type of Ser/Thr specific phosphatase, namely the type 2C protein phosphatases (PP2C), down-regulates the stress-responsive MAPK pathways in budding yeast, fission yeast, plants and mammals (Maeda et al., 1994; Shiozaki et al., 1995; Hanada et al., 1998; Meskiene et al., 1998; Takekawa et al., 1998). In human cells, PP2Cα inhibits both the p38 and JNK pathways by dephosphorylating MAPKKs (MKK6 and SEK1) and a MAPK (p38) (Takekawa et al., 1998; Fjeld and Denu, 1999). Because the PP2Cα gene is expressed constitutively, and because PP2Cα is evenly localized both in the cytoplasm and the nucleus, its major function may be to maintain low MAPK activities in the absence of external stimuli.
In this study, we initially observed that various stress conditions led to increased expression of Wip1 mRNA. Our results also demonstrated that Wip1 specifically dephosphorylates and inactivates p38 MAPK, both in vivo and in vitro. In addition, Wip1 was found to form a physical complex with p38 in vivo. Unlike PP2Cα, Wip1 is localized predominantly in the nucleus, and the expression of the Wip1 gene is highly inducible by DNA damage and other environmental stresses. Another important difference between Wip1 and PP2Cα is that the Wip1 phosphatase has a much narrower substrate specificity than PP2Cα. Wip1 displays little activity toward MAPKKs (MKK6, SEK1 and MEK1) in vivo, or toward JNK and ERK MAPKs. Thus, Wip1 specifically inactivates the p38 MAPK pathway, while PP2Cα inhibits both the JNK and p38 pathways. Our results provide the first evidence that members of the human PP2C family enzymes differentially regulate the stress responsive MAPK pathways.
Recent studies in the fission yeast (Schizosaccharo myces pombe) have revealed that, like the human Wip1, a yeast PP2C, Ptc1, is induced by stress and its expression depends on the stress-responsive Spc1 MAPK, a homolog of human p38 (Gaits et al., 1997). However, another yeast PP2C, Ptc3, like human PP2Cα, is constitutively expressed. Ptc1 and Ptc3 directly dephosphorylate the conserved Thr residue of Spc1, resulting in the attenuation of Spc1 activity. Interestingly, Nguyen and Shiozaki (1999) showed that Pyp1, the major tyrosine specific phosphatase that dephosphorylates and inactivates Spc1, is inhibited in heat-shocked cells, and, under such conditions, Ptc1 and Ptc3 are responsible for inactivation of Spc1. Similar negative regulatory roles were proposed for PP2C in the budding yeast stress-activated Hog1 MAPK cascade (Maeda et al., 1994; Wurgler-Murphy et al., 1997). Given the high level of conservation of MAPK pathways from yeast to mammals, these findings thus suggest that human PP2C enzymes may also play a central role in negative regulation of the p38 pathway, particularly when other MAPK phosphatases are not available.
Genotoxic stress agents, such as UV radiation, activate p53 as well as the p38 MAPK pathway (Kyriakis and Avruch, 1996; Giaccia and Kastan, 1998). Recent studies have shown that p38 plays a prominent role in UV-induced activation of p53 through its phosphorylation at Ser33 and Ser46. In this report, we demonstrate that Wip1 expression attenuates UV-induced phosphorylation of p53 on Ser33 and Ser46. However, purified GST–Wip1 was unable to dephosphorylate these residues in vitro, suggesting that Wip1 only indirectly effects dephosphorylation of p53 on Ser33 and Ser46. It is thus likely that Wip1 attenuates p53 phosphorylation at these sites through inhibition of p38.
While phosphorylation of p53 on Ser33 is inhibited by either SB203580 treatment or the Wip1 expression, our data revealed that Ser46 is inhibited only by Wip1 expression but not by the p38 inhibitor (Figure 7B and E). This apparent discrepancy may be due to the distinct sensitivities of p38 isoforms (p38α, -β, -γ and -δ) to the p38 inhibitor. Individual p38 isoforms have distinct substrate specificities and sensitivities to SB203580: p38α and -β are inhibited by SB203580, while p38γ and -δ are insensitive to the drug (Goedert et al., 1997). Thus, it is possible that p53 Ser46 is mainly phosphorylated by SB253080-insensitive p38 isoforms, although we cannot exclude the possibility that Ser46 is phosphorylated by another, hitherto unidentified kinase.
Another residue, Ser389 of murine p53 (equivalent to Ser392 in human), has also been described recently as a site for p38 phosphorylation (Huang et al., 1999; Keller et al., 1999). However, whether Ser392 is phosphorylated by p38 remains controversial. Bulavin et al., (1999) reported that Ser392 of human p53 is not phosphorylated by p38 in vitro. In addition, we observed that Wip1 did not significantly reduce phosphorylation of Ser392 in vivo (Figure 7B). Moreover, this site is not adjacent to a proline residue, representing an unlikely candidate for direct phosphorylation by proline-directed kinases such as p38. Thus, our data suggest that a p38-independent mechanism(s) may participate in the UV-induced phosphorylation of Ser392, at least in human cells.
Because Wip1 expression is induced in response to UV radiation by a process that requires both p38 and p53, our data indicate that Wip1 may mediate a negative feedback regulation of the p38-p53 signaling pathway. Thus, Wip1 may play an important role in negative regulation of DNA damage-induced apoptosis, in which both p38 and p53 have been shown to be involved (Levine, 1997; Kang et al., 2000; Sanchez-Prieto et al., 2000), through preventing both prolonged activation of p38 MAPK and hyperactivation of p53. Consistent with this model, we also showed that expression of Wip1 inhibits p53-mediated transcription and apoptosis in response to UV radiation. Eukaryotic cells respond to DNA damage by inducing temporary cell cycle arrest and DNA repair if the level of damage is such that it can be corrected without inflicting too many mutations. If, however, damage is too extensive, cells will undergo apoptotic cell death, which might be induced by prolonged activation of p38 and p53. Thus, the Wip1 phosphatase may play a critical role in making a fateful choice between cell survival and apoptotic cell death, by down-modulating the activities of p38 and p53.
Materials and methods
Media and buffers
Lysis buffer, phosphatase buffer, kinase buffer and immunoprecipitation (IP) buffer have been described previously (Takekawa et al., 1998).
Plasmids
The human Wip1 coding region was amplified by PCR using human placenta cDNA (Clontech) as a template, and subcloned into the mammalian expression plasmid pcDNA3 (Invitrogen) to generate pWip1. The phosphatase-deficient mutant Wip1(D/A) was generated by replacing the Asp314 with an Ala codon using a PCR mutagenesis method (Takekawa et al., 1998). A PCR-based procedure was used to attach a Flag-tag sequence to the N-terminus of the Wip1 ORFs to generate pFlag-Wip1 or pFlag-Wip1(D/A), respectively. GFP fusion constructs were made using the pEGFP-C3 vector (Clontech). Mammalian expression plasmids pMT3-HA-p38, pSRα-HA-JNK1, pSRα-HA-ERK2, pCMV-Flag-p38, pEBG-p38, pEBG-MKK6, pEBG-SEK1, pEBG-MEK1 and pSRα-MKK6DD are described (Takekawa et al., 1998). The reporter plasmid pGL3-p53RE was provided by S.Ishida and T.Tokino (Sapporo Medical University). pC53-SN3 expressing wild-type human p53 was provided by B.Vogelstein (Johns Hopkins University).
Northern blot analysis
Cells were exposed to various stresses as follows: γ-ray (20 Gly); UV-C (30 J/m2); MMS (100 µg/ml); anisomycin (10 µg/ml); H2O2 (0.5 mM) and sorbitol (0.3 M). Total RNA was then prepared using Trizol (Gibco-BRL). Twenty micrograms/lane of total RNA was fractionated by denaturing agarose gel electrophoresis, and transferred onto a nylon membrane. The blot was hybridized to a probe specific to Wip1 as described (Takekawa and Saito, 1998).
Expression and purification of GST fusion proteins
GST-fusion proteins were constructed using the pGEX-4T plasmid (Pharmacia). GST–ATF2, GST–c-jun and GST–Wip1 were expressed in Escherichia coli DH5 and purified using glutathione–Sepharose beads as described (Takekawa et al., 1997).
Tissue culture and transient transfection
Cells were transfected using the Effectene Transfection Reagent (Qiagen) as recommended by the manufacturer. Total amount of plasmid DNA was adjusted to 0.4 µg/plate with vector DNA (pcDNA3). The cells were harvested 30 h after transfection and, where indicated, the cells were treated with UV-C (60 J/m2 followed by a 1 h incubation at 37°C), MMS (100 µg/ml for 3 h), anisomycin (10 µg/ml for 30 min), sorbitol (0.4 M for 20 min) or PMA (100 nM for 20 min).
Immunoblotting
Either a 30 µg (for epitope-tagged MAPKs) or a 100 µg (for phosphorylated p53) aliquot of whole-cell lysates or appropriate amounts of glutathione–Sepharose affinity-purified proteins were resolved by SDS–PAGE and transferred onto nitrocellulose membranes. After blocking with 4% skimmed milk, membranes were probed with appropriate antibodies and visualized using enhanced chemiluminescence detection (Amersham). The following antibodies were used: an anti-HA monoclonal antibody (mAb) 12CA5 (Boehringer Mannheim); an anti-Flag mAb M2 (Sigma); rabbit polyclonal antisera specific to phosphorylated MKK6, SEK1, MEK1 and p38, respectively (New England Biolabs); rabbit polyclonal antibodies to MAPKAP-K2 (Sigma); goat anti-GST antibody (Pharmacia); and an anti-p53 mAb DO-1 (Santa Cruz Biotechnology).
Immunocomplex protein kinase assay
To assess the kinase activity of epitope-tagged MAPKs, transfected cells were lysed in lysis buffer containing 0.5% deoxycholate. Cell lysates were incubated with the appropriate antibody for 2 h at 4°C. Immunocomplexes were recovered with the aid of Protein G–Sepharose beads, washed twice with lysis buffer, once in 100 mM Tris–HCl pH 7.5, 0.5 M LiCl, and twice with kinase buffer. Immunoprecipitates were resuspended in 30 µl of the kinase buffer containing 3 µg of specific substrate (GST–ATF2 for p38, GST–jun for JNK1 or MBP for ERK2). The kinase reaction was initiated by addition of 10 µCi of [γ-32P]ATP and 50 µM cold ATP, as described (Takekawa et al., 1997). After 20 min incubation at 30°C, reactions were terminated by adding SDS loading buffer. The endogenous MAPKAP-kinase2 activity was monitored by an in vitro kinase assay as described (Iordanov et al., 1997) using purified Hsp25 (Sigma) as a substrate.
In vitro phosphatase reactions
To prepare phosphorylated epitope-tagged MAPKs, COS-7 cells were transfected with plasmid DNA encoding either HA-MAPK (pMT3-HA-p38, pSRα-HA-JNK1, pSRα-HA-ERK2) or GST–p38 (pEBG-p38). After stimulating the cells with UV radiation (60 J/m2), the activated and phosphorylated MAPKs were purified and used in in vitro phosphatase reactions as described (Takekawa et al., 1998). Briefly, phosphorylated HA-MAPKs were immunoprecipitated, and resuspended in 50 µl of phosphatase buffer supplemented with 1 mg/ml bovine serum albumin (BSA) with or without 30 mM MgCl2. Following addition of purified GST–Wip1, reaction mixtures were incubated at 30°C for 15 min, washed and resuspended in 30 µl of the kinase buffer. The remaining HA-MAPK activities were assayed by in vitro kinase reactions. Phosphorylated GST–p38 was immobilized on glutathione–Sepharose. After treatment with Wip1 in phosphatase buffer as described above, samples were washed with lysis buffer and resolved by SDS–PAGE. The phosphorylation status of p38 was monitored by immunoblotting with anti-dual phospho-p38 and an anti-phosphotyrosine antibody 4G10 (Transduction Laboratories).
Co-immunoprecipitation assay
Cell lysates were prepared in IP buffer containing 0.5% NP-40. Cell extracts (600 µg) were incubated with 3 µg of anti-Flag mAb M2 for 6 h at 4°C, mixed with 35 µl of Protein G–Sepharose suspension, and incubated for an additional hour. Immunoprecipitates were washed six times with IP buffer plus 0.1% NP-40, and subjected to SDS–PAGE. Immunoblot analysis was performed with the anti-HA mAb 12CA5.
Luciferase assay
Cells were transfected with a pCMV-β-galactoside reporter plasmid (0.07 µg) and a luciferase reporter plasmid (0.1 µg) driven by three copies of the p53RE motif (Tokino et al., 1994), together with 0.3 µg of the various expression plasmids as indicated using the Effectene Transfection Reagent. Following transfection the cells were incubated for 6 h, following which the medium was exchanged. Eighteen hours afterwards the cells were exposed to UV (25 J/m2) for p53 induction, and harvested 7 h after radiation for luciferase and β-galactosidase assays as described (Keller et al., 1999). Luciferase activity was measured using a luminometer (Mini Lumat LB9506) and normalized to β-galactosidase activity.
Immunofluorescence analysis
Cells growing on glass coverslips were transfected as described above. Twenty-four hours after transfection, cells were treated with UV radiation (60 J/m2), and then fixed with 3% paraformaldehyde/phosphate-buffered saline (PBS) for 10 min. Cells were permeabilized with 0.1% Triton X-100 for 5 min, and incubated in blocking solution (BlockAce; Nippon Gene) at 4°C for 2 h. Cells were then incubated with 2.5 µg/ml of anti-Flag mAb M2 and the anti-phospho-specific p38 antisera for 1 h in PBS containing 3% BSA, washed and incubated with Alexa Fluor 594-conjugated goat anti-mouse IgG (Molecular Probes) and FluoroLink Cy2-linked goat anti-rabbit IgG (Amersham) for an additional 45 min. To assess apoptotic cell death, transfected A549 cells were fixed, permeabilized and their nuclei stained with DAPI (0.025 µg/ml in PBS) as described (Takekawa and Saito, 1998).
Acknowledgments
Acknowledgements
We thank Ms Itoh and Ms Fujii for their excellent technical assistance, Drs S.Ishida, T.Tokino and B.Vogelstein for plasmids, and Drs H.Saito and P.O’Grady for critical reading of the manuscript. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan, and a grant from the Akiyama foundation.
References
- Alessi D.R., Gómez,N., Moorhead,G., Lewis,T., Keyse,S.M. and Cohen,P. (1995) Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100 in various cell lines. Curr. Biol., 5, 283–295. [DOI] [PubMed] [Google Scholar]
- Anderson N.G., Maller,J.L, Tonks,N.K. and Sturgill,T.W. (1990) Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature, 343, 651–653. [DOI] [PubMed] [Google Scholar]
- Bulavin D.V., Saito,S., Hollander,M.C., Sakaguchi,K., Anderson,C.W., Appella,E. and Fornace,A.J.,Jr (1999) Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J., 18, 6845–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari T. (2000) How to activate p53. Curr. Biol., 10, R315–R317. [DOI] [PubMed] [Google Scholar]
- Chen Y.-R., Wang,X., Templeton,D., Davis,R.J. and Tan,T.-H. (1996) The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation: duration of JNK activation may determine cell death and proliferation. J. Biol. Chem., 271, 31929–31936. [DOI] [PubMed] [Google Scholar]
- Eyers P.A., van den IJssel,P., Quinlan,R.A., Goedert,M. and Cohen P. (1999) Use of a drug-resistant mutant of stress-activated protein kinase 2a/p38 to validate the in vivo specificity of SB203580. FEBS Lett., 451, 191–196. [DOI] [PubMed] [Google Scholar]
- Fiscella M., Zhang,H., Fan,S., Sakaguchi,K., Shen,S., Mercer,W.E., Vande Woude,G.F., O’Connor,P.M. and Appella,E. (1997) Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl Acad. Sci. USA., 94, 6048–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjeld C.C. and Denu,J.M. (1999) Kinetic analysis of human serine/threonine protein phosphatase 2Cα. J. Biol. Chem., 274, 20336–20343. [DOI] [PubMed] [Google Scholar]
- Gaits F., Shiozaki,K. and Russell,P. (1997) Protein phosphatase 2C acts independently of stress-activated kinase cascade to regulate the stress response in fission yeast. J. Biol. Chem., 272, 17873–17879. [DOI] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Goedert M., Cuenda,A., Craxton,M., Jakes,R. and Cohen,P. (1997) Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); comparison of its substrate specificity with that of other SAP kinases. EMBO J., 16, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez N. and Cohen,P. (1991) Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature, 353, 170–173. [DOI] [PubMed] [Google Scholar]
- Gottlieb T.M. and Oren,M. (1996) p53 in growth control and neoplasia. Biochim. Biophys. Acta., 1287, 77–102. [DOI] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Hanada M. et al. (1998) Selective suppression of stress-activated protein kinase pathway by protein phosphatase 2C in mammalian cells. FEBS Lett., 437, 172–176. [DOI] [PubMed] [Google Scholar]
- Hazzalin C.A., Cano,E., Cuenda,A., Barratt,M.J., Cohen,P. and Mahadevan,L.C. (1996) p38/RK is essential for stress-induced nuclear responses: JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient. Curr. Biol., 6, 1028–1031. [DOI] [PubMed] [Google Scholar]
- Huang C., Ma,W.Y., Maxiner,A., Sun,Y. and Dong,Z. (1999) p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J. Biol. Chem., 274, 12229–12235. [DOI] [PubMed] [Google Scholar]
- Ip Y.T. and Davis,R.J. (1998) Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol., 10, 205–219. [DOI] [PubMed] [Google Scholar]
- Iordanov M., Bender,K., Ade,T., Schmid,W., Sachsenmaier,C., Engel,K., Gaestel,M., Rahmsdorf,H.J. and Herrlich,P. (1997) CREB is activated by UVC through a p38/HOG-1-dependent protein kinase. EMBO J., 16, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.J., Zhou,Z.X., Wang,G.W., Buridi,A. and Klein,J.B. (2000) Suppression by metallothionein of doxorubicin-induced cardio myocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J. Biol. Chem., 275, 13690–13698. [DOI] [PubMed] [Google Scholar]
- Keller D., Zeng,X., Li,X., Kapoor,M., Iordanov,M.S., Taya,Y., Lozano,G., Magun,B. and Lu,H. (1999) The p38MAPK inhibitor SB203580 alleviates ultraviolet-induced phosphorylation at serine 389 but not serine 15 and activation of p53. Biochem. Biophys. Res. Commun., 261, 464–471. [DOI] [PubMed] [Google Scholar]
- Keyse S.M. (2000) Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr. Opin. Cell Biol., 12, 186–192. [DOI] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054-1072. [DOI] [PubMed] [Google Scholar]
- Kyriakis J.M. and Avruch,J. (1996) Sounding the alarm: protein kinase cascades activated by stress and inflammation. J. Biol. Chem., 271, 24313–24316. [DOI] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Lu H., Taya,Y., Ikeda,M., and Levine,A.J. (1998) Ultraviolet radiation, but not γ radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc. Natl Acad. Sci. USA, 95, 6399–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T., Wurgler-Murphy,S.M. and Saito,H. (1994) A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature, 369, 242–245. [DOI] [PubMed] [Google Scholar]
- Marshall C.J. (1994) MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr. Opin. Genet. Dev., 4, 82–89. [DOI] [PubMed] [Google Scholar]
- Meek D.W. (1998) New developments in the multi-site phosphorylation and integration of stress signalling at p53. Int. J. Radiat. Biol., 74, 729–737. [DOI] [PubMed] [Google Scholar]
- Meskiene I., Bögre,L., Glaser,W., Balog,J., Brandstötter,M., Zwerger,K., Ammerer,G. and Hirt,H. (1998) MP2C, a plant protein phosphatase 2C, functions as a negative regulator of mitogen-activated protein kinase pathways in yeast and plants. Proc. Natl Acad. Sci. USA, 95, 1938–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A.N. and Shiozaki,K. (1999) Heat shock-induced activation of stress MAP kinase is regulated by threonine- and tyrosine-specific phosphatases. Genes Dev., 13, 1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K. et al. (2000) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell, 102, 849–862. [DOI] [PubMed] [Google Scholar]
- Prives C. and Hall,P.A. (1999) The p53 pathway. J. Pathol., 187, 112–126. [DOI] [PubMed] [Google Scholar]
- Sanchez-Prieto R., Rojas,J.M., Taya,Y. and Gutkind,J.S. (2000) A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res., 60, 2464–2472. [PubMed] [Google Scholar]
- Shiozaki K. and Russell,P. (1995) Counteractive roles of protein phosphatase 2C (PP2C) and a MAP kinase kinase homolog in osmoregulation of fission yeast. EMBO J., 14, 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B. and Karin,M. (1996) Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol., 8, 402–411. [DOI] [PubMed] [Google Scholar]
- Takekawa M. and Saito,H. (1998) A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell, 95, 521–530. [DOI] [PubMed] [Google Scholar]
- Takekawa M., Posas,F. and Saito,H. (1997) A human homolog of the yeast Ssk2/Ssk22 MAP kinase kinase kinases, MTK1, mediates stress-induced activation of the p38 and JNK pathways. EMBO J., 16, 4973–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa M., Maeda,T. and Saito,H. (1998) Protein phosphatase 2Cα inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J., 17, 4744–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokino T., Thiagalingam,S., El-Deiry,W.S., Waldman,T., Kinzler,K.W. and Vogelstein,B. (1994) p53 tagged sites from human genomic DNA. Hum. Mol. Genet., 3, 1537–1542. [DOI] [PubMed] [Google Scholar]
- Tournier C. et al. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science, 288, 870–874. [DOI] [PubMed] [Google Scholar]
- Waskiewicz A.J. and Cooper,J.A. (1995) Mitogen and stress response pathways: MAP kinase cascades and phosphatase regulation in mammals and yeast. Curr. Opin. Cell Biol., 7, 798–805. [DOI] [PubMed] [Google Scholar]
- Wurgler-Murphy S.M., Maeda,T., Witten,E.A. and Saito,H. (1997) Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol. Cell. Biol., 17, 1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.D., Kuan,C.Y., Whitmarsh,A.J., Rincon,M., Zheng,T.S., Davis,R.J., Rakic,P. and Flavell,R.A. (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature, 389, 865–870. [DOI] [PubMed] [Google Scholar]
- Zanke B.W., Boudreau,K., Rubie,E., Winnett,E., Tibbles,L.A., Zon,L., Kyriakis,J., Liu,F.F. and Woodgett,J.R. (1996) The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr. Biol., 6, 606–613. [DOI] [PubMed] [Google Scholar]